
Abstract
We report a case of Wilson disease (WD) with dilated cardiomyopathy in which whole-genome sequencing (WGS) revealed the rare co-occurrence of two novel compound heterozygous ATP7B pathogenic variants (NM_001005918.3:c.2250del/p.N751Tfs*9 and c.3496C>T/p.L1166F) and a known FLNC pathogenic variant. Our results highlight the usefulness of WGS, even in the diagnosis of well-characterized genetic diseases such as WD.
Similar content being viewed by others
Wilson disease (WD) is an autosomal recessive neurodegenerative disorder affecting copper metabolism (MIM #277900) via pathogenic variants of the ATP7B gene. The mutated gene prevents proper function of the copper transport protein, leading to copper accumulation in the liver, brain, kidneys, and skeletal system1. Notably, pathogenic variants that eliminate ATP7B tend to result in more severe and specific phenotypes than missense variants2,3.
The clinical features of WD are highly variable. Although most symptomatic patients present with hepatic and/or neuropsychiatric dysfunction, WD can affect almost any organ in the body, including the heart, resulting in a very complex clinical profile in individuals4. The variety of cardiac symptoms observed in some WD patients include arrhythmia, cardiomyopathy, and autonomic dysfunction. Structural and functional changes in the heart in WD patients have been reported to manifest primarily as mild left ventricular hypertrophy and diastolic functional changes, which are more apparent in patients with neurological WD5. WD patients with severe clinical manifestations may present with cardiac symptoms and are at greater risk for developing structural heart disease6. However, the possibility of complications of other genetic diseases that may be masked by the diverse clinical manifestations of WD patients should receive more attention. Here, we report a rare case of WD with FLNC-related cardiomyopathy.
A 50-year-old woman visited another clinic due to a 20-year history of exertional shortness of breath. Echocardiography revealed a dilated left ventricular (LV) end-diastolic diameter (65 mm), a dilated LV end-systolic diameter (55 mm), a diffusely hypokinetic left ventricle with a depressed ejection fraction (45–50%), and severe mitral valve regurgitation. Coronary angiography revealed no coronary artery stenosis. Although cardiovascular MRI and myocardial scintigraphy were not performed to assess the cardiac lesions, the patient was clinically diagnosed with dilated cardiomyopathy (DCM). Despite optimal medical therapies, the functional status of her DCM deteriorated (ejection fraction, 30–35%), and she underwent mitral valve repair at our hospital.
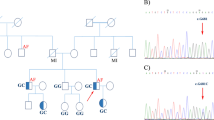
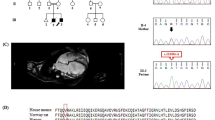
At 58 years of age, the patient noticed shaking in her hands and consulted a neurologist. During the neurological examination, she exhibited postural and kinetic tremors in her upper limbs. By age 68, she struggled with tasks such as opening or closing a water tap, operating a TV remote control/smartphone, and brushing her teeth. Additionally, she exhibited rigidity in all her limbs, leading to her admission to the neurological department. The patient’s tremors and rigidity were mild and did not significantly impair movement. However, she was unable to explain how to use necessities of daily living and could not use them correctly. Tests revealed low ceruloplasmin (8.8 mg/dL; reference range, 30–58 mg/dL) and copper (21 µg/dL; reference range, 70–140 µg/dL) blood concentrations and high urine copper excretion (2985 µg/day; reference range, 15–60 µg/day; 2000 mL urine). An ophthalmologist observed bilateral Kayser-Fleischer rings, and an MRI of the brain showed hyperintensity in the white matter, which is atypical in WD, and in the basal ganglia (Fig. 1A–C). Based on these findings, the patient was diagnosed with WD. While no family history of WD or other neurological or psychiatric disorders was reported, the patient’s son died at age 30 due to DCM, although medical details are unknown. Symptoms of WD and DCM developed earlier than the relatively late onset of neuropsychiatric symptoms, and the family history of DCM in the patient’s son prompted us to perform whole-genome sequencing (WGS) to diagnose the patient (Fig. 2A).

A FLAIR image showing periventricular hyperintensity and deep and subcortical white matter hyperintensity (axial; 1.5 T; TR, 8000 ms; TE, 100 ms). B DWI showing high-intensity signals (arrows) in the region of the corticomedullary junction in the bilateral occipital lobes (axial; 1.5 T; TR, 3732 ms; TE, 89 ms). C FLAIR image showing basal ganglia hyperintensity (axial; 1.5 T; TR, 8000 ms; TE, 100 ms).

A Family pedigree of the patient. The proband’s mother died at the age of 74 due to pancreatic cancer. The father died at the age of 54 due to unknown causes. The patient’s son died at the age of 30 due to DCM. B Sanger sequencing confirmed these variants.
WGS and variant filtering were performed as described previously7. DNA samples were obtained from the patient’s peripheral blood using a QIAamp® DNA Blood Midi/Maxi kit (Qiagen), and DNA libraries were prepared with the TruSeq DNA PCR-Free HT Library Prep Kit with a 550-bp insertion size. WGS was conducted using an Illumina NovaSeq 6000 instrument at Takara Bio Laboratory. Data processing was carried out using Parabricks® Pipeline version 3.5.0 (NVIDIA Clara™) at Takara Bio Inc., as previously described7. Variant annotation was conducted using ANNOVAR (https://annovar.openbioinformatics.org/en/latest/). WGS revealed heterozygous variants, NM_001005918.3:c.2250del/p.(N751Tfs*9) and c.3496C>T/p.(L1166F), in ATP7B responsible for WD. Sanger sequencing confirmed these variants (Fig. 2B). The variants were confirmed to be in trans by long-read sequencing (Supplementary Methods and Supplementary Fig. 1), suggesting that the WD patient was a compound heterozygote. Neither variant was reported in reference population databases or disease-related databases. NM_001005918:c.3496C>T was predicted to impair protein function according to in silico analyses. According to the guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP guidelines)8, we classified NM_001005918.3:c.2250del as “pathogenic” (PVS1, PM2, and PP4) and NM_001005918.3:c.3496C>T as “likely pathogenic” (PM2, PM3, PP3, and PP4). Therefore, the patient was determined to be a compound heterozygote with two different novel pathogenic variants in ATP7B. In addition, we identified a heterozygous canonical splice site variant, NM_001458.5:c.5199+1G>T, potentially resulting in protein truncation, in FLNC, which is responsible for cardiomyopathy (MIM #617047) and muscular diseases (MIM #609524 and #614065). DNA from the patient’s deceased son, who succumbed to DCM at the age of 30 years, was unavailable. This variant was rarely observed in population cohorts in the Genome Aggregation Database (gnomAD, 0.0009%) and was not found in the WGS database of 9850 Japanese control individuals7. Therefore, this variant can be classified as “likely pathogenic” (PM1, PM2 supportive, PP3, PP4, and PP5) according to the ACMG/AMP guidelines8. This variant was also reported as likely pathogenic by two submitters in ClinVar.
In conclusion, we reported a case illustrating the exceptionally rare co-occurrence of WD and FLNC-related cardiomyopathy. This report emphasizes the importance of comprehensive gene analysis, even in the diagnosis of well-characterized genetic diseases such as WD.
HGV database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.3403, https://doi.org/10.6084/m9.figshare.hgv.3406, https://doi.org/10.6084/m9.figshare.hgv.3409
References
Bull, P. C., Thomas, G. R., Rommens, J. M., Forbes, J. R. & Cox, D. W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat. Genet. 5, 327–337 (1993).
Deguti, M. M. et al. Wilson disease: novel mutations in the ATP7B gene and clinical correlation in Brazilian patients. Hum. Mutat. 23, 398 (2004).
Liu, X. Q. et al. Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease. World J. Gastroenterol. 10, 590–593 (2004).
Yuan, X. Z., Yang, R. M. & Wang, X. P. Management perspective of wilson’s disease: early diagnosis and individualized therapy. Curr. Neuropharmacol. 19, 465–485 (2021).
Buksinska-Lisik, M., Litwin, T., Pasierski, T. & Czlonkowska, A. Cardiac assessment in Wilson’s disease patients based on electrocardiography and echocardiography examination. Arch. Med. Sci. 15, 857–864 (2019).
Quick, S. et al. Cardiac manifestation of Wilson’s disease. J. Am. Coll. Cardiol. 72, 2808–2809 (2018).
Kaburagi, K. et al. A Novel NODAL variant in a young embolic stroke patient with visceral heterotaxy. BMC Neurol. 24, 119 (2024).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Acknowledgements
We are grateful to the patient for participating in our study. We thank Eri Nonoyama, Ritsuko Oikawa, and Yoshiko Miyake for their technical assistance. Additionally, we are thankful for the partial support provided by the supercomputer at the ROIS National Institute of Genetics for computation. This work was supported by grants from the Practical Research Project for Rare/Intractable. Diseases of the Japan Agency for Medical Research and Development (AMED, No.JP23ek0109529, JP23ek0109548, 24ek0109617, 24ek0109735, and 24ek0109759), Rare and Intractable Diseases from the Ministry of Health, Labour and Welfare of Japan (No. JPMH22FC1013), and Japan Society for the Promotion of Science (JSPS) KAKENHI (No. JP22H02987). We thank the patient and his parents for participating in our study. The computations were partially performed on the NIG supercomputer at the ROIS National Institute of Genetics.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the institutional review board of the St. Marianna University School of Medicine (#4983).
Informed consent
Written informed consent was obtained from the patient.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Imai, T., Mitsuhashi, S., Isahaya, K. et al. Wilson disease (novel ATP7B variants) with concomitant FLNC-related cardiomyopathy. Hum Genome Var 11, 34 (2024). https://doi.org/10.1038/s41439-024-00283-y
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-024-00283-y
- Springer Nature Limited