
Abstract
Atrial fibrosis occurs frequently with structural heart disease and is considered as a major cause of arrhythmia. Microarray-based profiling predicted the differential expression of SPP1 in atrial fibrosis. Herein, we aimed to analyze the role of shRNA-mediated SPP1 knockdown in the progression of atrial fibrosis as well as the downstream mechanism. In vivo model in mice and in vitro HL-1 cell model of atrial fibrosis were developed by the angiotensin II (Ang II) method, where SPP1 expression was validated by RT-qPCR. Gain- and loss-of-function experiments were performed in Ang II-induced mice and HL-1 cells to evaluate the effect of the SPP1/TGF-β/SREBP2/PCSK9 axis on cell viability, apoptosis, collagen production and mitochondrial DNA (mtDNA) damage in atrial fibrosis. Expression of SPP1, TGF-β, SREBP2 and PCSK9 was increased in Ang II-induced mice and HL-1 cells. Silencing of SPP1 inhibited the occurrence of atrial fibrosis, as reflected by attenuated cell viability and collagen production as well as increased cell apoptosis. Conversely, upregulated SPP1 enhanced atrial fibrosis, which was related to upregulation of TGF-β. In addition, TGF-β elevated the expression of SREBP2, which promoted mtDNA damage and the consequent atrial fibrosis by augmenting the expression of PCSK9. This study uncovers previously unrecognized pro-fibrotic activities of SPP1 in atrial fibrosis, which is achieved through activation of the TGF-β/SREBP2/PCSK9 signaling pathway and promotion of mtDNA damage.
Similar content being viewed by others


Introduction
Atrial fibrillation is a common type of sustained arrhythmia [1]. Atrial fibrosis is considered as the main substrate for atrial fibrillation maintenance and has been linked to fewer favorable outcomes of atrial fibrillation [2]. Therefore, atrial fibrosis remains a significant target of therapy for patients with atrial fibrillation, and a deepened understanding of the pathogenesis is thus needed.
Secreted phosphoprotein 1 (SPP1), also known as osteopontin, has been implicated in the fibrosis process, including atrial fibrosis [3, 4]. SPP1 expression increases in the heart during hypertrophy and heart failure [5] while silencing of SPP1 leads to reduced heart-to-body ratio, myocyte size and fibrosis in cardiac tissues [6]. SPP1 is capable of promoting the expression and activation of transforming growth factor-β 1 (TGF-β1), increasing the production of fibrous matrix proteins in lung cells exposed to multi-walled carbon nanotubes [7]. TGF-β has been detected to be upregulated in CD14++ CD16+ and CD14+ CD16++ monocytes in patients with atrial fibrillation and severe atrial fibrosis [8]. Meanwhile, activation of the TGF-β pathway contributes to atrial fibrosis in an atrial fibrillation experimental model and patients [9].
Additionally, treatment with TGF-β1 in the cultured chondrocytes induces an increase of expression of sterol regulatory element-binding protein 2 (SREBP2) [10]. SREBP2 has been involved in the development of cardiovascular diseases [11], during which, mitochondrial DNA (mtDNA) damage plays a central role [12]. More importantly, augmented cell mtDNA damage is essential for fibrosis [13]. SREBP2 pathway activation contributes to an increase in the expression of proprotein convertase subtilisin/kexin type 9 (PCSK9) in different organs [14]. PCSK9 has demonstrated a close correlation with cardiac diseases and their related risk factors, such as arrhythmia, atrial fibrillation and hypertension [15]. In addition, PCSK9 enhances mtDNA damage during chronic myocardial ischemia [16]. These published literature reports allowed us to hypothesize and then extensively validate an unexpected role for SPP1 in controlling the mtDNA damage in atrial fibrosis by regulating the TGF-β/SREBP2/PCSK9 axis.
Results
SPP1 is highly expressed in mouse and cell models of atrial fibrosis
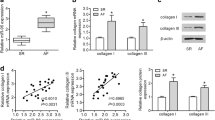
First, we constructed a mouse model of atrial fibrosis. The results of HE staining, Masson’s trichrome staining and IHC staining showed disordered atrial muscle fibers, enlarged internuclear space, and increased atrial fibrosis and collagen in the mice with atrial fibrosis compared with the normal mice (Fig. 1A–C). Additionally, results of CCK-8 assay, western blot analysis and TUNEL staining displayed augmented cell viability and collagen production as well as reduced apoptosis in Ang II-induced cells (Fig. 1D–F, Supplemental Fig. 1A). These results indicate that mouse and cell models of atrial fibrosis were successfully constructed.

A HE staining of pathological changes of atrial tissues in normal and Ang II-treated mice. B Masson’s trichrome staining of atrial fibrosis degree in normal and Ang II-treated mice. Collagen fibers were in blue and muscle fibers were in red. C IHC analysis of collagen I and collagen III proteins in atrial tissues of normal and Ang II-treated mice. D CCK-8 detection of cell viability in control and Ang II-induced HL-1 cells. E TUNEL detection of apoptosis of control and Ang II-induced HL-1 cells. F Western blot analysis of collagen I and collagen III proteins in control and Ang II-induced HL-1 cells. G A volcano plot of differential genes in the GSE27133 dataset. The abscissa indicates −log10 (p-value), and the ordinate indicates log2FC value. H Venn diagram of the DEGs and atrial fibrosis-related genes. I A heat map of key candidate gene expression. The color scale from blue to red indicates the increasing expression value. J SPP1 expression in atrial fibrosis samples in the GSE27133 dataset. K RT-qPCR detection of SPP1 expression in atrial tissues of normal and Ang II-treated mice. L RT-qPCR detection of SPP1 expression in control and Ang II-induced HL-1 cells. *p < 0.05, compared with the normal mice or control HL-1 cells. All experiments were repeated three times independently. n = 10 for mice in each group.
Profiling of the GSE27133 dataset revealed 201 DEGs, including 112 highly expressed genes and 89 poorly expressed genes (Fig. 1G). A total of 2089 atrial fibrosis-related genes were obtained from the GeneCards database, which were then subjected to intersection analysis with the DEGs, with 21 candidate key genes identified (Fig. 1H). A heat map of these candidate genes is shown in Fig. 1I, which contained LOX, RBP4 and SPP1, of which SPP1 was found to be significantly highly expressed in atrial fibrosis in the GSE27133 dataset (Fig. 1J). Meanwhile, RT-qPCR data also revealed upregulated SPP1 expression in the atrial tissues of mouse models and Ang II-induced HL-1 cells (Fig. 1K, L). The above results indicate that SPP1 is robustly induced in the mouse and cell models of atrial fibrosis.
Silencing of SPP1 arrests the development of atrial fibrosis
Next, we moved to determine the effect of SPP1 on atrial fibrosis. RT-qPCR results showed that the expression of SPP1 was reduced in cells treated with sh-SPP1-1 or sh-SPP1-2, of which sh-SPP1-1 had superior silencing efficiency (Fig. 2A) and was thus used for subsequent experiments.

A RT-qPCR detection of SPP1 expression in HL-1 cells treated with sh-SPP1-1 or sh-SPP1-2. B CCK-8 detection of cell viability in HL-1 cells treated with sh-SPP1. C TUNEL detection of apoptosis of HL-1 cells treated with sh-SPP1. D Western blot analysis of collagen I and collagen III proteins in HL-1 cells treated with sh-SPP1. E HE staining of pathological changes of atrial tissues of mice treated with sh-SPP1. F Masson’s trichrome staining of atrial fibrosis degree in mice treated with sh-SPP1. G IHC analysis of collagen I and collagen III proteins in atrial tissues of mice treated with sh-SPP1. *p < 0.05, compared with the Ang II-induced HL-1 cells or mice treated with sh-NC. All experiments were repeated three times independently. n = 10 for mice in each group.
Results of CCK-8, western blot analysis and TUNEL staining revealed declines in cell viability and collagen production yet an increase in cell apoptosis in response to the absence of SPP1 (Fig. 2B–D, Supplemental Fig. 1B). Moreover, the results of HE staining, Masson’s trichrome staining and IHC staining presented that SPP1 silencing reduced atrial fibrosis and collagen production in mice (Fig. 2E–G). The aforementioned results provide evidence that silencing of SPP1 can inhibit the occurrence of atrial fibrosis.
SPP1 promotes atrial fibrosis by upregulating TGF-β
The next focus was on analyzing the downstream signaling pathway of SPP1. The Coxpresdb database yielded 1000 co-expressed genes of SPP1 and following Venn diagram analysis with atrial fibrosis-related genes, 165 candidate genes were obtained (Fig. 3A). KEGG enrichment analysis on these candidate genes demonstrated that they were mainly involved in the PI3K-Akt, IL-17 and TGF-β signaling pathways (Fig. 3B).

A Venn diagram of co-expressed genes of SPP1 in the Coxpresdb database and atrial fibrosis-related genes. B KEGG enrichment analysis on candidate genes. The abscissa represents GeneRatio. C Correlation between SPP1 and TGF-β in heart tissues analyzed by the Chipbase v2.0 database (r = 0.6641, p-value = 3.65e−49). D RT-qPCR detection of TGF-β expression in atrial tissues of normal and Ang II-treated mice. E RT-qPCR detection of TGF-β expression in control and Ang II-induced HL-1 cells. F RT-qPCR detection of TGF-β expression in HL-1 cells treated with sh-SPP1. G Western blot analysis of TGF-β protein in HL-1 cells treated with sh-SPP1. H RT-qPCR detection of TGF-β expression in HL-1 cells treated with oe-TGF-β. I Western blot analysis of TGF-β protein in HL-1 cells treated with oe-TGF-β. J RT-qPCR detection of SPP1 and TGF-β expression in HL-1 cells treated with sh-SPP1 or combined with oe-TGF-β. K CCK-8 detection of cell viability in HL-1 cells treated with sh-SPP1 or combined with oe-TGF-β. L TUNEL detection of apoptosis of HL-1 cells treated with sh-SPP1 or combined with oe-TGF-β. M Western blot analysis of collagen I and collagen III proteins in HL-1 cells treated with sh-SPP1 alone or in combination with oe-TGF-β. N HE staining of pathological changes of atrial tissues of Ang II-induced mice treated with sh-SPP1 or combined with oe-TGF-β. O Masson’s trichrome staining of atrial fibrosis degree in Ang II-induced mice treated with sh-SPP1 or combined with oe-TGF-β. P IHC analysis of collagen I and collagen III proteins in atrial tissues of Ang II-induced mice treated with sh-SPP1 or combined with oe-TGF-β. *p < 0.05, compared with the normal mice, control HL-1 cells, Ang II-induced HL-1 cells treated with sh-NC, oe-NC or sh-NC + oe-NC or Ang II-induced mice treated with sh-NC + oe-NC. # p < 0.05, compared with the Ang II-induced HL-1 cells or mice treated with sh-SPP1 + oe-NC. All experiments were repeated three times independently. n = 10 for mice in each group.
Moreover, Chipbase v2.0 database retrieval indicated positive correlation between SPP1 and TGF-β in heart tissues (r = 0.6641, p-value = 3.65e−49) (Fig. 3C). Results of RT-qPCR showed upregulated TGF-β expression in both mouse and cell models of atrial fibrosis (Fig. 3D, E). Notably, TGF-β expression was reduced in the sh-SPP1-treated cells (Fig. 3F–H, Supplemental Fig. 1C). These results indicate that SPP1 can upregulate TGF-β expression.
As confirmed by RT-qPCR and western blot analyses, TGF-β level was up-regulated in the oe-TGF-β-treated cells (Fig. 3H, I, Supplemental Fig. 1D). In addition, the mRNA expression of SPP1 and TGF-β was decreased in the absence of SPP1, while further overexpression of TGF-β enhanced the TGF-β mRNA expression (Fig. 3J). As depicted in Fig. 3K–M (Supplemental Fig. 1E), the inhibiting effects of SPP1 silencing on cell viability and collagen production along with the promoting effect on cell apoptosis were abolished by further overexpression of TGF-β.
The results of HE staining, Masson’s trichrome staining and IHC staining exhibited that SPP1 silencing weakened atrial fibrosis and collagen production whereas additional overexpression of TGF-β increased them (Fig. 3N–P). Overall, these findings support that SPP1 may promote the occurrence of atrial fibrosis by upregulating TGF-β.
TGF-β promotes atrial fibrosis by upregulating SREBP2
Subsequently, we focused on exploring the downstream factor of TGF-β. In the Chipbase v2.0 database, TGF-β and SREBP2 were found to be positively correlated with each other in heart tissues (r = 0.3088, p-value = 9.47e−10) (Fig. 4A). RT-qPCR results displayed amplified SREBP2 expression in both mouse and cell models of atrial fibrosis (Fig. 4B, C). In addition, overexpression of TGF-β in Ang II-induced HL-1 cells could up-regulate SREBP2 protein and mRNA expression (Fig. 4D, E, Supplemental Fig. 1F).

A Correlation between TGF-β and SREBP2 in heart tissues analyzed by the Chipbase v2.0 database (r = 0.3088, p-value = 9.47e−10). B RT-qPCR detection of SREBP2 expression in atrial tissues of normal and Ang II-treated mice. C RT-qPCR detection of SREBP2 expression in control and Ang II-induced HL-1 cells. D RT-qPCR detection of SREBP2 expression in HL-1 cells treated with oe-SREBP2. E Western blot analysis of SREBP2 protein in HL-1 cells treated with oe-SREBP2. F RT-qPCR detection of TGF-β expression in HL-1 cells treated with sh-TGF-β-1 or sh-TGF-β-2. G RT-qPCR detection of SREBP2 expression in HL-1 cells treated with oe-SREBP2. H RT-qPCR detection of SREBP2 and TGF-β expression in HL-1 cells treated with sh-TGF-β or combined with oe-SREBP2. I CCK-8 detection of cell viability in HL-1 cells treated with sh-TGF-β or combined with oe-SREBP2. J TUNEL detection of apoptosis of HL-1 cells treated with sh-TGF-β or combined with oe-SREBP2. K Western blot analysis of collagen I and collagen III proteins in HL-1 cells treated with sh-TGF-β alone or in combination with oe-SREBP2. L HE staining of pathological changes of atrial tissues of Ang II-induced mice treated with sh-TGF-β or combined with oe-SREBP2. M Masson’s trichrome staining of atrial fibrosis degree in Ang II-induced mice treated with sh-TGF-β or combined with oe-SREBP2. N IHC analysis of collagen I and collagen III proteins in atrial tissues of Ang II-induced mice treated with sh-TGF-β or combined with oe-SREBP2. *p < 0.05, compared with the normal mice, control HL-1 cells, Ang II-induced HL-1 cells treated with sh-NC, oe-NC or sh-NC + oe-NC or Ang II-induced mice treated with sh-NC + oe-NC. # p < 0.05, compared with the Ang II-induced HL-1 cells or mice treated with sh-TGF-β + oe-NC. All experiments were repeated three times independently. n = 10 for mice in each group.
Silencing efficiency of two sh-TGF-β sequences was validated by RT-qPCR, which revealed a decline of TGF-β expression in cells treated with sh-TGF-β−1 or sh-TGF-β−2, with sh-TGF-β−1 having better silencing efficiency (Fig. 4F), and was therefore used for follow-up experiments. Additionally, the mRNA expression of SREBP2 was increased in the oe-SREBP2-treated cells (Fig. 4G). In contrast, the mRNA expression of TGF-β and SREBP2 was downregulated in the absence of TGF-β while SREBP2 expression was elevated in response to dual treatment with sh-TGF-β and oe-SREBP2 (Fig. 4H). Moreover, as shown in Fig. 4I–K and Supplemental Fig. 1G, treatment with sh-TGF-β suppressed cell viability and collagen production, and promoted cell apoptosis, effects of which were negated by further overexpression of SREBP2.
Furthermore, the results of HE staining, Masson’s trichrome staining and IHC staining exhibited that TGF-β silencing reduced atrial fibrosis and collagen production whereas additional overexpression of SREBP2 resulted in opposite results (Fig. 4L–N). Cumulatively, TGF-β can promote atrial fibrosis by elevating SREBP2 expression.
SREBP2 triggers mtDNA damage by upregulating PCSK9 expression in vitro
The detection results of mtDNA damage and mtROS showed an increase in Ang II-induced HL-1 cells (Fig. 5A, B). PCSK9 overexpression occurred in both mouse and cell models of atrial fibrosis (Fig. 5C, D). The expression of PCSK9 was enhanced following SREBP2 overexpression (Fig. 5E). As illustrated in Fig. 5F, t the expression of SREBP2 was reduced in the presence of sh-SREBP2-1 or sh-SREBP2-2, and sh-SREBP2-1 was selected for subsequent experiments due to a superior silencing efficiency.

A mtDNA damage measurement in control and Ang II-induced HL-1 cells. B mtROS production measurement in control and Ang II-induced HL-1 cells. C RT-qPCR detection of PCSK9 expression in atrial tissues of normal and Ang II-treated mice. D RT-qPCR detection of PCSK9 expression in control and Ang II-induced HL-1 cells. E RT-qPCR detection of PCSK9 expression in HL-1 cells treated with oe-SREBP2. F RT-qPCR detection of SREBP2 expression in HL-1 cells treated with sh-SREBP2-1 or sh-SREBP2-2. G RT-qPCR detection of PCSK9 expression in HL-1 cells treated with oe-PCSK9. H RT-qPCR detection of SREBP2 and PCSK9 expression in HL-1 cells treated with sh-SREBP2 or combined with oe-PCSK9. I mtDNA damage measurement in HL-1 cells treated with sh-SREBP2 or combined with oe-PCSK9. J mtROS production measurement in HL-1 cells treated with sh-SREBP2 or combined with oe-PCSK9. *p < 0.05, compared with the normal mice, control HL-1 cells, Ang II-induced HL-1 cells treated with sh-NC, oe-NC or sh-NC + oe-NC. # p < 0.05, compared with the Ang II-induced HL-1 cells treated with sh-SREBP2 + oe-NC. All experiments were repeated three times independently. n = 10 for mice in each group.
In addition, the mRNA expression of PCSK9 was increased in the oe-PCSK9-treated cells (Fig. 5G). A decline was noted in the mRNA expression of SREBP2 and PCSK9 in the absence of SREBP2 while PCSK9 mRNA expression was elevated in the presence of sh-SREBP2 + oe-PCSK9 (Fig. 5H). Besides, silencing of SREBP2 led to inhibited mtDNA damage and mtROS production while contrary results were found following co-treatment with sh-SREBP2 and oe-PCSK9 (Fig. 5I, J). These results indicate that SREBP2 may induce mtDNA damage by upregulating PCSK9 expression.
SREBP2 enhances atrial fibrosis by upregulating PCSK9 expression in vivo
The following experiments focused at exploring whether SREBP2 enhanced atrial fibrosis in mice by elevating PCSK9. The results of CCK-8, western blot analysis and TUNEL staining showed declines in cell viability and collagen production yet an elevation in cell apoptosis upon silencing of SREBP2, while combined treatment with sh-SREBP2 and oe-PCSK9 reversed the effects of sh-SREBP2 treatment alone (Fig. 6A–C, Supplemental Fig. 1H). Additionally, dual treatment with sh-SREBP2 and oe-PCSK9 abolished the inhibitory role of SREBP2 silencing alone in the atrial fibrosis and collagen production (Fig. 6D–F). Collectively, these data evidence that the promoting effect of SREBP2 on the atrial fibrosis may be attributed to the upregulation of PCSK9.

A CCK-8 detection of cell viability in HL-1 cells treated with sh-SREBP2 or combined with oe-PCSK9. B TUNEL detection of apoptosis of HL-1 cells treated with sh-SREBP2 or combined with oe-PCSK9. C Western blot analysis of collagen I and collagen III proteins in HL-1 cells treated with sh-SREBP2 alone or in combination with oe-PCSK9. D HE staining of pathological changes of atrial tissues of Ang II-induced mice treated with sh-SREBP2 or combined with oe-PCSK9. E Masson’s trichrome staining of atrial fibrosis degree in Ang II-induced mice treated with sh-SREBP2 or combined with oe-PCSK9. F IHC analysis of collagen I and collagen III proteins in atrial tissues of Ang II-induced mice treated with sh-SREBP2 or combined with oe-PCSK9. *p < 0.05, compared with Ang II-induced mice treated with sh-NC + oe-NC. # p < 0.05, compared with the Ang II-induced mice treated with sh-SREBP2 + oe-NC. All experiments were repeated three times independently. n = 10 for mice in each group.
SPP1 induces mtDNA damage in atrial fibrosis by inactivating the TGF-β/SREBP2/PCSK9 axis
Finally, we aimed to characterize whether SPP1 induces mtDNA damage and the resultant atrial fibrosis by promoting the TGF-β/SREBP2/PCSK9 axis. RT-qPCR results showed that the expression of SPP1, TGF-β, SREBP2, and PCSK9 was reduced following silencing of SPP1 while PCSK9 expression was augmented upon further overexpression of PCSK9 (Fig. 7A).

A RT-qPCR detection of SPP1, TGF-β, SREBP2 and PCSK9 expression in Ang II-induced HL-1 cells treated with sh-SPP1 or combined with oe-PCSK9. B mtDNA damage measurement in Ang II-induced HL-1 cells treated with sh-SPP1 or combined with oe-PCSK9. C mtROS production measurement in Ang II-induced HL-1 cells treated with sh-SPP1 or combined with oe-PCSK9. D CCK-8 detection of cell viability in HL-1 cells treated with sh-SPP1 or combined with oe-PCSK9. E TUNEL detection of apoptosis of HL-1 cells treated with sh-SPP1 or combined with oe-PCSK9. F Western blot analysis of collagen I and collagen III proteins in HL-1 cells treated with sh-SPP1 alone or in combination with oe-PCSK9. G HE staining of pathological changes of atrial tissues of Ang II-induced mice treated with sh-SPP1 or combined with oe-PCSK9. H Masson’s trichrome staining of atrial fibrosis degree in Ang II-induced mice treated with sh-SPP1 or combined with oe-PCSK9. I IHC analysis of collagen I and collagen III proteins in atrial tissues of Ang II-induced mice treated with sh-SPP1 or combined with oe-PCSK9. *p < 0.05, compared with Ang II-induced cells or mice treated with sh-NC + oe-NC. # p < 0.05, compared with the Ang II-induced cells or mice treated with sh-SPP1 + oe-NC. All experiments were repeated three times independently. n = 10 for mice in each group.
Moreover, silencing of SPP1 reduced mtDNA damage and mtROS production, which were abrogated following combined treatment with sh-SPP1 and oe-PCSK9 (Fig. 7B, C). The results of CCK-8, western blot analysis and TUNEL staining revealed declines in cell viability and collagen production yet an increase of cell apoptosis in response to SPP1 silencing, the effect of which was counterweighed by further treatment with oe-PCSK9 (Fig. 7D–F, Supplemental Fig. 1I).
The results of HE staining, Masson’s trichrome staining and IHC staining indicated a reduction in atrial fibrosis and collagen production in the presence of SPP1 silencing while a contrasting result was observed following dual treatment with sh-SPP1 and oe-PCSK9 (Fig. 7G–I). Taken together, these lines of evidence indicate that SPP1 can result in mtDNA damage by activating the TGF-β/SREBP2/PCSK9 axis, thereby promoting atrial fibrosis.
Discussion
Atrial fibrosis is recognized as a hallmark of atrial fibrillation-related structural remodeling and a contributor to the perpetuation of atrial fibrillation [17]. With the aim to further elucidate the pathogenesis of atrial fibrillation, this study illuminate a mechanism by which SPP1 leads to mtDNA damage via the TGF-β/SREBP2/PCSK9 axis and thereby promotes atrial fibrosis.
Our initial results demonstrated robustly induced SPP1 in the mouse and cell models of atrial fibrosis and that silencing of SPP1 could arrest the development of atrial fibrosis as shown by declines in cell viability and collagen production yet an increase in cell apoptosis in the absence of SPP1. A recent study revealed an increased expression of SPP1 in atrial fibrillation patients with higher degree of atrial fibrosis, and furthermore, OPN can promote the proliferation of human atrial fibroblasts and increase the production of collagen I, thus inducing atrial fibrosis [4]. In addition, previously reported data also suggested markedly upregulated SPP1 expression in mice after Ang II infusion and that SPP1 deficiency leads to lessened cardiac fibrosis and reduced collagen production after Ang II infusion [18]. The aforementioned results indicate that SPP1 inhibition may serve as a contributor to alleviating atrial fibrosis.
Further analysis showed that SPP1 promoted atrial fibrosis by upregulating expression of TGF-β. Consistently, recently published data have highlighted that SPP1 promotes atrial fibrosis in mdx fibroblasts by enhancing TGF-β signaling [19]. Moreover, the protein expression of TGF-β1 is significantly augmented in atrial muscle tissues of Ang II-treated mice and TGF-β1 activation has the potential to facilitate atrial fibrosis [20]. Notably, the promoting effect of TGF-β on the atrial fibrosis was demonstrated in the current study to be associated with the elevated expression of SREBP2. In line with our results, TGF-β has been documented to induce SREBP2 pathway activation through promotion of ITGAV and PI3K in chondrocytes [21]. SREBP2 is upregulated in patients with coronary artery disease [22], and furthermore, its promoting role in fibrosis has been largely reported [23, 24]. However, little is known in its correlation with the atrial fibrosis, which requires further investigation.
Another important observation in the present study revealed that SREBP2 triggered mtDNA damage and the ensuing atrial fibrosis by upregulating PCSK9 expression. It has been well-documented that mtDNA damage is necessary for the development of fibrosis [25, 26]. SREBP2 inhibition may be related to the decreased mitochondrial cholesterol accumulation and cytosolic mtDNA damage in bone-marrow-derived macrophages treated with lipopolysaccharide [27]. In agreement with the present results, attenuated PCSK9 expression in HepG2 cells has been associated with activation of SREBP2 [28]. Meanwhile, siRNA-mediated inhibition of PCSK9 reduces mtDNA damage while its enhancement increases mtDNA damage [29]. Besides, aortic calcification is accompanied by elevated SPP1 expression in aortic tissues and policosanol has anti-calcifying effect through inhibition of PCSK9 [30], and thus, it could be concluded a positive correlation between SPP1 and PCSK9. Therefore, targeting the SPP1/TGF-β/SREBP2/PCSK9 axis could be a future direction to develop a novel intervention tool for atrial fibrosis.
Taken together, we provide the evidence that SPP1 contributed to mtDNA damage and the resultant atrial fibrosis. The underlying regulatory mechanism is that SPP1 exited its function through acting as a promoting factor for TGF-β and increasing the activity of SREBP2/PCSK9, thus forming a SPP1/TGF-β/SREBP2/PCSK9 axis to participate in atrial fibrosis (Fig. 8). These findings provide a new molecular theoretical basis for the understanding into the pathogenesis of atrial fibrosis.

SPP1 activates the TGF-β/SREBP2 signaling pathway, thus elevating the expression of PCSK9. By this mechanism, the mtDNA damage is induced and the subsequent atrial fibrosis is ultimately promoted.
Materials and methods
Ethics statement
The current study was approved by the Animal Ethics Committee of Ningbo First Hospital and performed in strict accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. Extensive efforts were made to ensure minimal suffering of the animals used in the study.
Establishment of mouse models of atrial fibrosis
Totally, 120 male C57BL/6 J mice (aged 6–7 weeks old, Vital River Laboratory Animal Technology Co., Ltd., Beijing, China) were raised in a specific pathogen-free mouse room at 22–24 °C with a 12 h light/dark cycle. The mice used for model establishment were injected intraperitoneally with 1.5 μg/g angiotensin II (Ang II; HY-13948, MedChemExpress) every day, and the normal mice were injected with the same amount of phosphate-buffered saline (PBS). After 4 weeks of Ang II treatment, the mice were injected with 20 μL of 3 × 1011 v.g/mL corresponding adeno-associated virus (AAV) via tail vein. Atrial fibrosis mice were treated with AAV expressing short hairpin RNA-negative control (sh-NC), sh-SPP1, sh-NC + overexpression (oe)-NC, sh-SPP1 + oe-NC, sh-SPP1 + oe-TGF-β, sh-TGF-β + oe-NC, sh-TGF-β + oe-SREBP2, sh-SREBP2 + oe-NC, sh-SREBP2 + oe-PCSK9 and sh-SPP1 + oe-PCSK9, with 10 mice following each treatment. Two weeks after AAV injection, the mice were euthanized, and atrial muscle tissues were collected for subsequent experiments. AAV was purchased from Shanghai Genechem (Shanghai, China). AAV-targeted short hairpin RNA (shRNA) was constructed using the GV478 vector (Genechem) while overexpression of AAV was constructed by the GV388 vector (Genechem). The primer sequence, vector construction, virus packaging and purification were completed by Genechem.
Cell culture and treatment
HL-1 mouse atrial myocytes purchased from American Type Culture Collection (Manassas, VA) were cultured (37 °C, 5% CO2) in DMEM medium (10569044, Gibco, Carlsbad, CA) containing 10% fetal bovine serum (FBS, 10099141, Gibco), 100 U/mL penicillin and 100 μg/mL streptomycin (15070063, Gibco). Cells were treated with 1 μM Ang II for 24 h to establish atrial fibrosis cell models.
The cells were then grouped and transduced with lentivirus carrying oe-NC, oe-TGF-β, oe-SREBP2, oe-PCSK9, sh-NC, sh-SPP1-1, sh-SPP1-2, sh-TGF-β-1, sh-TGF-β-2, sh-SREBP2-1, sh-SREBP2-2, sh-NC + oe-NC, sh-SPP1 + oe-NC, sh-SPP1 + oe-TGF-β, sh-TGF-β + oe-NC, sh-TGF-β + oe-SREBP2, sh-SREBP2 + oe-NC, sh-SREBP2 + oe-PCSK9 or sh-SPP1 + oe-PCSK9.
The core plasmid (PLKO.1) and adjuvant plasmid (RRE, REV and Vsvg) inserted into the target gene shRNA sequence were used to package shRNA lentivirus. The core plasmid (Fugw-GFP, Plx304) and adjuvant plasmid (RRE, REV and Vsvg) inserted into the target gene cDNA sequence were employed to package overexpression lentivirus, with the titer of 1 × 109 TU/mL. The cells were treated with 40 μL/mL lentivirus for 6 h, and continued to culture in the renewed medium. The lentivirus was commercially synthesized (Sangon Biotechnology, Shanghai, China). The shRNA sequences are shown in Supplemental Table 1.
RNA isolation and quantitation
Extraction of total RNA from tissues or cells utilizing the TRIzol reagent (16096020, Thermo Fisher Scientific Inc., Waltham, MA), which was then reversely transcribed into cDNA using First Strand cDNA Synthesis kit (D7168L, Shanghai Beyotime Biotechnology, Shanghai, China). RT-qPCR was conducted using the RT-qPCR kit (Q511-02, NanJing Vazyme Biotech, Nanjing, China) on the Bio-Rad CFX96 Real-time PCR system (Bio-Rad Laboratories, Hercules, CA). With β-actin serving as internal control, the fold changes were calculated by the 2−△△Ct method. The primer sequences (shown in Supplemental Table 2) were provided by Sangon.
Western blot analysis
Total protein was extracted from tissues and cells with radio-immunoprecipitation assay lysis buffer (P0013B, Beyotime) containing 100 mM phenylmethylsulphonyl fluoride. The nuclear and cytoplasmic protein was extracted with kit (P0028, Beyotime) according to the instructions, followed by measurement of protein concentration with a bicinchoninic acid kit (P0011, Beyotime). After separation by 8–12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, the protein was transferred onto a polyvinylidene fluoride membrane (1620177, Bio-Rad). Next, the membrane was blocked with 5% skimmed milk powder for 1 h and underwent overnight incubation at 4 °C with primary rabbit antibodies against β-actin (4970, 1:5000, Cell Signaling Technology, Beverly, MA), TGF-β (ab215715, 1:1000, Abcam, Cambridge, UK) and SREBP2 (ab228653, 1:1000, Abcam). The following day, the membrane was incubated with horseradish peroxidase-labeled secondary antibody goat anti-rabbit IgG (ab6721, 1:5000, Abcam) at room temperature for 1 h. Afterwards, the immunocomplexes on the membrane were visualized using enhanced chemiluminescence reagent (1705062, Bio-Rad). Image exposure was performed on Image Quant LAS 4000 C gel imager (GE company, Fairfield, CT). With β-actin serving as the internal reference, protein expression was quantified utilizing the Image J software.
Hematoxylin-eosin (HE) staining
Prepared sections of mouse atrial muscle tissues were fixed, permeabilized and rehydrated. Then, the sections were stained with hematoxylin for 7 min and counterstained with eosin staining for 1 min. Thereafter, the sections were rehydrated in descending series of alcohol, cleared in xylene, dried in a ventilator cabinet, and mounted with neutral gum before observation under an optical microscope (BX63, Olympus Optical, Tokyo, Japan).
Masson’s trichrome staining
Sections were soaked in 10% trichloroacetic acid and 10% potassium dichromate liquids for 40 min each, stained with hematoxylin (PT001, Shanghai Bogoo Biological Technology, Shanghai, China) for 8 min, and immersed in 1% ponceau (HL12202, Shanghai Haling Biotechnology, Shanghai, China) and 1% magenta mixture (HPBIO-SJ820, Shanghai Hepeng Biotechnology, Shanghai, China) for 40 min, which was halted by 1% glacial acetic acid and then 1% molybdic acid solution. Finally, sections were observed under a microscope (BX63, Olympus).
Immunohistochemistry (IHC)
Paraffin sections were heated at 60 °C for 20 min, cleared in xylene, and rehydrated. Following washing with double distilled water, and immersed in 3% H2O2 for 25 min to block the endogenous peroxidase. The sections were subjected to microwave-stimulated antigen retrieval in citric acid buffer for 10 min, allowed to stand at room temperature for 10 min and treated with normal goat serum blocking solution (Sangon) at room temperature for 20 min. Next, sections were immunostained with the diluted primary antibody against Collagen I (rabbit anti-mouse, ab88147, 1:100, Abcam) and Collagen III (ab7778, 1:100, Abcam) at 4 °C overnight, followed by 30 min incubation with secondary antibody goat anti-rabbit IgG (ab6721, 1:5000, Abcam). Subsequently, sections were treated with streptavidin biotin peroxidase complex (Vector Labs, Burlingame, CA) at 37 °C for 30 min, and the blots were visualized with DAB kit (P0203, Beyotime) for 6 min. After 25 min staining with hematoxylin, sections were subjected to observation under an upright microscope (BX63, Olympus).
Cell counting kit-8 (CCK-8) assay
CCK-8 kit (C0037, Beyotime) was used for the detection of cell viability. Cells were seeded into a 96-well plate (2 × 103 cells/well, 100 μL) and incubated for 1 h with 10 μL of CCK-8 reagent. Subsequently, the optical density (OD) values at 450 nm were determined.
TUNEL assay
Cells (5 × 107 cells/mL) were pre-treated with 0.3% Triton X-100 in PBS. The TUNEL detection solution was prepared following the protocols of the one-step TUNEL Assay Apoptosis Detection Kit (C1088, Beyotime). Next, each sample was incubated with 50 μL TUNEL detection solution at 37 °C in the dark for 60 min to make the TUNEL detection solution evenly cover the sample. Afterwards, cells were observed under a fluorescence microscope (BX63, Olympus).
mtDNA damage analysis
Genomic DNA extraction kit (K280-50, BioVision, Milpitas, CA) was used to isolate mtDNA, and specific primers were applied to amplify mtDNA fragment. The relative amplification of large DNA fragments ~10 kb) between the treated sample and the control sample was compared and normalized to the amplification of smaller fragments (~110 bp) to quantify DNA damage. The primers used were as follows: long fragment primer with 10,235 bp product (forward sequence: ACATACCCATGGCCAACCT and reverse sequence: TATGTTTGCGGTTTCGATGA) and short primer with 113 bp product (forward sequence: ACATACCCATGGCCAACCT and reverse sequence: GGGCTTTGCGTAGTTGTAT). Poisson transformation was used to calculate mtDNA damage.
Mitochondrial reactive oxygen species (mtROS) measurement
MitoSOX™Red (M36008, Invitrogen) is an indicator of mitochondrial superoxide. Flow cytometry was used to detect the level of mtROS. The data were recorded as the “M2 percentage” fluorescence change (mean fluorescence intensity), which indicated an enhanced proportion of cells produced by mtROS.
Microarray-based gene expression profiling
Atrial fibrosis-related dataset GSE27133 (annotation files: GPL11661) was retrieved from the Gene Expression Omnibus (GEO) database. GSE27133 contained 3 normal samples and 2 atrial fibrosis samples. Differential analysis was conducted using R language “limma” package with |log2FC | > 1 and p < 0.05 set as the threshold to screen differentially expressed genes (DEGs). A heat map of DEGs was plotted using R “pheatmap” package. Atrial fibrosis-related genes were searched from the GeneCards database with “atrial fibrosis” set as the keyword and subjected to intersection analysis with the DEGs using the venny tool. The Coxpresdb database was applied to identify the co-expressed genes of key genes, which were intersected with atrial fibrosis-related genes. The obtained genes were subjected to KEGG enrichment analysis using KOBAS website. Correlation analysis of the two genes in heart tissues was retrieved in the Chipbase v2.0 database.
Statistical analysis
All data were processed using SPSS 21.0 statistical software (IBM Corp. Armonk, NY). Measurement data were expressed as mean ± standard deviation. Data obeying normal distribution and homogeneity of variance between two groups were analyzed using unpaired t-test, while those among multiple groups were analyzed by one-way analysis of variance (ANOVA) or repeated measures ANOVA, followed by Tukey’s post hoc tests with corrections for multiple comparisons. p < 0.05 was considered as statistically significant.
Data availability
The datasets generated/analyzed during the current study are available.
Change history
30 June 2023
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41420-023-01516-9
References
Zimetbaum P. Atrial fibrillation. Ann Intern Med. 2017;166:ITC33–48.
Lau DH, Schotten U, Mahajan R, Antic NA, Hatem SN, Pathak RK, et al. Novel mechanisms in the pathogenesis of atrial fibrillation: practical applications. Eur Heart J. 2016;37:1573–81.
Latoche JD, Ufelle AC, Fazzi F, Ganguly K, Leikauf GD, Fattman CL. Secreted phosphoprotein 1 and sex-specific differences in silica-induced pulmonary fibrosis in mice. Environ Health Perspect. 2016;124:1199–207.
Lin R, Wu S, Zhu D, Qin M, Liu X. Osteopontin induces atrial fibrosis by activating Akt/GSK-3beta/beta-catenin pathway and suppressing autophagy. Life Sci. 2020;245:117328.
Xie Z, Singh M, Singh K. Osteopontin modulates myocardial hypertrophy in response to chronic pressure overload in mice. Hypertension 2004;44:826–31.
Caballero EP, Santamaria MH, Corral RS. Endogenous osteopontin induces myocardial CCL5 and MMP-2 activation that contributes to inflammation and cardiac remodeling in a mouse model of chronic Chagas heart disease. Biochim Biophys. Acta Mol Basis Dis. 2018;1864:11–23.
Dong J, Ma Q. Osteopontin enhances multi-walled carbon nanotube-triggered lung fibrosis by promoting TGF-beta1 activation and myofibroblast differentiation. Part Fibre Toxicol. 2017;14:18.
Heinzmann D, Fuss S, Ungern-Sternberg SV, Schreieck J, Gawaz M, Gramlich M, et al. TGFbeta is specifically upregulated on circulating cd14++ cd16+ and cd14+ cd16++ monocytes in patients with atrial fibrillation and severe atrial fibrosis. Cell Physiol Biochem. 2018;49:226–34.
Shen H, Wang J, Min J, Xi W, Gao Y, Yin L, et al. Activation of TGF-beta1/alpha-SMA/Col I profibrotic pathway in fibroblasts by galectin-3 contributes to atrial fibrosis in experimental models and patients. Cell Physiol Biochem. 2018;47:851–63.
Kostopoulou F, Malizos KN, Papathanasiou I, Tsezou A. MicroRNA-33a regulates cholesterol synthesis and cholesterol efflux-related genes in osteoarthritic chondrocytes. Arthritis Res Ther. 2015;17:42.
Schiano C, Benincasa G, Franzese M, Della Mura N, Pane K, Salvatore M, et al. Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases. Pharm Ther. 2020;210:107514.
Quan Y, Xin Y, Tian G, Zhou J, Liu X. Mitochondrial ROS-Modulated mtDNA: a potential target for cardiac aging. Oxid. Med Cell Longev. 2020;2020:9423593.
Jablonski RP, Kim SJ, Cheresh P, Williams DB, Morales-Nebreda L, Cheng Y, et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. 2017;31:2520–32.
Xu RX, Liu J, Li XL, Li S, Zhang Y, Jia YJ, et al. Impacts of ezetimibe on PCSK9 in rats: study on the expression in different organs and the potential mechanisms. J Transl Med. 2015;13:87.
Guo Y, Yan B, Tai S, Zhou S, Zheng XL. PCSK9: associated with cardiac diseases and their risk factors? Arch Biochem Biophys. 2021;704:108717.
Wang X, Li X, Liu S, Brickell AN, Zhang J, Wu Z, et al. PCSK9 regulates pyroptosis via mtDNA damage in chronic myocardial ischemia. Basic Res Cardiol. 2020;115:66.
Dzeshka MS, Lip GY, Snezhitskiy V, Shantsila E. Cardiac fibrosis in patients with atrial fibrillation: mechanisms and clinical implications. J Am Coll Cardiol. 2015;66:943–59.
Collins AR, Schnee J, Wang W, Kim S, Fishbein MC, Bruemmer D, et al. Osteopontin modulates angiotensin II-induced fibrosis in the intact murine heart. J Am Coll Cardiol. 2004;43:1698–705.
Kramerova I, Kumagai-Cresse C, Ermolova N, Mokhonova E, Marinov M, Capote J, et al. Spp1 (osteopontin) promotes TGFbeta processing in fibroblasts of dystrophin-deficient muscles through matrix metalloproteinases. Hum Mol Genet. 2019;28:3431–42.
Cao F, Li Z, Ding WM, Yan L, Zhao QY. LncRNA PVT1 regulates atrial fibrosis via miR-128-3p-SP1-TGF-beta1-Smad axis in atrial fibrillation. Mol Med. 2019;25:7.
Kostopoulou F, Gkretsi V, Malizos KN, Iliopoulos D, Oikonomou P, Poultsides L, et al. Central role of SREBP-2 in the pathogenesis of osteoarthritis. PLoS One. 2012;7:e35753.
Li P, Yan X, Xu G, Pang Z, Weng J, Yin J, et al. A novel plasma lncRNA ENST00000416361 is upregulated in coronary artery disease and is related to inflammation and lipid metabolism. Mol Med Rep. 2020;21:2375–84.
Lin YC, Wang JC, Wu MS, Lin YF, Chen CR, Chen CY, et al. Nifedipine Exacerbates Lipogenesis in the Kidney via KIM-1, CD36, and SREBP Upregulation: Implications from an Animal Model for Human Study. Int J Mol Sci. 2020;21:4359.
Oteng AB, Loregger A, van Weeghel M, Zelcer N, Kersten S. Industrial trans fatty acids stimulate SREBP2-mediated cholesterogenesis and promote non-alcoholic fatty liver disease. Mol Nutr Food Res. 2019;63:e1900385.
Kim SJ, Cheresh P, Jablonski RP, Morales-Nebreda L, Cheng Y, Hogan E, et al. Mitochondrial catalase overexpressed transgenic mice are protected against lung fibrosis in part via preventing alveolar epithelial cell mitochondrial DNA damage. Free Radic Biol Med. 2016;101:482–90.
Sung PH, Lin KC, Chai HT, Chiang JY, Shao PL, Luo CW et al. Losing Regulation of the Extracellular Matrix is Strongly Predictive of Unfavorable Prognostic Outcome after Acute Myocardial Infarction. Int J Mol Sci. 2020;21:6219.
Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell 2017;171:1057–71 e1011.
Wu YR, Li L, Sun XC, Wang J, Ma CY, Zhang Y, et al. Diallyl disulfide improves lipid metabolism by inhibiting PCSK9 expression and increasing LDL uptake via PI3K/Akt-SREBP2 pathway in HepG2 cells. Nutr Metab Cardiovasc Dis. 2021;31:322–32.
Ding Z, Liu S, Wang X, Mathur P, Dai Y, Theus S, et al. Cross-Talk between pcsk9 and damaged mtDNA in vascular smooth muscle cells: role in apoptosis. Antioxid. Redox Signal. 2016;25:997–1008.
Elseweidy MM, Mohamed HE, Elrashidy RA, Atteia HH, Elnagar GM. Inhibition of Aortic calcification by policosanol in dyslipidemic rabbits is enhanced by pentoxifylline: potential role of PCSK9. J Cardiovasc Pharm Ther. 2018;23:551–60.
Funding
This study was supported by Medical Health Science and Technology Project of Zhejiang Province Health Commission (2020KY821 and 2022KY302).
Author information
Authors and Affiliations
Contributions
XD, TL and CS wrote the paper; BH, MF and JL conceived the experiments; WZ, GF and BW analyzed the data; YX and HC collected and provided the sample for this study. All authors have read and approved the final submitted manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41420-023-01516-9
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, X., Liu, T., Shen, C. et al. RETRACTED ARTICLE: Anti-fibrotic mechanism of SPP1 knockdown in atrial fibrosis associates with inhibited mitochondrial DNA damage and TGF-β/SREBP2/PCSK9 signaling. Cell Death Discov. 8, 246 (2022). https://doi.org/10.1038/s41420-022-00895-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-022-00895-9
- Springer Nature Limited