
Abstract
CD4+ T-cell large granular lymphocyte leukemia (T-LGLL) is a rare subtype of T-LGLL with unknown etiology. In this study, we molecularly characterized a cohort of patients (n = 35) by studying their T-cell receptor (TCR) repertoire and the presence of somatic STAT5B mutations. In addition to the previously described gain-of-function mutations (N642H, Y665F, Q706L, S715F), we discovered six novel STAT5B mutations (Q220H, E433K, T628S, P658R, P702A, and V712E). Multiple STAT5B mutations were present in 22% (5/23) of STAT5B mutated CD4+ T-LGLL cases, either coexisting in one clone or in distinct clones. Patients with STAT5B mutations had increased lymphocyte and LGL counts when compared to STAT5B wild-type patients. TCRβ sequencing showed that, in addition to large LGL expansions, non-leukemic T cell repertoires were more clonal in CD4+ T-LGLL compared to healthy. Interestingly, 25% (15/59) of CD4+ T-LGLL clonotypes were found, albeit in much lower frequencies, in the non-leukemic CD4+ T cell repertoires of the CD4+ T-LGLL patients. Additionally, we further confirmed the previously reported clonal dominance of TRBV6-expressing clones in CD4+ T-LGLL. In conclusion, CD4+ T-LGLL patients have a typical TCR and mutation profile suggestive of aberrant antigen response underlying the disease.
Similar content being viewed by others

Introduction
T-cell large granular lymphocyte leukemia (T-LGLL) is a rare lymphoproliferative disease characterized by chronic expansion of clonal, mature cytotoxic T cells in the peripheral blood and bone marrow [1]. Two subtypes of T-LGLL are commonly recognized: the most common CD8+ T-LGLL (70% of cases) and the less frequent CD4+ T-LGLL (30% of cases) [1]. Clinically, CD4+ T-LGLL is usually an indolent disease and unlike in its CD8+ counterpart, these patients rarely have cytopenias or autoimmune symptoms. However, CD4+ T-LGLL has been reported more frequently associated with secondary neoplasms such as monoclonal B-cell lymphocytosis and plasma cell disorders [2, 3]. In CD4+ T-LGLL, the leukemic T-LGLs express CD4 (either alone or together with CD8) and the α/β T-cell receptor (TCR) together with a typical mature cytotoxic (Granzyme B+, CD56+, CD57+) and activated/memory T-cell (CD2bright, CD7dim, CD11abright, CD28−, CD62L−) phenotype [3].
Up to 55% of CD4+ T-LGLL patients have been shown to harbor STAT5B mutations [4, 5]. In CD8+ T-LGLL, the most common mutated gene is STAT3 [6], whereas STAT5B mutations are rare and often associated with an aggressive disease form [7,8,9]. All reported STAT5B mutations in CD4+ T-LGLL are point mutations within the SH2 or transactivation domains of STAT5B. N642H and Y665F are the most common STAT5B mutations, and they both have been shown to increase STAT5B protein activity [4, 10, 11].
The etiology of CD4+ T-LGLL remains unknown. An initial antigen-driven expansion of CD4+ T cells, followed by the occurrence of oncogenic events (i.e., somatic mutations), has been suggested to lead to the persistence of abnormal T-cell clones [11]. Non-self-antigen(s) instead of autoantigens [5] are proposed targets of CD4+ T-LGL clones as CD4+ T-LGLL is not associated with autoimmune diseases. In some earlier reports, CD4+ T-LGL clones have been implied to recognize cytomegalovirus (CMV) antigens [12, 13]. In CD4+ T-LGLL [3], the enrichment of the Vβ13.1 gene usage has also been reported, differentiating it from CD8+ T-LGLL where no enrichment of specific Vβ gene usage has been observed [14, 15]. Moreover, CD4+ T-LGLL patients with a monoclonal expansion of TCRVβ13.1 display a common HLA-DRB1*07:01 genotype and are reported to display an identical motif (QG) in the middle of the CDR3 sequence [16, 17]. These observations suggest that the evolution of the expanded CD4+ T-LGLL clones is not a stochastic process, but rather a result of an antigen-driven immune response [18,19,20,21,22].
As CD4+ T-LGLL is a rare disease, previous studies evaluating the clinical impact of STAT5B mutations have been limited in size, and no deep TCR profiling has been performed with modern sequencing and bioinformatic tools [23,24,25]. Therefore, we aimed to collect a large cohort of CD4+ T-LGLL patients (n = 35) and examine by deep amplicon sequencing the STAT5B mutation status and correlate the genotype information with the clinical data. Additionally, by deep TCRβ sequencing, we studied the landscape of both the leukemic and non-leukemic T-cell repertoires in CD4+ T-LGLL and compared that to the landscapes from the healthy controls and patients with CD8+ T-LGLL.
Materials and methods
Patient cohort
CD4+ T-LGLL patients (n = 35) were recruited from the hematology units of 3 academic institutions, namely Padova (Italy; n = 22 patients), Helsinki (Finland; n = 6 patients) and Shinshu (Japan; n = 7 patients). Buffy coats from healthy blood donors (n = 37) were provided by the Finnish Red Cross Blood service (Helsinki, Finland). The study was conducted in accordance with the Ethics Committees (Padova University Hospital ethics committee, approval number 4213/AO/17; Helsinki University Hospital ethics committee, approval number 303/12/03/01/2011; Shinshu University Hospital ethics committee, approval number 581). All patients gave written informed consent prior to their inclusion in the study in accordance with the Declaration of Helsinki.
Sample preparation
Peripheral blood mononuclear cells (PBMC) were isolated with Ficoll-Hypaque (Sigma Aldrich) gradient centrifugation. Leukemic clones were purified using magnetic micro-beads coated with monoclonal anti-human CD4, CD57, or CD56 antibodies (Miltenyi Biotec). When available, cryopreserved cells (n = 15) were first subjected to depletion of CD8+ cells, after which the CD8-negative population (or CD8+ in case of CD4+ CD8+ double-positive LGL clone) was further purified with anti-human CD4-conjugated microbeads. Alternatively, CD57 (n = 8) or CD56 (n = 3) purified LGLs or PBMC (n = 9) were used depending on sample availability. A similar approach was used to purify CD4+ cells from buffy coats obtained from healthy controls (n = 37). Flow cytometry analysis was conducted to control the purity of the obtained cell fractions (>95% among lymphocytes) with FACSCanto II in Padova and FACS Verse in Helsinki; data were analyzed with FACS Diva and FACS Suite software, respectively (all Becton Dickinson). Surface staining was performed according to the purification: CD4 FITC CD8 PE or CD4 FITC CD57 PE or CD4 FITC CD56 APC (all Becton Dickinson). DNA was extracted using Gentra Puregene Cell and Tissue kit (Qiagen) and quantified using the Qubit 2.0 fluorometer with DNA high sensitivity kit (ThermoFisher Scientific).
Amplicon sequencing of STAT5B
STAT5B sequencing was performed from LGL or PBMC fraction (Fig. 1) to detect somatic mutations. Firstly, targeted sequencing was performed starting from 50 ng of DNA. Libraries, generated according to the manufacturer’s instructions, were sequenced on an Illumina MiSeq instrument using MiSeq Reagent kit v2 or v3 and following the TruSeq Custom Amplicon Assay (Illumina) pipeline. Bioinformatic variant calling was performed using MiSeq Reporter software (version 2.5.1) with default settings while for variant annotation Illumina Variant Studio 2.0 was used. Next, the observed mutations were confirmed using amplicon sequencing as previously described in Kim et al. [26]. Amplicon primers used for both techniques are reported in Supplementary Table 1. Germline variants were excluded by examination of a non-leukemic cell population from each patient. All identified variants were visually confirmed using the Integrative Genome Viewer (IGV) [27].

STAT5B mutations and the corresponding VAFs were evaluated in the LGLs or PBMC populations by deep amplicon sequencing. Novel STAT5B mutations are in light blue squares. The patients are divided into those with multiple STAT5B mutations (above) and with single STAT5B mutation (below). The square colors indicate mutation VAF percentages, as reported in the legend. The protein level localization of the identified STAT5B mutations is given on the top row. CCD coiled coil domain, DBD DNA-binding domain, TAD transactivation domain, VAF variant allele frequency.
Western blot assay and STAT5B luciferase reporter assay
Please refer to Supplementary methods and Supplementary Table 2.
TCRβ sequencing and data analysis
TCRβ sequencing from genomic DNA (the same DNA as used for amplicon sequencing, Fig. 1 and Table 1) of CD4+ T-LGLL patients (n = 27) was conducted with Adaptive Biotechnologies’ ImmunoSEQ assay with “Survey” resolution [28]. Only productive, i.e., complete, in-frame, TCRβ rearrangements were included in the analyses. Sample clonality was assessed using the Simpson clonality as provided by the Adaptive Immunoseq Analyzer (ver 3.0). Healthy controls (n = 785) from peripheral blood [29] were also used after downsampling to 40,000 reads.
Clones, defined as unique rearranged receptor sequences in the immunoSEQ Analyzer, were classified as very rare (≤0.01%), small (0.01–0.1%), medium (0.1–1%), large (1–20%), and hyperexpanded (>20%). The lowest detectable productive frequency in the TCR-sequencing data was 2.59 × 10−5. OLGA [25] (ver 1.2.3) was used to calculate the generation probabilities of the TCR rearrangements with default parameters.
VDJtools was used to generate circus plots for V gene usage from TCRβ-sequencing data [30]. The query against the VDJ database (VDJdb; ver 2021-01-10) [24] was done for all available TCRs from CD4+ cells. To evaluate possible amino acid level similarities of TCRs, GLIPH (ver 1.0.0 and 2.0.0) [23, 31] was used on a remote server with default parameters.
Statistical methods
Statistical analysis for the evaluation of differential STAT5B transcriptional activities through luciferase reporter assay was performed with a two-way ANOVA followed by Dunn’s multiple correction test. Fisher’s exact, Chi-square, or Bonferroni corrected Mann–Whitney tests were used to analyze the relationship between STAT5B mutational status, TCRβ repertoire covariates, and relative clinical features of the cohort of patients profiled (see Table 2 for more details). Fisher’s exact test with Bonferroni multiple correction was used to calculate V genes enrichment for CD4+ T-LGLL clones. All the analyses were performed with GraphPad Prism 6 (ver 6.07) or R (ver 4.0.2) and p < 0.05 was considered significant.
Results
STAT5B mutation status is associated with clinical features in CD4+ T-LGLL
Clinical characteristics of the entire CD4+ T-LGLL cohort (n = 35) are shown in Table 2a. The median age at diagnosis was 72 years (ranging from 41 to 86 years) and no gender bias was observed (ratio Males/Females = 1.06). White blood cell (WBC) and LGL counts were overall high (9.4 ± 4 × 109/l and 3.45 ± 2.40 × 109/l, respectively). Absolute neutrophil count (ANC), hemoglobin (Hb), and platelets (PLT) were mostly within the normal range. Half of the patients (49%, n = 17) presented a CD4+CD8− leukemic expansion while 31% (n = 11) had a CD4+CD8+ leukemic clone. Moreover, 20% (n = 7) of the patients harbored two coexisting clonal expansions, six presenting one CD4+CD8− and one CD4+CD8+ clone, whereas patient #29 presented one CD4+CD8+ and one CD4−CD8+ clone (Supplementary Table 3). All CD4+ T-LGLL clones were CD57+ and the majority of them (79%, n = 26 patients) were CD56+CD16−. Three patients harbored an expanded CD56−CD16− clone. Conversely, none of the patients presented a CD56−CD16+ T-LGLL clone. A detailed table of the clones and associated clinical features is presented in Supplementary Table 3.
By targeted amplicon sequencing, we discovered that 23 out of 35 patients (66%) harbored somatic STAT5B point mutations (Fig. 1). No differences were detected in the median age between the two groups, whereas STAT5B mutations were more frequent in males (Fisher’s exact test, p = 0.035; Fig. 2a and Table 2b). Patients with STAT5B mutations displayed increased LGL counts and lymphocytosis when compared to the STAT5B wild-type patients (Mann–Whitney test, p = 0.006 for both variables; Fig. 2b, c and Table 1b). There was no statistically significant difference in the platelet counts between the mutated and wild-type patients, while a tendency towards higher ANC and Hb levels in the STAT5B mutated patients was noted (p = 0.07 and p = 0.06, respectively; Fig. 2d, e).

Box and bar plots of the clinical features in STAT5B mutated and wild-type patients: a Sex, b LGL count, c WBC, d ANC, e Hb All clinical features were analyzed with Mann–Whitney U-test except for sex prevalence which was analyzed with Fisher’s exact test. WBC white blood cells, LGL large granular lymphocytes, ANC absolute neutrophil count, Hb hemoglobin, wt wild-type.
STAT5B mutations were particularly common in patients with CD4+CD8+ phenotype (82%, 9 of 11 patients). Furthermore, all patients (n = 4) having CD57+CD56+CD16+ phenotype had STAT5B mutation, whereas in patients with CD57+CD56+CD16− phenotype mutation prevalence was 62% (16 of 26 patients).
Identification of multiple and novel STAT5B mutations
Among the mutated cases (n = 23), 78% of the patients (n = 18) harbored STAT5B mutations with a variant allele frequency (VAF) over 15%, whereas the remaining 22% of cases (n = 5) presented lower VAFs ranging from 1% to 6% (Fig. 1 and Table 1).
Multiple STAT5B mutations were detected in five patients (#9, #11, #12, #18, #29) (Fig. 1), corresponding to 22% of the mutated CD4+ T-LGLL cases. Careful examination of the sequencing reads revealed that in two cases (#12 and #18) the closely located genetic lesions were present in different alleles. For patient #29, amplicon sequencing was performed independently in the two purified leukemic clones. In the CD4+CD8+ clone, we identified the N642H variant (VAF 49%) and in the CD4−CD8+ clone, we identified the E433K mutation (VAF 34%). Patient #9 harbored P685R, N642H, and E433G mutations with almost identical VAF (47%), thus suggesting that these heterozygous mutations co-localized in the dominant leukemic clone (97%). In patient #11, the 2 mutations (Y665F, V712E) were sequenced in separate amplicons and although the exact clonal structure was not possible to establish, significantly varying VAFs (32% and 11%) suggest clonal heterogeneity.
Besides the previously identified STAT5B mutations in CD4+ T-LGLL (N642H, Y665F, Q706L, and S715F [4]), we discovered additional six novel somatic STAT5B mutations in the cohort. T628S (n = 5) and P685R (n = 1) mutations are located in the SH2 domain, whereas the V712E mutation (n = 2) is in the transactivation domain (TAD), both of which are mutational hotspot regions of the STAT5B gene. In addition, we identified four additional mutations that are located in the coiled-coil domain (CCD; Q220H, n = 1), DNA binding domain (DBD; E433G/K, n = 2), and in the inter-domain region (P702A, n = 1).
The most frequent mutation was N642H (9/23, 39%), followed by T628S (5/23, 22%), S715F (4/23, 17%) and Y665F (3/23, 13%). Two different base substitutions led to the same missense T628S mutation: the wild-type nucleotide triplet ACC was found to mutate to either AGC or TCC, both inducing the conversion from tyrosine (T) to serine (S). The first nucleotide alteration has already been listed in COSMIC [32], whereas the latter is novel. Interestingly, the two possible substitutions leading to the same amino acid conversion were both identified in different alleles (i.e., in different reads) of patient #12.
E433K and V712E are novel activating STAT5B mutations associated with CD4+ T-LGLL
Functional characterization of E433K, V712E, and P685R mutations was performed to determine their impact on the transcriptional activity of the STAT5B gene; known activating variant N642H and the wild-type STAT5B constructs were used as positive and negative controls, respectively (Fig. 3a). Q220H and P702A mutations were excluded from the analysis because of their low VAF (2% and 3%, respectively). T628S mutation with gain-of-function property has already been described before in T-prolymphocytic leukemia (T-PLL) [33].

a Luciferase reporter assay revealed the STAT5B transcriptional activity induced by the E433K, V712E, and P685R mutations. By using STAT5B mutants-transfected HeLa cells, luciferase activities of STAT5B mutations were compared to the wt STAT5B data and are reported as luminescence counts. WT STAT5B: a negative control. STAT5B N642H mutation: a positive control. Statistical analysis was performed using two-way ANOVA. WT: wild-type; **p < 0.001; ***p < 0.0001. b Protein expression in HeLa cells transiently carrying STAT5B mutants. The cells were serum-starved for 6 h after the transfection. Protein extracts were subjected to western blot assay with p-STAT5, STAT5, c-MYC, Bcl-2, and Pim-1 specific antibodies. β-actin served as loading control. Data are representative of the three independent experiments.
As shown in Fig. 3a, both E433K and V712E were found to be highly activating when compared to wild-type STAT5B (p < 0.001, two-way ANOVA followed by Dunn’s multiple correction test). There was at least a 10-fold increase in the luminescent signal for the E433K mutation and a 15-fold increase for the V712E mutation when compared to the wild-type STAT5B. The signal from the P685R mutated plasmid did not differ from the wild-type construct. This mutation was detected in the patient with co-existing N642H and E433K mutations. In addition, HeLa cells transiently carrying the STAT5B GOF mutants exhibited upregulated phosphorylation of STAT5B protein compared to the cells carrying WT STAT5B (Fig. 3b). Furthermore, STAT5B mutated cells had increased protein expression levels of c-MYC, Bcl-2, and Pim-1 which are downstream targets of STAT5 [34].
Both the leukemic and non-leukemic TCRβ repertoires are highly clonal in CD4+ T-LGLL
TCRβ sequencing (TCRβ-seq) was performed on samples from CD4+ T-LGLL patients (n = 27) and healthy controls (n = 37). From T-LGLL patients, genomic DNA was isolated from bead-separated cells (CD4+, CD4+CD8+, CD56+ or CD57+) or PMBC depending on sample availability (Table 1). In healthy controls, CD4+ selected fractions were used. As expected, CD4+ T-LGLL patients harbored larger T-cell clones than in healthy controls (Fig. 4a). Both monoclonal and oligoclonal expansion patterns were observed. By focusing on clones with clone size exceeding 5% of the repertoire, we identified a total of 59 possible T-LGLL clones from the 27 patients (Supplementary Table 4).

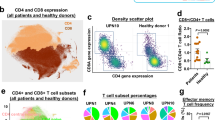
a The ten most expanded clones as proportion of total repertoire from all samples are shown. The top 3 clones are shown in individual colors while the sum of the rest (clones 4–10) is shown in one color. The cell isolation method (CD4, CD4 CD8, CD57, CD56, or non-selected PBMC) of the analyzed cell population and the mutational STAT5B status of the patients are given below the sample numbers. b Total CD4+ repertoire clonality as normalized Shannon–Wiener clonality index from CD4+ T-LGLL (patient samples purified for CD4, n = 6) and CD4+ cells from healthy controls (n = 37). c Non-leukemic CD4+ repertoire clonality (selecting the CD4+ T-LGL clones with a size < 5%) as normalized Shannon–Wiener clonality index from CD4+ T-LGLL (patient samples purified for CD4, n = 6) and CD4+ cells from healthy controls (n = 37). d Total repertoire clonality as normalized Shannon–Wiener clonality index from CD4+ T-LGLL (patient samples purified for CD4, n = 6) and CD8+ cells from CD8+ T-LGLL (patient samples purified for CD8, n = 9). e Non-leukemic repertoire clonality (selecting the CD4+ T-LGL clones with a size < 5%) as normalized Shannon–Wiener clonality index from CD4+ T-LGLL (patient samples purified for CD4, n = 6) and CD8+ cells from CD8+ T-LGLL (patient samples purified for CD8, n = 9). All p-values were calculated with a Mann–Whitney test and corrected with Bonferroni.
For the CD4+ sorted samples, the overall TCRβ clonality was higher in CD4+ T-LGLL (n = 6) as compared to healthy controls (n = 37) (Fig. 4b, padj < 0.05, Bonferroni corrected Mann–Whitney test). Even after removing the leukemic T-LGLL clones from the patient samples, the observed difference in the clonality persisted (Fig. 4c, padj < 0.05, Bonferroni corrected Mann–Whitney test). Thus, the non-leukemic TCR repertoires in CD4+ T-LGLL patients are less diverse than the CD4+ TCR repertoires in the healthy controls. By repeating the analysis with varying thresholds of 3–7% for selecting the putative T-LGLL clones, we showed that this was invariant to our chosen threshold of 5% (Supplementary Fig. 1).
We also compared a similarly profiled CD8+ T-LGLL cohort [35] (n = 9) with the CD4+ T-LGLL cohort. Surprisingly, the overall clonality in CD4+ T-LGLL samples was higher than in the CD8+ T-LGLL samples (Fig. 4d, padj < 0.05, Bonferroni corrected Mann–Whitney test), although it is known that CD8+ TCR repertoires are more clonal than CD4+ repertoires [36]. This was however not seen in the non-leukemic repertoires, where the non-leukemic compartments from CD8+ T-LGLL samples had higher clonality than the non-leukemic compartments from CD4+ T-LGLL samples (Fig. 4e, padj < 0.05, Bonferroni corrected Mann–Whitney test).
Most CD4+ T-LGLL-associated STAT5B mutations are clonally restricted
For most of the patients (71%) presenting with a STAT5B mutated clone over 15%, the VAF of the STAT5B mutation corresponded to the size of the main T-LGLL clone: the mutation VAFs were half of the size of the TCR clones suggesting heterozygous mutations (Table 1). Only in patients #2 and #15, the mutation VAFs were slightly higher, suggesting that there was more than one T-cell clone harboring the same mutation. In patient #21 the mutation VAF (23%) matched with the combined size of the two main T-LGLL clones (24% and 22%), suggesting that the mutation is present either in one clone in homozygous/hemizygous fashion or in both clones as heterozygous (Table 1). Interestingly, patients with low VAF (<5%) STAT5B mutations (#8, #13, #17, and #24) had nevertheless large T-LGLL clones (37–71%) suggesting that they had either subclonal STAT5B mutations or the mutations were located outside the main clone (Table 1).
In patients with multiple STAT5B mutations, the clonal structure was evaluated based on the VAF and TCR clone size. In patient #9, the three mutations co-existed in the same clone based on their similar VAFs (47%) matching with the size of the main clone (97%) (Table 1). In patient #18, the Y665F (VAF 34%) and N642H (VAF 5%) mutations were detected in different subclones, and accordingly, also two expanded TCR clonotypes were detected (Table 1). Patient #11 had one large main TCR clone (90%) but two different STAT5B mutations with different VAFs (Y665F: 32% and V712E: 11%) (Fig. 1 and Table 1). This suggests possible clonal evolution and the presence of different genetic lesions in individual cells bearing the same TCR.
CD4+ T-LGLL clonotypes are predominantly restricted to individual patients
To understand whether the 59 identified CD4+ T-LGLL clonotypes (Supplementary Table 4) could be a result of convergent evolution, we examined if these clones were shared between patients. In concordance with what has been reported before in CD8+ T-LGLL [14], none of the patients had the same TCR rearrangement in their main leukemic CD4+ T-LGLL clonotypes (Fig. 5a). However, 25% (15/59) of T-LGLL clonotypes were found in the non-leukemic TCR repertoires of other patients, albeit in smaller frequencies (ranging between frequencies of 1.41 × 10−5–1.27 × 10−2, Supplementary Table 5, Fig. 5a). Of these 59 clones, 27% (16/59) were found in healthy CD4+ sorted TCR repertoire (n = 37), again in much smaller frequencies (ranging between 8.24 × 10−5–2.59 × 10−5) (Supplementary Table 5, Fig. 5a). We further looked for the presence of the CD4+ T-LGLL clonotypes in a larger publicly available cohort (n = 786) of healthy donors’ unsorted TCR repertoire from Emerson et al. [29]. We observed that 36/59 of the patient clones (around 61%) were found at least once in the healthy donors with the most common clone being found in 163 healthy donors (Fig. 5b). However, most of these clones were found in extremely rare frequencies (ranging between 8.2 × 10−5 and 1.05 × 10−5).

a CD4+ T-LGLL clonotypes are shared between CD4+ T-LGLL patients (n = 27) and healthy cohort (n = 37) in extremely rare frequencies. Shared clonotypes are at the bottommost rows and the colors refer to the size of the clonotype. b CD4+ T-LGLL clonotypes found in smaller frequencies in publicly available, unsorted, healthy cohort data (n = 786) [29] where the colors refer to the size of the clonotype.
We performed a similar analysis with our CD8+ T-LGLL cohort. None of the CD8+ leukemic clones (defined similarly as clones above 5%) was shared between the patients (Supplementary Fig. 2a). 50% of these clones were found in the Emerson et al. [29] cohort in low frequencies (Supplementary Fig. 2b).
Next, we calculated the generation probabilities of the leukemic TCRs with OLGA [25] as high generation probabilities could indicate a bias in the recombination and low generation probabilities towards convergent evolution. The TCR generation probabilities were higher in clones that were shared between healthy individuals. Conversely, the generation probabilities in clones that were exclusive to CD4+ T-LGLL were low (Supplementary Fig. 3).
Skewed Vβ gene usage in CD4+ T-LGLL clones
Previous studies using Vβ flow cytometry analysis have suggested a biased usage of the Vβ13.1 family (consisting of TRBV06-05, TRBV06-06, and TRBV06-09 genes) in CD4+ T-LGLL [16]. In our data, the predominant V-gene of CD4+ T-LGLL clonotypes belonged to the TRBV06 family in 20% (12/59) of CD4+ T-LGLL clonotypes (Supplementary Table 6, Supplementary Fig. 4). By selecting the single most expanded clones, we found that 8 out of 27 T-LGLL clones belonged to the TRBV06 family, in comparison to only 2 out of the 35 clones belonging to the TRBV06 family in healthy (Fig. 6a, p = 0.01, Fisher’s two-sided exact test). Representative examples from CD4+ T-LGLL, with both monoclonal and polyclonal expansions, and healthy are shown in Fig. 6b.

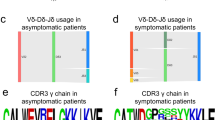
a Comparison of V gene family usage between CD4+ T-LGLL clones and healthy controls. 20% of the leukemic clones express the TRBV06 gene. b Circos plots showing skewed V gene usage in two representative CD4+ T-LGLL as compared to one representative healthy sample. c Logo plot showing the non-conservative middle part of TCRs CDR3, which is usually within 5 Å of the antigen. The “QG” motif that was found in the middle CDR3 in multiple CD4+ T-LGLL clones is shown in CDR3s that are 13 amino acids long.
To test whether the CD4+ T-LGLL clones target a known viral antigen (from e.g., CMV, EBV, or Influenza A), we queried the 59 T-LGLL clones against the largest database containing known antigen specificities (VDJdb [24]) but no exact matches for TCRs recognizing epitope bound to class II HLAs were found. The antigen specificities of the non-leukemic TCRs returned multiple matches, with Influenza A as the most common target (Supplementary Fig. 5a).
Since structurally similar TCRs are known to recognize similar antigens [23, 31, 37], we used GLIPH to identify significantly enriched motifs within CD4+ T-LGLL clonotypes in comparison to healthy naïve repertories. Interestingly, we identified two conserved motifs within the 59 T-LGLL clonotypes. The first one, “SDP”, in three T-LGLL clones all of which shared the TRBV24 gene, and the second one, “SLRG”, in three other T-LGLL clones with different V genes (Supplementary Table 7). No correlation was observed between motifs and the mutational statuses of the patients. Next, we analyzed whether the T-LGLL clonotypes shared motifs within their repertoire, i.e., whether the T-LGLL responses could be polyclonal antigen-specific responses. In 33% (2/6) of CD4+ sorted patients’ samples, we could identify a motif that was shared between leukemic and non-leukemic repertoires. Structurally similar TCRs from patient #18 are shown in Supplementary Fig. 5b, where the leukemic clone can be seen to share similarities with the multiple non-leukemic TCRs.
Although GLIPH does not consider motifs less than three amino acids long, Garrido et al. have previously reported the presence of the “QG” motif in the middle of the CDR3 region in patients with monoclonal Vβ13.1 expansion and HLA-DRB1*07:01 genotype [16]. We found this “QG” motif in 18.6% (11/59) of T-LGLL clones (of which only two expressed Vβ13.1), but also in 13.4% (20/144) of top 5 expanded clonotypes in healthy CD4+ repertoire (Fig. 6c, Supplementary Table 4, p = 0.39, Fisher’s two-sided exact test). To address whether “QG” is specific to CD4+ T-LGLL or specific to HLA-DRB1*07:01 in general, we analyzed whether “QG” was more common in CD4+ clones of HLA-DRB1*07:01 positive (n = 5) than negative (n = 32) healthy donors. We found that “QG” was enriched in HLA-DRB1*07:01 positive healthy donors (3.5% of CD4+ clones) in comparison to negative donors (0.54% of CD4+ clones; p = 7.53 × 10−122, Fisher’s one-sided exact test), further suggesting its association with CD4+ cells and HLA-DRB1*07:01 in general and not specifically to CD4+ T-LGLL.
Discussion
Somatic STAT5B mutations were initially described in CD8+ T-LGLL [9], but follow-up analyses have revealed that they more often occur in CD4+ and immature T-cell malignancies [4, 33, 38,39,40,41,42]. Here, we performed a detailed molecular characterization of 35 CD4+ T-LGLL patients, which is the largest cohort described so far. We identified novel gain of function STAT5B mutations and found that leukemic clonotypes are predominantly private to the patients.
At least one STAT5B mutation was detected in 66% of the 35 CD4+ T-LGLL patients. This is the highest frequency of STAT5B mutations thus far reported in the literature. While the previously published studies have mostly focused on the mutational hotspot regions of STAT5B [5, 9], our sequencing assay covered the whole protein-coding sequence of STAT5B. Moreover, the use of a highly sensitive method allowed us to detect low-frequency mutations down to 1% VAF.
Functional analyses of the newly discovered STAT5B mutations confirmed STAT5 pathway activation, as the downstream targets of STAT5 (c-MYC, Bcl-2, and Pim-1 proteins) [34] were overexpressed. Similarly, a previous study has shown that STAT5B mutant-transduced cell lines upregulate Bcl-2 [43]. c-MYC is an essential regulator of cell proliferation and survival [34], and Pim-1, an anti-apoptotic gene, is known to synergize with c-MYC in leukemogenesis [44]. Altogether, this data suggests that the STAT5B GOF mutations are likely to be involved in clonal dominance and maintenance, probably by conferring proliferation advantage for the mutated cells. Accordingly, STAT5B mutated CD4+ T-LGLL patients had higher lymphocyte and LGL counts compared to wild-type patients.
In addition to the earlier described N642H, S715F, and Y665F mutations, we discovered the T628S mutation to be common in CD4+ T-LGLL (22% of our cohort). This mutation has been previously reported as a highly activating mutation in T-PLL [33, 45] and hepatosplenic T-cell lymphoma (HSTCL) [42]. Although SH2 and TAD domains are the known mutation hotspot area in the STAT5B gene, we also identified novel variants in the CCD (Q220H) and DBD (E433K/G) of STAT5B. Similarly, the majority of the STAT3 mutations are in the SH2 domain in CD8+ T-LGLL but additional mutations are also found in other domains [4] underlining the importance of covering all mutation hotspots in the diagnostic tests of LGLL.
Among the STAT5B mutated CD4+ T-LGLL patients, 22% harbored multiple STAT5B mutations either in the main T-LGLL clone or in separate smaller clones. Interestingly, mutation VAFs did not always correlate with the T-cell clone size based on TCRβ sequencing, suggesting sub-clonal heterogeneity. Similarly, 17% of CD8+ T-LGLL patients have been reported to harbor multiple STAT3 mutations [46]. Our results are consistent with previous data, suggesting that in T-LGLL additional somatic mutations can arise in the pre-expanded clonotypes [47].
In CD8+ T-LGLL, STAT3 mutations have been associated with autoimmune manifestations such as rheumatoid arthritis and neutropenia [2, 5, 8, 48, 49]. In CD4+ T-LGLL, neutropenia is uncommon, and similarly, in our cohort STAT5B mutated patients had normal ANC levels but increased lymphocyte and LGL counts. Irrespective of the mutation status, the phenotype of the CD4+ T-LGLL clone was in most (75%) cases CD16−CD56+. Although CD16+CD56+ phenotype was detected in only four cases, all these had mutated STAT5B. CD56+ immunophenotype has also been associated with STAT5B mutations in CD8+ T-LGLL [9] in which the LGLs are usually CD56-.
TCRβ-seq has provided new insights into the nature of clonal expansions in CD8+ T-LGLL [14] but has not been performed in CD4+ T-LGLL earlier. In accordance with the previous flow cytometry-based data, in CD4+ LGLL the TRBV06 family was enriched in the most hyper-expanded clones. Interestingly, 25% of T-LGLL clonotypes were also found among the non-expanded TCR repertoire in CD4+ T-LGLL patients and 27% were found in healthy individuals. This was not observed in the CD8+ T-LGLL disease, consistent with previous results [14]. Although our data was not HLA-matched, this could hint that the eliciting antigen in CD4+ T-LGLL could be commonly encountered. Further, in CD4+ T-LGLL also the non-leukemic T-cell compartment was significantly more clonal than in healthy controls. It has been shown previously, that CD4+ T cells in healthy controls are much richer in their diversity as compared to CD8+ T cells [50]. Hence, it was interesting to note that the overall clonality was higher in CD4+ T-LGLL compared to CD8+ T-LGLL, but this could also be due to the relatively small sample size. However, the eliciting antigen(s) remain unknown, as no exact matches were found for the known antigen-specific TCRs, which could be due to the small amount of known class II pMHC–TCR pairs and potential bias in the HLA genotypes of our samples. Additionally, since previous literature has indicated that a significant proportion of CD4+ T-LGLL patients has an underlying disease or malignancy [3], it cannot be excluded that the triggering event is abnormal antigen stimulation such as tumor antigen.
More research is warranted to clarify whether CD4+ T-LGLL is a true hematological malignancy or a reactive disorder of the immune system as clonal T cell proliferations and somatic mutations are also observed in healthy controls and non-malignant diseases [26, 51]. Following the initial event, activating STAT5B mutations may boost the aberrant proliferation and clonal persistence. Despite the STAT5B hyperactivation and the presence of multiple STAT5B mutations, the clinical course of CD4+ T-LGLL is indolent, and patients rarely have symptoms, differentiating it from CD8+ T-LGLL and other mature and immature CD4+ T-cell leukemias and lymphomas.
Data availability
TCRβ sequencing data is freely available at https://clients.adaptivebiotech.com/pub/bhattacharya-2022-BCJ (https://doi.org/10.21417/DB2022BCJ).
References
Matutes E. The 2017 WHO update on mature T- and natural killer (NK) cell neoplasms. Int J Lab Hematol. 2018;40:97–103.
Kuwahara N, Kodaka T, Zushi Y, Sasaki M, Goka T, Maruoka H, et al. T-cell large granular lymphocytic (LGL) leukemia consists of CD4/CD8 and CD4/CD8 LGL populations in association with immune thrombocytopenia, autoimmune neutropenia, and monoclonal B-cell lymphocytosis. J Clin Exp Hematop. 2019;59:202–6.
Lima M, Almeida J, dos Anjos Teixeira M, del Carmen Alguero M, Santos AH, Balanzategui A, et al. TCRαβ/CD4 large granular lymphocytosis. Am J Pathol. 2003;163:763–71.
Andersson EI, Tanahashi T, Sekiguchi N, Gasparini VR, Bortoluzzi S, Kawakami T, et al. High incidence of activating STAT5B mutations in CD4-positive T-cell large granular lymphocyte leukemia. Blood. 2016;128:2465–8.
Barilà G, Teramo A, Calabretto G, Vicenzetto C, Gasparini VR, Pavan L, et al. Stat3 mutations impact on overall survival in large granular lymphocyte leukemia: a single-center experience of 205 patients. Leukemia. 2020;34:1116–24.
Rajala HLM, Porkka K, Maciejewski JP, Loughran TP Jr, Mustjoki S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann Med. 2014;46:114–22.
Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905–13.
Teramo A, Barilà G, Calabretto G, Ercolin C, Lamy T, Moignet A, et al. STAT3 mutation impacts biological and clinical features of T-LGL leukemia. Oncotarget 2017;8:61876–89.
Rajala HLM, Eldfors S, Kuusanmäki H, van Adrichem AJ, Olson T, Lagström S, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121:4541–50.
Teramo A, Barilà G, Calabretto G, Vicenzetto C, Gasparini VR, Semenzato G, et al. Insights into genetic landscape of large granular lymphocyte leukemia. Front Oncol. 2020;10:152.
Barilà G, Calabretto G, Teramo A, Vicenzetto C, Gasparini VR, Semenzato G, et al. T cell large granular lymphocyte leukemia and chronic NK lymphocytosis. Best Pract Res Clin Haematol. 2019;32:207–16.
Rodríguez-Caballero A, García-Montero AC, Bárcena P, Almeida J, Ruiz-Cabello F, Tabernero MD, et al. Expanded cells in monoclonal TCR-alphabeta+/CD4+/NKa+/CD8−/+dim T-LGL lymphocytosis recognize hCMV antigens. Blood. 2008;112:4609–16.
Mutreja D, Sharma RK, Kotru M, Saxena R. CD4 positive/CD8 negative/CD56 positive T cell large granular lymphocyte proliferations; clonal disorders of uncertain significance. BMC Hematol. 2014;14:9.
Clemente MJ, Przychodzen B, Jerez A, Dienes BE, Afable MG, Husseinzadeh H, et al. Deep sequencing of the T-cell receptor repertoire in CD8+ T-large granular lymphocyte leukemia identifies signature landscapes. Blood. 2013;122:4077–85.
Sandberg Y, Kallemeijn MJ, Dik WA, Tielemans D, Wolvers-Tettero ILM, van Gastel-Mol EJ, et al. Lack of common TCRA and TCRB clonotypes in CD8(+)/TCRαβ(+) T-cell large granular lymphocyte leukemia: a review on the role of antigenic selection in the immunopathogenesis of CD8(+) T-LGL. Blood Cancer J. 2014;4:e172.
Garrido P, Ruiz-Cabello F, Bárcena P, Sandberg Y, Cantón J, Lima M, et al. Monoclonal TCR-Vbeta13.1+/CD4+/NKa+/CD8−/+dim T-LGL lymphocytosis: evidence for an antigen-driven chronic T-cell stimulation origin. Blood. 2007;109:4890–8.
Rodriguez-Caballero A, Garcia-Montero AC, Almeida J, Balanzategui A, Munoz-Criado S, Orfao A. Association between the HLA haplotype and the TCR-Vbeta repertoire of anti-hCMV specific memory T-cells in immunocompetent healthy adults. Cytom B Clin Cytom. 2007;72:371–9.
Nearman ZP, Wlodarski M, Jankowska AM, Howe E, Narvaez Y, Ball E, et al. Immunogenetic factors determining the evolution of T-cell large granular lymphocyte leukaemia and associated cytopenias. Br J Haematol. 2007;136:237–48.
O’Byrne KJ, Dalgleish AG. Chronic immune activation and inflammation as the cause of malignancy. Br J Cancer. 2001;85:473–83.
Cesarman E, Mesri EA. Kaposi sarcoma-associated herpesvirus and other viruses in human lymphomagenesis. Curr Top Microbiol Immunol. 2007;312:263–87.
Rezk SA, Weiss LM. Epstein-Barr virus-associated lymphoproliferative disorders. Hum Pathol. 2007;38:1293–304.
Yasunaga J-I, Matsuoka M. Human T-cell leukemia virus type I induces adult T-cell leukemia: from clinical aspects to molecular mechanisms. Cancer Control. 2007;14:133–40.
Huang H, Wang C, Rubelt F, Scriba TJ, Davis MM. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat Biotechnol. 2020;38:1194–202.
Bagaev DV, Vroomans RMA, Samir J, Stervbo U, Rius C, Dolton G, et al. VDJdb in 2019: database extension, new analysis infrastructure and a T-cell receptor motif compendium. Nucleic Acids Res. 2020;48:D1057–D1062.
Sethna Z, Elhanati Y, Callan CG, Walczak AM, Mora T. OLGA: fast computation of generation probabilities of B- and T-cell receptor amino acid sequences and motifs. Bioinformatics. 2019;35:2974–81.
Kim D, Park G, Huuhtanen J, Lundgren S, Khajuria RK, Hurtado AM, et al. Somatic mTOR mutation in clonally expanded T lymphocytes associated with chronic graft versus host disease. Nat Commun. 2020;11:2246.
Robinson JT, Thorvaldsdóttir H, Wenger AM, Zehir A, Mesirov JP. Variant review with the integrative genomics viewer. Cancer Res. 2017;77:e31–e34.
Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–107.
Emerson RO, DeWitt WS, Vignali M, Gravley J, Hu JK, Osborne EJ, et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat Genet. 2017;49:659–65.
Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, et al. VDJtools: unifying post-analysis of T cell receptor repertoires. PLoS Comput Biol. 2015;11:e1004503.
Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, et al. Identifying specificity groups in the T cell receptor repertoire. Nature 2017;547:94–98.
López C, Bergmann AK, Paul U, Murga Penas EM, Nagel I, Betts MJ, et al. Genes encoding members of the JAK-STAT pathway or epigenetic regulators are recurrently mutated in T-cell prolymphocytic leukaemia. Br J Haematol. 2016;173:265–73.
Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, Bailey NG, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124:1460–72.
Alvarez JV, Frank DA. Genome-wide analysis of STAT target genes: elucidating the mechanism of STAT-mediated oncogenesis. Cancer Biol Ther. 2004;3:1045–50.
Savola P, Kelkka T, Rajala HL, Kuuliala A, Kuuliala K, Eldfors S, et al. Somatic mutations in clonally expanded cytotoxic T lymphocytes in patients with newly diagnosed rheumatoid arthritis. Nat Commun. 2017;8:15869.
Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee J-Y, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci USA 2014;111:13139–44.
Pogorelyy MV, Minervina AA, Shugay M, Chudakov DM, Lebedev YB, Mora T, et al. Detecting T cell receptors involved in immune responses from single repertoire snapshots. PLoS Biol. 2019;17:e3000314.
Kontro M, Kuusanmäki H, Eldfors S, Burmeister T, Andersson EI, Bruserud O, et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia. 2014;28:1738–42.
de Araujo ED, Erdogan F, Neubauer HA, Meneksedag-Erol D, Manaswiyoungkul P, Eram MS, et al. Structural and functional consequences of the STAT5B driver mutation. Nat Commun. 2019;10:2517.
Liu Y, Easton J, Shao Y, Maciaszek J, Wang Z, Wilkinson MR, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017;49:1211–8.
Wahnschaffe L, Braun T, Timonen S, Giri AK, Schrader A, Wagle P, et al. JAK/STAT-activating genomic alterations are a hallmark of T-PLL. Cancers. 2019;11:1833.
McKinney M, Moffitt AB, Gaulard P, Travert M, De Leval L, Nicolae A, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 2017;7:369–79.
Küçük C, Jiang B, Hu X, Zhang W, Chan JKC, Xiao W, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat Commun. 2015;6:6025.
van Lohuizen M, Verbeek S, Krimpenfort P, Domen J, Saris C, Radaszkiewicz T, et al. Predisposition to lymphomagenesis in pim-1 transgenic mice: cooperation with c-myc and N-myc in murine leukemia virus-induced tumors. Cell. 1989;56:673–82.
Andersson EI, Pützer S, Yadav B, Dufva O, Khan S, He L, et al. Discovery of novel drug sensitivities in T-PLL by high-throughput ex vivo drug testing and mutation profiling. Leukemia. 2018;32:774–87.
Rajala HLM, Olson T, Clemente MJ, Lagström S, Ellonen P, Lundan T, et al. The analysis of clonal diversity and therapy responses using STAT3 mutations as a molecular marker in large granular lymphocytic leukemia. Haematologica. 2015;100:91–99.
Kerr CM, Clemente MJ, Chomczynski PW, Przychodzen B, Nagata Y, Adema V, et al. Subclonal STAT3 mutations solidify clonal dominance. Blood Adv. 2019;3:917–21.
Mariotti B, Calabretto G, Rossato M, Teramo A, Castellucci M, Barilà G, et al. Identification of a -Fas ligand axis in the development of neutropenia in T large granular lymphocyte leukemia. Haematologica. 2020;105:1351–60.
Muñoz-García N, Jara-Acevedo M, Caldas C, Bárcena P, López A, Puig N, et al. Mutations in T/NK-cell chronic lymphoproliferative disorders of large granular lymphocytes (LGL): association with disease features. Cancers. 2020;12:3508.
Li HM, Hiroi T, Zhang Y, Shi A, Chen G, De S, et al. TCRβ repertoire of CD4+ and CD8+ T cells is distinct in richness, distribution, and CDR3 amino acid composition. J Leukoc Biol. 2016;99:505–13.
Mustjoki S, Young NS. Somatic mutations in ‘benign’ disease. N Engl J Med. 2021;384:2039–52.
Acknowledgements
The research was funded by the European Research Council (Project: M-IMM 647355), Academy of Finland Heal-Art consortium (314442) and ERA PerMed (JAKSTAT-TARGET consortium), Sigrid Juselius Foundation, Signe and Ane Gyllenberg Foundation, Helsinki Institute for Life Science, and Cancer Foundation Finland to SM; Italian Association for Cancer Research (AIRC #20216) to GS; Società Italiana Ematologia Sperimentale (SIES) fellowship to AT. Amplicon sequencing was performed at the Institute for Molecular Medicine Finland FIMM Technology Centre, which is supported by Biocenter Finland. We acknowledge IT Center for Science Ltd and the Aalto Science-IT project for computational resources and Finnish Red Cross Blood Service for providing samples from healthy donors. We acknowledge Emmi Rehn for providing a modified script of GLIPH that was used for the previous version of the findings. We thank Jay Klievink for technical expertise in mutagenesis assays.
Author information
Authors and Affiliations
Contributions
DB, AT, VRG, TKe, FI, RZ, and SM conceived the study; DB, AT, VRG, GSc, CV, GC, TKa, HN, BF and ET prepared the samples and performed sequencings; DB, DK and VRG performed functional assays; DB, JH and JT performed bioinformatic analyses; GB, MF, FI, GSe, RZ and SM provided the patient samples; DB, JH, AT, and VRG wrote the paper; GSe and SM provided funding; TKe, GSe, RZ and SM critically revised the paper. All authors revised the paper.
Corresponding author
Ethics declarations
Competing interests
SM has received honoraria and research funding from Novartis, Bristol-Myers Squibb, Incyte, and Pfizer. GSe has received honoraria from Novartis and Roche. Others report no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Co-last authors: Tiina Kelkka, Renato Zambello, Satu Mustjoki.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bhattacharya, D., Teramo, A., Gasparini, V.R. et al. Identification of novel STAT5B mutations and characterization of TCRβ signatures in CD4+ T-cell large granular lymphocyte leukemia. Blood Cancer J. 12, 31 (2022). https://doi.org/10.1038/s41408-022-00630-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-022-00630-8
- Springer Nature Limited
This article is cited by
-
Diagnostic and prognostic molecular pathology of lymphoid malignancies
Virchows Archiv (2024)
-
All that glitters is not LGL Leukemia
Leukemia (2022)
-
Defining TCRγδ lymphoproliferative disorders by combined immunophenotypic and molecular evaluation
Nature Communications (2022)