
Abstract
Aberrant somatic hypermutation (aSHM) can target proto-oncogenes and drive oncogenesis. In mantle cell lymphoma (MCL), CCND1 is targeted by aSHM in the non-nodal subtype (nnMCL), giving rise to exon1 encoded mutant proteins like E36K, Y44D, and C47S that contribute to lymphomagenesis by virtue of their increased protein stability and nuclear localization. However, the vast majority of somatic variants generated by aSHM are found in the first intron of CCND1 but their significance for mantle cell lymphomagenesis is unknown. We performed whole-genome and whole-transcriptome sequencing in 84 MCL patients to explore the contribution of non-coding somatic variants created by aSHM to lymphomagenesis. We show that non-coding variants are enriched in a MCL specific manner in transcription factor-binding sites, that non-coding variants are associated with increased CCND1 mRNA expression, and that coding variants in the first exon of CCND1 are more often synonymous or cause benign amino acid changes than in other types of lymphomas carrying a t(11;14) translocation. Therefore, the increased frequency of somatic variants due to aSHM might be a consequence of selection pressure manifested at the transcriptional level rather than being a mere mechanistic consequence of misguided activation-induced cytidine deaminase (AID) activity.
Similar content being viewed by others


Introduction
Mantle cell lymphoma (MCL) is an aggressive B cell neoplasm genetically characterized by the translocation t(11;14)(q13;q32), leading to CCND1 overexpression [1, 2]. Two molecular subtypes are currently recognized: (1) Classical MCL (cMCL) is composed of B cells with minimally mutated or unmutated immunoglobulin heavy chain variable (IGHV) region that express SOX11. Patients have generalized lymphadenopathy and the outcome is adverse. (2) In non-nodal MCL (nnMCL), B cells do not express SOX11 and often carry mutated IGHV. Here the involved organs are peripheral blood, bone marrow, and spleen. Cases are often clinically indolent [1]. The cells of origin are believed to be naive B cells that do not undergo germinal center reactions as in the case of cMCL and memory B cells in nnMCL [3].
Frequent molecular alterations in MCL are found in ATM, TP53, NSD2, KMT2D, NOTCH1/2, UBR5, BIRC3, TRAF2, MAP2K14, CARD11, SMARCA4, and BTK [3]. The most characteristic alteration in MCL is, however, rearrangement of the CCND1 locus, which leads to the juxtaposition of the strong immunoglobulin heavy-chain enhancer on chr14, and results in CCND1 overexpression [4, 5]. The CCND1 transcript can also be stabilized by deletions or point mutations in the 3’UTR eliminating miRNA binding sites or creating premature polyadenylation sites [6, 7]. Stabilization of CCND1 at the protein level has also been observed in MCL. Alternatively, spliced isoforms lacking the T286 phosphorylation site needed for CCND1 degradation [8,9,10] and amino acid changes linked to increased protein stability have been described [11].
Widespread occurrence of CCND1 mutations caused by aberrant somatic hypermutation has first been detected by [12] using whole-transcriptome sequencing (WTS). Other screens have confirmed these findings [13,14,15]. However, the biological significance of these mutations has not been fully clarified. One reason may be found in the mechanism of aberrant somatic hypermutation (aSHM) generation by activation-induced cytidine deaminase (AID), whose activity is tightly regulated and restricted to 1–2 kb from the transcriptional start site of its target genes [16]. Another reason may be that the effects of aSHM are still incompletely understood.
Here we present the analysis of the mutation spectrum of 84 MCL patients whose genomes and transcriptomes have been fully sequenced. We report that CCND1 mutations generated by aSHM are particularly frequently observed in MCL as compared to other lymphomas carrying the t(11;14)(q13;q32) translocation. Our results are compatible with the hypothesis that CCND1 mRNA expression levels are a rate-limiting factor for MCL lymphomagenesis and that non-coding mutations generated by aSHM are selected for their impact on CCND1 mRNA expression.
Materials and methods
Patients and samples
The cohort includes 84 patients diagnosed with MCL among 4610 samples (5k data set) from a wide variety of hematological malignancies (Table 1 and Supplementary Table 1). The selection of samples for the 5k data set was based on the following criteria:
-
1.
Consent of patients for research use of their data and good prospects for collaborative follow-up studies with clinicians.
-
2.
Uniform coverage of molecular subtypes of master cohorts as defined by WHO [2] criteria.
-
3.
Percentage of aberrant white blood cells at least 30%.
Bone marrow (BM), and peripheral blood (PB) samples from patients had been sent to MLL Leukemia Laboratory between 2006 and 2020 for immediate diagnostic work-up. The respective diagnosis was established based on cytomorphology, immunophenotyping, cytogenetics, fluorescence in situ hybridization (FISH), and molecular genetics following WHO guidelines [2]. All patients gave their written informed consent for scientific evaluations. The study was approved by the Internal Review Board and adhered to the tenets of the Declaration of Helsinki. DNA samples from BM and/or PB, at diagnosis or before treatment, were collected from all cases and DNA and total RNA extracted using the MagNA Pure 96 Instrument and the MagNAPure96 DNA and Viral NA LV Kit and MagNA Pure 96 Cellular RNA LV Kit, respectively (Roche LifeScience, Mannheim, Germany) [17].
Whole-genome sequencing (WGS) and analysis
WGS libraries were prepared from 1 µg of DNA with the TruSeq PCR free library prep kit following the manufacturer’s recommendations (Illumina, San Diego, CA, USA) and 2×150 bp paired-end sequences were generated on a NovaSeq 6000 or HiSeqX instrument with 100× coverage (Illumina, San Diego, CA, USA) and processed as described previously [17, 18]. A so-called Tumor/Unmatched normal workflow was used for variant calling to reduce technical artifacts and germline calls. Each variant was queried against the gnomAD database (v2.1.1) and variants with global population frequencies >0.5% were excluded. The final analysis was performed only on PASS filtered variants. Mutational signatures were analyzed using the R package MutSignatures [19]. Oncoprint representations of mutation data were generated using the R package ComplexHeatmap [20].
WTS and analysis
For transcriptome analysis, the TruSeq Total Stranded RNA kit was used, starting with 250 ng of total RNA, to generate RNA libraries following the manufacturer’s recommendations (Illumina, San Diego, CA, USA). In all, 2×100 bp paired-end reads were sequenced on the NovaSeq 6000 with a median of 50 million reads per sample (Illumina, San Diego, CA, USA) and processed as described previously [17]. Counts were extracted with Cufflinks (v2.2.1) [21]. Counts were normalized applying the variance stabilizing transformation normalization method. Genes were kept if they were expressed (>5 CPM) in at least 66% of the samples. Gene expression differences were assessed using the DESeq2 package [22]. Model parameters included sequencing run, aSHM mutation status, material (peripheral blood or bone marrow), gender, chr11 breakpoint distance from CCND1 TSS, and mutation status of ATM, KMT2D, NSD2, and TP53. The significance of coefficients in a negative binomial generalized linear model was estimated using the Wald test. Gene set enrichment analysis was performed with the GSEA package [23, 24] using a CCND1 pre-ranked list based on DESeq2 normalized expression counts. Cluster analysis was carried out using the R package pheatmap (https://github.com/raivokolde/pheatmap).
Variant enrichment analysis
Enrichment analysis using bipartite graph models was performed essentially as described [25, 26]. Briefly, bipartite graph models consisting of variant bins on one side of the graph and samples on the other side were constructed. The degree of bin vertices in this graph reflects the number of variants in that bin. Randomization of the graph yields a statistical null model. Two subgraphs were then constructed: one containing only MCL samples and the other containing only non-MCL samples. The significance of deviation of bin vertex degrees in the subgraphs from the predictions of the null model is then estimated using Poisson-binomial Z-scores and the bi-binomial approximation [26] of Poisson binomial P values.
Motif searches
Motif searches were performed using the AME and the Centrimo apps of the MEME suite [27] and by motif consensus matching to motifs listed in Jaspar [28] and HOCOMOCO [29]. Motif matching results for all variants using wild-type and mutated sequences with 15 bases of padding to the left and to the right of the mutated position are listed in Supplementary Table 1.
IGHV mutation status
IGHV mutation status was analyzed by Sanger sequencing of corresponding PCR fragments, by IGCaller software [30] analysis of WGS data, or both.
In silico prediction of pathogenicity
The pathogenicity of amino acid changes was estimated using HePPY (https://doi.org/10.1182/blood-2019-128488).
Results
The landscape of coding and non-coding variants in MCL
Our cohort of MCL patients comprises 84 cases. We performed enrichment analysis for the number of variants in bins of 10, 100, 1000, and 10,000 bases to identify genomic regions that carry significantly more variants in MCL patients as compared to our cohort of 4610 cases of leukemia and lymphoma (30 different entities, 5k data set, details listed in Table 1 and Supplementary Table 1) with whole-genome sequence data. We analyzed coding and non-coding variants separately.
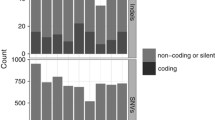
Figure 1A shows the results for the analysis of coding variants. We detected three highly significant (−log10(P) ≥ 10) regions corresponding to CCND1, ATM, and NSD2. Coding mutations in these genes have been reported before for MCL [2] and this result validates our general approach. It is worth noting that our analysis is relative to other cohorts in our 5k data set (Table 1). Thus, genes like TP53 and KMT2D, although mutated not only in MCL but also in many other malignancies, will not score as enriched in MCL in this analysis. Mutation data for ATM, NSD2, KMT2D, and TP53 for all cohorts are listed in Supplementary Table 1.

Circos plot of negative log10(P) for the number of variants in bins of different sizes. High values indicate hotspots of somatic mutation specific for MCL. The outer circle represents a genomic density plot of somatic mutations in the 10 bp bin (red). A Enrichment analysis for coding variants. B Enrichment analysis for non-coding variants.
The enrichment analysis for non-coding variants is shown in Fig. 1B. The complete list of genomic regions with significantly elevated densities of non-coding mutations in MCL can be found in Supplementary Table 2. The most significant region is found on chromosome 11 and corresponds to the CCND1 locus. We therefore turned our attention to the CCND1 locus for a more detailed analysis.
MCL samples are prone to mutagenesis in the CCND1 transcription regulatory region
We generated histograms for the number of non-coding variants in 100 bp bins covering the entire CCND1 locus and 5 kb upstream of the TSS (chr11:69455872, Fig. 2A). In this work, we refer to the 5 kb region upstream of the TSS as promoter region [31] whereas the region chr11:69450872-69458025 (hg19) encompassing the promoter, exon 1, and parts of intron 1 of CCND1 is denoted as transcription regulatory region. We noticed a strong increase of non-coding variants in the promoter region and the first intron in MCL samples as compared to other cohorts. The UCSC genome browser annotations of these regions show high levels of H3K27 acetylation, DNase 1 hypersensitivity, and transcription factor-binding sites (TFBSs). The bar plot at the bottom of Fig. 2A also shows that the variants in the first intron of the CCND1 gene are not uniformly distributed. Rather, they appear to be clustered in some regions while others remain essentially void of variants.

A Non-coding variants in the genomic region chr11:69450872-69469242 (hg19) covering the CCND1 locus including 5 kb upstream of the TSS were collected for all MCL and non-MCL samples. Red bars indicate the relative frequency of non-coding variants in the MCL cohort. Black bars refer to the relative frequency of non-coding variants in non-MCL samples. A screenshot from the UCSC genome browser of the CCND1 locus shows the density of H3K27 acetylation marks, the density of DNase 1 hypersensitivity sites, and transcription factor-binding regions, as well as the level of sequence conservation. The bars at the bottom of the panel indicate the distribution of individual variants in the first intron. The color of the bars indicates the number of samples a particular variant was observed in. B Plot of the percentage of samples with five or more non-coding variants in the CCND1 transcription regulatory region (chr11: 69450872-69458025, hg19) for indicated cohorts. Abbreviations for cohort names are described in Table 1. C Plot of the fraction of samples by the minimum number of non-coding variants in the CCND1 transcription regulatory region for Multiple Myeloma (MM) and MCL samples. Only samples carrying a t(11;14)(q13;q32) translocation and mutated IGHV status were included.
The accumulation of variants in the promoter, the first exon, and the first intron of CCND1 is not entirely specific for MCL. It is assumed that they are a byproduct of somatic hypermutation occurring in the germinal center microenvironment [12]. We detected such mutations in other B cell-derived malignancies such as multiple myeloma, albeit at a less pronounced level (see below).
However, MCL seems to be particularly prone to accumulate mutations in these regions. Figure 2B shows the percentage of samples with at least five non-coding mutations in the CCND1 transcription regulatory region for cohorts where such mutations were detected. The percentage of CCND1 mutated samples in the MCL cohort is two to three times higher than in other cohorts. The majority of these variants is found in intron 1 (Fig. 2A and Supplementary Table 2).
The nearly universal presence of the t(11;14)(q13;q32) translocation in MCL might be a confounding factor when comparing the percentage of CCND1 mutated samples between different types of lymphoma. Therefore, we analyzed only samples carrying a t(11;14)(q13;q32) translocation. Only samples with mutated IGHV were included to ensure that SHM has taken place. We found 28 MCL samples and 45 multiple myeloma samples satisfying these criteria. Also in this more stringent comparison, MCL samples consistently carry more than twice as many non-coding variants in the CCND1 locus as multiple myeloma samples (Fig. 2C). The majority of these variants are found in MCL samples with mutated IGHV, as can be seen from the data in Supplementary Table S1. MCL samples with mutated IGHV harbor nearly six times as many non-coding mutations than samples with unmutated IGHV. We conclude that MCL samples are particularly prone to accumulate non-coding variants in the regulatory region of the CCND1 locus.
Mutational signature analysis
The higher propensity of MCL for mutations in the CCND1 locus might originate from differences in the mutational processes generating these mutations. We performed mutational signature analysis on mutations in the CCND1 locus to address this question. We included all MCL samples in this analysis. For comparison, we used samples carrying the t(11;14)(q13;q32) translocation. We refer to this artificial cohort as non-MCL. It is mainly composed of multiple myeloma samples (45) and some samples from our marginal zone lymphoma (12), IgM monoclonal gammopathy of undetermined significance (3), and B cell non-Hodgkin lymphoma (5) cohorts.
Figure 3 shows the results of extracting two mutational signatures by non-negative matrix factorization from the MCL and the non-MCL cohorts. The domineering type of mutation is a C-T transition in both cohorts. This observation is compatible with the activity of AID during somatic hypermutation. Comparison of our signatures to known COSMIC signatures [32] confirms that the mutational processes at work in both cohorts are largely similar (Table 2). We conclude that differences in mutational processes are unlikely to account for a large number of mutations in the CCND1 locus in MCL.

Non-negative matrix factorization was applied to extract mutational signatures from MCL and non-MCL samples.
Cohort-specific enrichment of variant clusters
Next, we addressed the question of whether the clustered appearance of variants bears cohort specificity, which might be a reflection of an underlying biological significance. We performed enrichment analysis for the number of non-coding variants in bins from size five to ten base pairs using bipartite graph models (see “Materials and methods” section for details) in the region covering the CCND1 promoter and the first intron. This analysis yields Z-score estimates for the significance of enrichment of variants in each bin for the MCL and the non-MCL cohorts. For each base position, the maximum Z-score was determined and plotted using a color scale. Figure 4A shows the results of this analysis. Some variant clusters are enriched in both the MCL and the non-MCL cohorts. However, the majority of clusters appear to display MCL cohort-specific enrichment.

A Enrichment analysis of non-coding variants in the transcription regulatory region of the CCND1 locus. Analysis was performed using bins of size 5–10 bp. The maximum enrichment Z-score for the number of variants per bin is plotted for each base on a color scale for the MCL and the non-MCL samples. Motif searches were performed using either wild-type or mutated cluster sequence. Best matching motifs are indicated in green or red letters. Red letters indicate motif matches to the wild-type sequence. Green letters indicate motif matches to the mutated sequence. MCL-specific enrichment of clusters is marked by underlining the name of the matching motif. B, C Motif matches to enriched clusters. For each cluster, the cluster sequence and the best matching motif sequence logo are shown. Mutated bases are shown in bold. Brown and blue sequence letters indicate mutations matching c-AID and nc-AID sequence context, respectively. Genomic positions refer to hg19 coordinates. Motif matches to the wild-type cluster sequence are shown with red framing (B) indicating that the cluster is associated with destructive mutations. Green framing indicates that the cluster is subject to constructive mutations (C). Base changes for individual variants are shown underneath the sequence, in red for destructive mutations, in green for constructive mutations, and in black for neutral mutations with regard to the quality of the motif match. Reverse complemented genomic sequence is marked with a “*”. D Cluster analysis using the gene signature identified by [34] to classify MCL samples as cMCL or nnMCL. Gene expression levels are based on DESeq2 normalized counts. The criteria to further subclassify nnMCL samples as nnMCL_MUT or nnMCL_WT are described in the main text. E Wald test for differential expression of CCND1 mRNA depending on the mutation status of the transcription regulatory region of CCND1. F Gene set enrichment analysis for the mutation status of the transcription regulatory region of CCND1. A pre-ranked nnMCL sample list based on CCND1 expression levels as measured by DESeq2 normalized counts was used. Black bars indicate the rank of nnMCL_MUT samples.
We used motif searches to investigate whether the variant clusters co-localize with TFBS. The effect of mutations in TFBSs can be twofold: Some might serve to destabilize factor binding to a given site (destructive mutation) while others might change the binding specificity or increase the affinity of sites for some factors (constructive mutation). Constructive mutations are harder to link to a specific transcription factor as both the optimal binding motif and the factor best suited to bind to that site are not known. However, if an enriched cluster is subject to constructive mutations, the collection of mutations observed in that cluster should be a reflection of the unknown motif. Therefore, we used the mutations observed for each cluster for the construction of a set of mutated cluster sequences. Thus, for each cluster, the wild type and the mutated sequences were used for motif searches. Furthermore, for each variant, the sequence context was analyzed for matches compatible with classical AID (c-AID, C to T/G at WRCY motifs) and non-classical AID (nc-AID, A to C/G at WA motifs) mediated mutagenesis [33] (Supplementary Table 1). Both the wild type as well as the mutated sequence of each variant padded with 15 bases to the left and to the right were subjected to motif searches individually. The results are shown in Fig. 4B, C. We observed constructive mutations for NFAT, CEBP, E2F, and SOX motifs. MEF2, STAT, and HOX motifs were associated with destructive mutations. Interestingly, some motifs were identified more than once in different locations with a consistent preference for destructive or constructive mutations.
Next, we asked whether the enrichment of mutations in MCL-specific clusters had an impact on CCND1 expression levels. To exclude confounding effects from cMCL cases, we used the gene signature identified by [34] to classify samples as either cMCL or nnMCL as shown in Fig. 4D. Twenty-eight samples were identified as nnMCL. nnMCL samples with at least two mutations in MCL-specific TFBS mutation clusters and 10 or more non-coding mutations in the CCND1 regulatory region were assigned to the mutated group (nnMCL_MUT). The remaining nnMCL samples were assigned to the wild-type group (nnMCL_WT). Next, we used whole transcriptome data for two types of analysis: First, we tested for differential expression of CCND1 in the set of cMCL, nnMCL_MUT, and nnMCL_WT samples and, second, we performed gene set enrichment analysis using a list of nnMCL samples ranked according to CCND1 expression level. The results of these tests are shown in Fig. 4E, F. We observed that nnMCL_MUT samples carrying mutations in the transcription regulatory region of CCND1 have higher levels of CCND1 mRNA than nnMCL_WT samples.
Mutual exclusivity of ATM and CCND1 mutations
We investigated the relationship between CCND1 mutations and mutation patterns of known and potential driver genes in MCL with Oncoprint [35] (Fig. 5). Non-coding, missense, and synonymous CCND1 mutations were analyzed separately. All types of CCND1 mutations are found mainly in samples without mutations in ATM (P < 0.044, Fisher’s exact test). While it is a possibility that mutual exclusivity between ATM and CCND1 mutations is based on biological differences in the cell of origin giving rise cMCL (naive B cells) and nnMCL (memory B cells), we did detect ATM mutations also in nnMCL cases. Therefore, this mutation pattern may be a reflection of a common endpoint of ATM and CCND1 mutations, namely increased levels of CCND1 activity as ATM directly activates FBXO31 needed for CCND1 degradation [36].

Oncoprint representation of selected genes found mutated in MCL and CCND1 mutations. Mutated genes are depicted in rows, and cases are displayed in columns. Mutual exclusivity of ATM mutations and CCND1 coding (missense) (P = 0.003), non-coding (P = 0.044), and synonymous (P = 0.009) mutations was detected (Fisher’s exact test).
The abundance of synonymous or benign amino acid changes in CCND1 exon1
As a further test of the hypothesis that aSHM generated mutations of the CCND1 locus are driving CCND1 mRNA expression levels and that CCND1 is a rate-limiting factor for MCL lymphomagenesis, we analyzed the spectrum of coding mutations in the first exon of CCND1. The first exon as part of the CCND1 transcription regulatory region is potentially subject to selection pressure at the level of transcription as well as protein function. If protein function plays a crucial role, one would expect a mutation spectrum that is biased against synonymous mutations and mutations coding for benign amino acid changes. However, our results shown in Fig. 6A indicate that, in MCL, synonymous and benign mutations with low pathogenicity scores as estimated by HePPY (https://doi.org/10.1182/blood-2019-128488) are more abundant as compared to non-MCL samples, where deleterious mutations with high HePPY scores prevail.

A The number of samples with mutations in exon1 of CCND1 by HePPY score is shown. HePPY is a predictor of missense variant pathogenicity (https://doi.org/10.1182/blood-2019-128488). Low HePPY scores indicate benign amino acid changes. Deleterious mutations are associated with HePPY scores close to 1. B The sequence coding for CCND1 L32 to S41 is shown. Two frequently observed mutations in the first exon of CCND1 are overlapping an E2F-binding site. The red guanine bases are often changed to adenine, which favors E2F binding. C Significance of the proportion of synonymous mutations in the sample groups displayed on the x-axis. P values were calculated using the cumulative hypergeometric distribution. The negative decadic logarithm of (1−P) is shown. Values >2 are considered significant.
Interestingly, among the most frequently occurring mutations are 11-69456186-G-A and 11-69456187-G-A. As can be seen in Fig. 6B, both mutations stabilize an E2F-binding site, and in three out of eight samples carrying these mutations, they co-occur (Supplementary Table 3). 11-69456186-G-A is a synonymous mutation. 11-69456187-G-A codes for the previously reported E36K mutation [11]. It is possible that E36K adds a selective advantage to cells carrying this change. However, the frequent occurrence of the nearby synonymous mutation supports the hypothesis that the efficiency of transcription of the CCND1 gene also plays a role. This notion is further supported by the significant enrichment of synonymous mutations observed in nnMCL samples as compared to cMCL and non-MCL samples shown in Fig. 6C.
Discussion
We performed a genome-wide survey assessing the prevalence of non-coding mutations in MCL as compared to other types of leukemia and lymphoma. The CCND1 locus emerges as the major hotspot of non-coding mutagenesis. MCL is characterized by a near-universal presence of a t(11;14)(q13;q32) translocation bringing the CCND1 locus into close proximity with the immunoglobulin heavy-chain enhancer [4, 5], which can drive SHM in a subset of MCL cases [37]. SHM can occur outside of Ig loci. The first examples of SHM at non-physiological targets (aSHM) with a proven impact on target gene expression were MYC, BCL6, and CD95 [38,39,40]. aSHM at the CCND1 locus in MCL has first been reported by [12]. However, the restriction of AID activity mediating aSHM to about 1-2 kb downstream of the transcription start site [16] makes it difficult to distinguish bystander mutations from drivers on statistical grounds alone in the absence of mechanistic evidence of an impact on the transcription regulation of a candidate target of aSHM.
We addressed this difficulty in two ways. First, we analyzed relative enrichment of variants in the 5’ region of CCND1 as compared to other types of lymphoma carrying the t(11;14)(q13;q32) translocation with evidence for mutated IGHV. To exclude variants selected at the level of protein function, we restricted our analysis to the set of non-coding variants. We observed MCL-specific enrichment of mutations in TFBSs. Spatial clustering of non-coding mutations at the CCND1 locus in MCL samples has recently been reported by [15] using the tool OncodriveCLUST [41]. However, the mechanistic consequences of this clustering and the relative enrichment in MCL were not investigated in this study.
Second, we used motif searches to investigate the potential impact of clustered non-coding mutations on CCND1 expression. While the largely overlapping motifs for TFBSs belonging to the same family make it difficult to pinpoint the precise factor bound at a specific site, a consistent picture seems to emerge. We find that both destructive and constructive mutations can be identified. Destructive mutations prevent transcription factor binding that would normally occur. We detected destructive mutations in MEF2, STAT, and HOX motifs. A sizable fraction of these mutations occurs in sequence contexts that are compatible with c-AID mediated mutagenesis, whereas nc-AID mediated mutagenesis did not seem to play a major role. MEF2 transcription factors can activate or repress transcription depending on their association with activating or repressive co-factors [42]. MEF2B mutations have been reported in mantle cell lymphoma [13]. STAT6 mutations in the DNA binding region have been described in Hodgkin lymphoma [43]. HOX transcription factors are involved in T cell differentiation and are known as drivers of T-ALL [44, 45]. In mantle cell lymphoma, HOX genes are silenced by H3K27me3 chromatin methylation mediated by overexpression of EZH2 [46].
We also observed constructive mutations that change the binding specificity of a site or create new binding sites. NFAT-binding sites were created in five different locations in the first intron of CCND1. NFAT proteins have long been known as major targets of antigen receptors expressed on T and B cells [47]. Recently, elevated levels of NFATC1 have been observed in CLL caused by DNA hypomethylation of the NFATC1 locus, and NFATC1 expression levels were shown to correlate with higher expression of CCND1 [48]. In the CCND1 promoter region as well as in the first intron, we observe the creation of CEBP binding sites. CEBP is known to be a major activator of CCND1 transcription [49]. Additionally, SOX binding sites are created in more than one location. SOX11 has been recognized as a major oncogene in mantle cell lymphoma [1, 3, 50]. In the 5’UTR an E2F site is created in close proximity to a pre-existing E2F binding site. E2F transcription factors are major drivers of cell cycle progression and are activated by CCND1 expression in a self-regulatory loop via CCND1 mediated phosphorylation of RB1 [51]. Interestingly, we observed co-occurrence of two mutations in the first exon (11-69456186-G-A, 11-69456187-G-A) that also create an E2F binding site. Both variants are among the most frequently observed mutations in the CCND1 locus. 11-69456186-G-A is a synonymous mutation while 11-69456187-G-A codes for E36K. The creation of an underlying E2F binding site may be an additional explanation for the frequently observed CCND1 E36K mutation. In support of this transcriptional perspective on the origin of E36K, we find that the Y44D mutation, which has been reported to increase CCND1 stability [11], is not a preferred mutation target in our MCL data set. Consistent with the view that in MCL the CCND1 transcription level is increased by aSHM mediated mutagenesis, we observed more synonymous and benign amino acid changes in exon1 as compared to non-MCL samples with t(11;14)(q13;q32) translocation and mutated IGHV.
We observed statistically significant differential regulation of CCND1 transcription depending on CCND1 mutation status in 28 nnMCL cases present in our cohort. This number does not allow us to determine whether aberrations in the 3’UTR are mutually exclusive with aSHM-generated mutations in CCND1. Alternative polyadenylation [7], aberrations of miRNA target sites [6], or alternative splicing [52] could have an impact on CCND1 mRNA levels. All samples used in this analysis are lymphomas and express CCND1 mRNA at a sufficient level to sustain lymphomagenesis. This circumstance negatively influences the significance of aSHM-mediated upregulation of CCND1 expression levels. Furthermore, we have not been able to establish whether the CCND1 mutation status has an impact on the severity of the disease. The utility of non-coding CCND1 mutations as markers for the diagnosis of nnMCL is another open question.
In conclusion, we find that non-coding mutations in the CCND1 locus are clustered to TFBSs in a MCL-specific manner. These mutations are associated with higher levels of CCND1 transcript. Assuming that CCND1 activity is a rate-limiting factor in MCL lymphomagenesis, aSHM-generated mutations at the CCND1 locus may be actively selected for their impact on transcription regulation. The selection process may be particularly pronounced in MCL, which could explain the abundance of non-coding CCND1 mutations in this disease.
References
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer; 2017.
Jain P, Wang M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am J Hematol. 2019;94:710–25.
Raffeld M, Jaffe ES. bcl-1, t(11;14), and mantle cell-derived lymphomas. Blood. 1991;78:259–63.
Williams ME, Meeker TC, Swerdlow SH. Rearrangement of the chromosome 11 bcl-1 locus in centrocytic lymphoma: analysis with multiple breakpoint probes. Blood. 1991;78:493–8.
Deshpande A, Pastore A, Deshpande AJ, Zimmermann Y, Hutter G, Weinkauf M, et al. 3’UTR mediated regulation of the cyclin D1 proto-oncogene. Cell Cycle. 2009;8:3592–600.
Wiestner A, Tehrani M, Chiorazzi M, Wright G, Gibellini F, Nakayama K, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood. 2007;109:4599–606.
Gladden AB, Woolery R, Aggarwal P, Wasik MA, Diehl JA. Expression of constitutively nuclear cyclin D1 in murine lymphocytes induces B-cell lymphoma. Oncogene. 2006;25:998–1007.
Lu F, Gladden AB, Diehl JA. An alternatively spliced cyclin D1 isoform, cyclin D1b, is a nuclear oncogene. Cancer Res. 2003;63:7056–61.
Krieger S, Gauduchon J, Roussel M, Troussard X, Sola B. Relevance of cyclin D1b expression and CCND1 polymorphism in the pathogenesis of multiple myeloma and mantle cell lymphoma. BMC Cancer. 2006;6:238.
Mohanty A, Sandoval N, Das M, Pillai R, Chen L, Chen RW, et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget. 2016;7:73558–72.
Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119:1963–71.
Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110:18250–5.
Nadeu F, Martin-Garcia D, Clot G, Diaz-Navarro A, Duran-Ferrer M, Navarro A, et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood. 2020;136:1419–32.
Pararajalingam P, Coyle KM, Arthur SE, Thomas N, Alcaide M, Meissner B, et al. Coding and noncoding drivers of mantle cell lymphoma identified through exome and genome sequencing. Blood. 2020;136:572–84.
Longerich S, Basu U, Alt F, Storb U. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol. 2006;18:164–74.
Stengel A, Shahswar R, Haferlach T, Walter W, Hutter S, Meggendorfer M, et al. Whole transcriptome sequencing detects a large number of novel fusion transcripts in patients with AML and MDS. Blood Adv. 2020;4:5393–401.
Hollein A, Twardziok SO, Walter W, Hutter S, Baer C, Hernandez-Sanchez JM, et al. The combination of WGS and RNA-Seq is superior to conventional diagnostic tests in multiple myeloma: ready for prime time? Cancer Genet. 2020;242:15–24.
Fantini D, Vidimar V, Yu Y, Condello S, Meeks JJ. MutSignatures: an R package for extraction and analysis of cancer mutational signatures. Sci Rep. 2020;10:18217.
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–9.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Finocchiaro G, Mancuso FM, Cittaro D, Muller H. Graph-based identification of cancer signaling pathways from published gene expression signatures using PubLiME. Nucleic Acids Res. 2007;35:2343–55.
Muller H, Mancuso F. Identification and analysis of co-occurrence networks with NetCutter. PLoS ONE. 2008;3:e3178.
Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015;43:W39–49.
Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92.
Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov RN, Fedorova AD, Rumynskiy EI, et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018;46:D252–D9.
Nadeu F, Mas-de-Les-Valls R, Navarro A, Royo R, Martin S, Villamor N, et al. IgCaller for reconstructing immunoglobulin gene rearrangements and oncogenic translocations from whole-genome sequencing in lymphoid neoplasms. Nat Commun. 2020;11:3390.
Gagniuc P, Ionescu-Tirgoviste C. Eukaryotic genomes may exhibit up to 10 generic classes of gene promoters. BMC Genomics. 2012;13:512.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–D7.
Kasar S, Kim J, Improgo R, Tiao G, Polak P, Haradhvala N, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat Commun. 2015;6:8866.
Clot G, Jares P, Gine E, Navarro A, Royo C, Pinyol M, et al. A gene signature that distinguishes conventional and leukemic nonnodal mantle cell lymphoma helps predict outcome. Blood. 2018;132:413–22.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459:722–5.
Thorselius M, Walsh S, Eriksson I, Thunberg U, Johnson A, Backlin C, et al. Somatic hypermutation and V(H) gene usage in mantle cell lymphoma. Eur J Haematol. 2002;68:217–24.
Cesarman E, Dalla-Favera R, Bentley D, Groudine M. Mutations in the first exon are associated with altered transcription of c-myc in Burkitt lymphoma. Science. 1987;238:1272–5.
Muschen M, Re D, Jungnickel B, Diehl V, Rajewsky K, Kuppers R. Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. J Exp Med. 2000;192:1833–40.
Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci USA. 1998;95:11816–21.
Tamborero D, Gonzalez-Perez A, Lopez-Bigas N. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics. 2013;29:2238–44.
Di Giorgio E, Hancock WW, Brancolini C. MEF2 and the tumorigenic process, hic sunt leones. Biochim Biophys Acta Rev Cancer. 2018;1870:261–73.
Tiacci E, Ladewig E, Schiavoni G, Penson A, Fortini E, Pettirossi V, et al. Pervasive mutations of JAK-STAT pathway genes in classical Hodgkin lymphoma. Blood. 2018;131:2454–65.
Ferrando AA, Neuberg DS, Dodge RK, Paietta E, Larson RA, Wiernik PH, et al. Prognostic importance of TLX1 (HOX11) oncogene expression in adults with T-cell acute lymphoblastic leukaemia. Lancet. 2004;363:535–6.
Soulier J, Clappier E, Cayuela JM, Regnault A, Garcia-Peydro M, Dombret H, et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood. 2005;106:274–86.
Kanduri M, Sander B, Ntoufa S, Papakonstantinou N, Sutton LA, Stamatopoulos K, et al. A key role for EZH2 in epigenetic silencing of HOX genes in mantle cell lymphoma. Epigenetics. 2013;8:1280–8.
Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–84.
Wolf C, Garding A, Filarsky K, Bahlo J, Robrecht S, Becker N, et al. NFATC1 activation by DNA hypomethylation in chronic lymphocytic leukemia correlates with clinical staging and can be inhibited by ibrutinib. Int J Cancer. 2018;142:322–33.
Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, et al. A mechanism of cyclin D1 action encoded in the patterns of gene expression in human cancer. Cell. 2003;114:323–34.
Vegliante MC, Palomero J, Perez-Galan P, Roue G, Castellano G, Navarro A, et al. SOX11 regulates PAX5 expression and blocks terminal B-cell differentiation in aggressive mantle cell lymphoma. Blood. 2013;121:2175–85.
Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer. 2019;19:326–38.
Betticher DC, Thatcher N, Altermatt HJ, Hoban P, Ryder WD, Heighway J. Alternate splicing produces a novel cyclin D1 transcript. Oncogene. 1995;11:1005–11.
Acknowledgements
The authors would like to thank all co-workers at the MLL Munich Leukemia Laboratory for approaching together many aspects in the field of leukemia diagnostics and research by their dedicated work, as well as all physicians for providing samples and caring for patients as well as collecting data.
Author information
Authors and Affiliations
Contributions
HM performed the analyses and wrote the manuscript. WW, SH, NN, and MM generated the WGS and WTS data. WK, TH, and CH supervised and coordinated the research.
Corresponding author
Ethics declarations
Competing interests
TH, CH, and WK declare part ownership of Munich Leukemia Laboratory (MLL). HM, WW, NN, SH, and MM are employed by the MLL.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Müller, H., Walter, W., Hutter, S. et al. Aberrant somatic hypermutation of CCND1 generates non-coding drivers of mantle cell lymphomagenesis. Cancer Gene Ther 29, 484–493 (2022). https://doi.org/10.1038/s41417-022-00428-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-022-00428-7
- Springer Nature America, Inc.