
Abstract
Antenatal synthetic glucocorticoid (sGC) treatment is a potent modifier of the hypothalamic-pituitary-adrenal (HPA) axis. In this context, epigenetic modifications are discussed as potential regulators explaining how prenatal exposure to GCs might translate into persistent changes of HPA axis “functioning”. The purpose of this study was to investigate whether DNA methylation and gene expression profiles of stress-associated genes (NR3C1; FKBP5; SLC6A4) may mediate the persistent effects of sGC on cortisol stress reactivity that have been previously observed. In addition, hair cortisol concentrations (hairC) were investigated as a valid biomarker of long-term HPA axis activity. This cross-sectional study comprised 108 term-born children and adolescents, including individuals with antenatal GC treatment and controls. From whole blood, DNA methylation was analyzed by targeted deep bisulfite sequencing. Relative mRNA expression was determined by RT-qPCR experiments and qBase analysis. Acute stress reactivity was assessed by the Trier Social Stress Test (TSST) measuring salivary cortisol by ELISA and hairC concentrations were determined from hair samples by liquid chromatography coupled with tandem mass spectrometry. First, no differences in DNA methylation and mRNA expression levels of the stress-associated genes between individuals treated with antenatal sGC compared to controls were found. Second, DNA methylation and mRNA expression levels were neither associated with cortisol stress reactivity nor with hairC. These findings do not corroborate the belief that DNA methylation and mRNA expression profiles of stress-associated genes (NR3C1; FKBP5; SLC6A4) play a key mediating role of the persistent effects of sGC on HPA axis functioning.
Similar content being viewed by others


Introduction
Prenatal maternal stress and exposure to antenatal synthetic glucocorticoids (sGC) can have long-term consequences on major stress response systems and mental health [1, 2]. Importantly, women at risk for preterm delivery are routinely treated with sGCs such as betamethasone (BETA) and dexamethasone (DEX) which directly cross the placenta and promote fetal lung maturation [3]. Apart from their clear benefits, several studies have identified antenatal sGC exposure as a potent modifier of the hypothalamus-pituitary-adrenal (HPA) axis (for a review see [1]), possibly leading to an increased risk for stress-related disorders later in life [2]. For example, our group observed increased cortisol stress responses in sGC-treated term-born children compared with untreated controls [4] that persisted into late adolescence [5].
Epigenetic modifications such as DNA methylation (DNAM) are discussed as a central mechanism explaining how in utero exposure to GCs might translate into persistent changes of HPA axis functioning [6, 7]. In the promoter region, together with histone modifications DNAM serves as a transcriptional regulator, e.g., by influencing transcription factors from binding to regulatory elements [7]. There is now considerable evidence that the fetal epigenome is responsive to a broad range of intrauterine environmental exposures, including prenatal maternal stress and in utero sGC exposure [8]. To date, the majority of findings derive from candidate gene studies investigating DNAM states at genes involved in neurotransmitter (SLC6A4 [9]) and stress hormone regulation (NR3C1 [10]) The most prominent example involves the glucocorticoid receptor gene (NR3C1), where different forms of prenatal stress have been associated with increased DNAM at one single CpG site (meta-analysis [11]). Given the pivotal role of glucocorticoid receptor (GR) signaling in negative feedback regulation of the HPA-axis, respective epigenetic changes may account for permanently altered glucocorticoid levels as it has been suggested by landmark rodent studies [12]. Another candidate region constitutes the FK506 binding protein 5 (FKBP5) gene that also acts as an important modulator of the HPA axis. FKBP5 provides an ultrashort negative feedback loop for GR signaling by reducing its cortisol binding affinity and impeding translocation of the receptor complex to the nucleus [13]. Several studies reported associations between early adversity (e.g., childhood trauma, institutionalized care) and a demethylation of a glucocorticoid response element (GRE) located in intron 7 of the FKBP5 gene (e.g. Klengel et al. [14]), although not without inconsistences (e.g. Alexander et al. [15]). In turn, this epigenetically-induced upregulation of FKBP5 presumably leads to a persistent GR resistance and a disruption of the HPA-axis feedback control [16]. First studies on prenatal stress exposures such as maternal affective disorders [17] and perceived distress [18] have also produced mixed findings by investigating a range of different CpG sites within the FKBP5 gene. Another stress-related gene that has also received considerable attention in epigenetic studies on early adversity (for a review see [9]) and HPA-axis functioning (e.g. refs. [19, 20]) is the serotonin transporter gene (SLC6A4). Regarding prenatal stress, one study revealed a negative association of maternal depressive mood during pregnancy and SLC6A4 DNAM levels [21] while other studies reported no effects of maternal prenatal stressors (e.g. Wankerl et al. [22]). Given the pivotal impact of serotonergic signaling on HPA-axis regulation [23], SLC6A4 DNAM profile may well account for long-term changes in cortisol output.
Only few studies have so far investigated epigenetic correlates of prenatal GC exposure, although GC reflects a key mechanism of how prenatal stress translates into persistent DNAM changes [24]. In an elegant model of human hippocampal progenitor cell (HPC) lines, GC exposure was found to induce widespread changes of DNAM, with the most pronounced effects observed during the proliferation and differentiation stage [25]. Regarding stress-related candidate genes in specific, together with another set of in vitro studies a demethylation of FKBP5 intron 7 in HPC cell lines following DEX treatment was demonstrated [14, 25]. While evidence from living humans is still rare, a first genome-wide association study identified 9672 differentially methylated probes (DMPs) associated with DEX treatment during the first trimester of children at risk for, but not having, congenital adrenal hyperplasia [26]. For instance, they observed hypermethylation of specific CpG sites of NR3C1, FKBP5, and SLC6A4 in DEX-treated individuals [26].
Following this line of research, the present study aimed to investigate whether DNAM profiles and respective changes in the gene expression of stress-related candidate genes (NR3C1, FKBP5, SLC6A4) mediate the persistent effects of sGC on cortisol reactivity to a standardized laboratory stressor that have been observed previously by our group [4, 5] and others (e.g. Edelmann et al. [27]). For this, we collected blood samples for DNAM and gene expression analyses from our children and adolescent cohort of mothers with a pathophysiological pregnancy (PP) who had received sGC treatment during pregnancy (PP/GC group) and controls from physiological pregnancies without complications. A second control group was further recruited to separate effects of sGC treatment from those related to maternal stress induced by the threat of preterm delivery and antepartum hospitalization. This group comprised children/adolescents of mothers who had been admitted to the hospital for serious pregnancy complications but had never received sGC therapy (PP/non-GC group). In contrast to most prior studies that included preterm infants with low birth weight (LBW), our group further aimed to disentangle the direct effects of antenatal sGC on DNAM profiles from the confounding effects of preterm delivery [28] and LBW [29]. To this end, only term-born individuals with normal birth weight were enrolled in the current study. With regard to the assessment of HPA axis functioning, prior research in this field was mostly limited to acute markers reflecting short-term hormone levels. Addressing the gap, we aimed to determine both acute cortisol stress reactivity and hair cortisol concentrations (hairC), which have been proven a reliable and valid marker of long-term HPA-axis activity over a period of approximately three months [30]. Interestingly, these two markers are only poorly correlated and thus reflect different aspects of HPA-axis functioning [31, 32].
Material and methods
Participants
The actual sample consisted of two cohorts including a total of 57 term-born children (7–12 years) and 51 adolescents (14–18 years), both comprising PP/GC, PP/non-GC, and controls.
Recruitment procedure
Children’s cohort
In cooperation with the Department of Gynecology and Obstetrics at the TU Dresden, medical reports from all mothers who delivered their babies between 2005 and 2010 were screened. From these 8421 mother/child dyads, only term-born offspring (≥37 weeks of gestation) who were not exposed to pediatric intensive care were considered as potential participants. Mothers were excluded from the study if the following exclusion criteria were fulfilled, i.e., serious diseases during pregnancy (metabolic diseases, gestational diabetes, placental insufficiency, preeclampsia, or known addiction), in order to avoid confounding effects of fetal lung maturation. Among these mothers, all who had been hospitalized for specific pregnancy complications, i.e., premature labor pain and/or vaginal bleeding and/or cervical insufficiency, were invited to take part in the study (n = 765). The remaining sample of hospitalized women consisted of two groups, namely mothers who received sGC therapy to accelerate fetal lung maturation (PP/GC, n = 523) and mothers who did not receive sGC treatment (PP/non-GC, n = 241). The PP/GC group was treated with either a single course of 12 mg of BETA administered twice over a 24 h interval or with DEX administered in four doses of 6 mg every 12 h during the 30th week of gestation on average. Additionally, mothers with physiological pregnancies were invited as controls (n = 502). In total, 81 mother/child dyads participated in the main study [33]. From this sample, 57 mother/child dyads agreed to donate blood for DNAM and mRNA expression analyses (which was an optional part of the main study), including 19 PP/GC, 13 PP/non-GC, and 25 controls.
Adolescent cohort
Medical reports from all mothers who delivered their babies between 1997 and 2003 were screened (for a detailed recruitment protocol, see [5]). Considering the above-mentioned inclusion and exclusion criteria, the PP/GC group included 304 mothers and the PP/non-GC group included 212 mothers (controls, n = 372). The PP/GC group was treated with the same BETA or DEX administration as described for the children’s cohort. In the first wave of testing (e.g., for the assessment of acute cortisol stress reactivity [4]), a total of 209 children took part in the study (PP/GC group, n = 81; PP/non-GC group, n = 43; controls, n = 85). In the actual second wave of testing, a subsample was re-invited in their adolescence for the analysis of acute cortisol stress reactivity [5], long-term hair steroids [33], and DNA methylation and mRNA expression of stress-associated genes (NR3C1, FKBP5, SLC6A4). Thus, all participants from the first study wave who (i) fulfilled inclusion criteria, and (ii) were willing to participate in the second study wave, and (iii) agreed to donate blood for DNA methylation and mRNA expression analysis (which was an optional part of the main study) were tested, including 23 PP/GC group, 7 PP/non-GC, and 21 controls.
Study procedure
For the assessment of demographic characteristics (age, sex), birth-related characteristics (birth weight, birth length, APGAR score 5 minutes after birth, length of gestation), health-related variables (smoking, oral contraceptive, prenatal stress exposure), and hair related characteristics (number of hair washes per week, hair color), children/adolescents and their parents filled out a set of questionnaires. All participants gave written informed consent and the study was approved by the local ethic committee of the TU Dresden (EK 235062014) following the principles of the Declaration of Helsinki.
Standardized laboratory stress test (TSST)
For stress induction, adolescents were exposed to the Trier Social Stress Test (TSST [34]), which is a standardized protocol that reliably elicits significant subjective and endocrine stress responses, including cortisol. Likewise, the children cohort underwent the Trier Social Stress Test for Children [35]. Detailed protocols have been published elsewhere [4, 5]. Saliva samples were collected 5 minutes before the TSST (baseline) as well as 10, 20, and 30 min after TSST onset. All test sessions were scheduled in the afternoon (1400–1800 h) to reduce variance in cortisol concentrations due to circadian secretion rhythms. Participants were asked to reschedule the session if feeling significantly impaired due to any reason and to refrain from physical exercising, smoking, eating, and drinking anything but water 1 h before the TSST.
Saliva cortisol analysis
Saliva samples were collected with swabs (Cortisol Salivette®; Sarstedt, Nürmbrecht, Germany). Participants chew on the swabs for 30–60 s to stimulate saliva flow. Saliva samples were stored at −20 °C until the samples were thawed and centrifuged at 3000 rpm for 3 min. Salivary cortisol was analyzed using a commercial chemiluminescence immunoassay (CLIA; IBL, Hamburg, Germany) with intra-assay precision of 3.0% and inter-assay precision of 4.2%.
Hair cortisol analysis
For the analyses of hair cortisol concentrations (hairC) scalp-near hair strands (~3 mm in diameter) were taken from a posterior vertex position. Hair strands were wrapped in aluminum foil and stored dry and dark until analysis. For the assessment of cumulated cortisol secretion over a 3-months period prior to sampling, hair segments of 3 cm were analyzed. HairC was analyzed by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) as described in detail elsewhere [36].
DNA methylation analysis
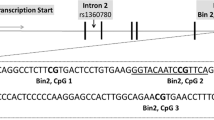
DNA from all participants was extracted from frozen EDTA-blood according to a simple salting out procedure [37] before 1 µg of DNA from each subject was bisulfite-treated using the EZ DNA Methylation-GoldTM kit (Zymo, USA). DNAM levels at specific genomic sites were investigated by targeted deep bisulfite sequencing as described elsewhere [38]. We focused on three regulatory regions of stress-associated candidate genes: 42 CpGs from NR3C1-1F promoter region (e.g. refs. [12, 39]), 84 CpGs from the SLC6A4 5′ regulatory region (e.g. refs. [19, 20]) and 5 CpGs located in intron 7 of FKBP5 (e.g. Klengel et al. [14]) (see Supplementary Information 1 including Suppl. Fig. 1A–C and Suppl. Table 1A–C for exact chromosomal positions). The pair-end sequencing was performed on a MiSeq system (Illumina; San Diego – USA) using the Illumina MiSeq reagent Kit v2 (500 cycles- 2 × 250 paired end) in collaboration with the BioChip Labor of the Center of Medical Biotechnology (ZMB, University Essen/ Duisburg). A detailed laboratory protocol and primer sequences have been published elsewhere [38].
RNA quantification by rt-qPCR
Blood was collected in PAXgene Blood RNA Tubes and total RNA extracted using the PAXgene Blood RNA Kit (PreAnalytiX GmbH, Switzerland). Subsequently, cDNA synthesis was conducted from 500 ng RNA using the iScript cDNA Synthesis Kit (Bio-Rad, USA) followed by real-time quantitative PCR analyses on a CFX384 realtime cycler (Bio-Rad, USA). A detailed protocol and primer sequences specially targeting mRNA of different housekeeping genes and three genes of interest (NR3C1, FKBP5, SLC6A4) are provided in Supplementary Information 2 and Suppl. Table 2. With respect to the retest reliability of mRNA expression profiles, a pilot study by our group already indicated a substantial trait component regarding SLC6A4 mRNA expression [22]. Doing geNorm analysis [40], a set of nine candidate housekeeping genes in a representative set of 18 independent samples was assayed for stable mRNA expression. Result of this analysis was the selection of the most stably expressed set of three housekeeping genes: Beta-2-Microglobulin (B2M), Peptidylprolyl Isomerase A (PPIA), and Transferrin Receptor (TFRC) (Suppl. Fig. 2). Relative mRNA quantification analysis was performed using the qBase method [41]. All RT-qPCR analyses were performed in triplicates.
Statistical analysis
Analyses were performed using R statistical programming (version 4.1). All statistical tests were two-tailed and a P-value of <0.05 was defined as statistically significant. Chi-squared tests for dichotomous and analyses of variance (ANOVAs) for continuous outcomes were used to examine group differences (PP/GC, PP/non-GC, controls) regarding demographic, as well as birth- and health-related characteristics. DNAM at single CpG sites as well as mean DNAM levels across the investigated genomic regions were analyzed. Given the large number of CpG sites quantified for SLC6A4, principal component analysis was conducted to reduce data into co-methylated factors (Supplementary Information 3 including Suppl. Fig. 3 and Suppl. Table 3). Tests for potential confounders regarding DNAM, mRNA expression, and cortisol values were assessed using correlations and ANOVAs (Supplementary Information 4 and Suppl. Table 4). Identified confounders, i.e., variables that were significantly associated with the exposure or the outcome, were included as covariates in the following analyses. In case of deviation from normal distribution (assessed with Kolmogorov–Smirnov test) of absolute DNAM, mRNA expression and/or cortisol values, subsequent analyses were based on natural log-transformed values. Non-parametrical statistical methods were used if log-transformation did not result in normality. In case of outliers, i.e., individuals who showed studentized residuals ≥ |3| regarding mean DNAM, mRNA expression and/or cortisol values, analyses will be run with and without outlier values to examine whether the outliers change the main findings. For descriptive purposes, mean data in figures were presented in original units.
First, we compared the mean DNAM levels over all corresponding CpG sites per analyzed gene and mRNA expression levels between the three groups. The equality of variances was tested with Levene’s test. Next, separate ANOVAs or Kruskal–Wallis tests, respectively, were used to compare the groups. Second, we considered differences in DNAM levels between the three groups at single CpG sites and co-methylated factors (in case of SLC6A4) by performing separate ANOVAs. All p-values were adjusted by Benjamini and Hochberg [42] correction for multiple testing by the number of CpG sites tested for each candidate gene (factors for SLC6A4). Further, linear regression analyses were conducted to test whether DNAM predicted mRNA expression levels. In addition, covariate-adjusted regression models were performed to test whether (1) DNAM (mean DNAM, at single CpG sites, co-methylated factors) and (2) mRNA expression were related to differences in cortisol stress reactivity (indexed by the cortisol area under the curve with respect to increase (AUCi) according to [43]) Further, covariate-adjusted regression models were set up to test whether (1) DNAM (mean DNAM, at single CpG sites, co-methylated factors) and (2) mRNA expression was related to differences in hairC. In addition to frequentist analyses, Bayes factors were calculated for all tested hypotheses to examine the likelihood of the alternative (H1) compared to the null (H0) hypothesis [44]. Bayes factor above 1 is considered in favor of the H1 and a common convention is to interpret a Bayes factor ≥3 as moderate (and ≥10 as strong) evidence for the H1 [44, 45]. Contrarily, Bayes factor below 1 is considered as evidence for the H0, with values ≤1/3 indicating moderate (and ≤1/10 strong) evidence for the H0.
Preregistration
This study was preregistered prior to analysis of the data (Open Science Framework; https://osf.io/akdtv/).
Results
Sample characteristics
The present study cohort included 108 healthy, term-born children and adolescents (42 PP/GC, 20 PP/non-GC, 46 controls). Group differences regarding demographic, birth- and health-related characteristics are shown in Table 1. The groups did not significantly differ with respect to sex (p = 0.349), age (p = 0.649) or BMI (p = 0.366). Although all participants were term-born (>37 weeks of gestation), significant group differences were observed in length of gestation (p = 0.038), showing that individuals in the PP/GC group were born earlier than individuals in the comparison groups. The groups did not differ in terms of other birth-related characteristics (birth weight, birth length, APGAR index at 5 min after birth; all p ≥ 0.190), or prenatal stress exposure (p = 0.673). Thus, length of gestation was included as a covariate into the following analyses testing for group differences. In previous studies, we had already shown that the PP/GC group was characterized by higher cortisol levels in response to the TSST both during childhood [4] and late adolescence [5] as compared to controls, whereas no group differences were found with regard to hairC [33].
DNA methylation analysis
On average, assays showed around 8247.26 (±5777.06) reads per sample and gene (Max = 28814, Min = 467). The DNAM analysis resulted in a mean methylation value of 91.36% (SD = 1.53%) for FKBP5 and 5.12% (SD = 0.65%) for SLC6A4. As also observed by others (e.g. refs. [38, 39, 46]), the analyzed NR3C1 stretch was generally unmethylated, with mean DNAM values below 1% (Suppl. Fig. 1A). Given the low mean DNAM level (M = 0.66%) as well as the low variability (SD = 0.10%) of NR3C1, no further statistical analyses were performed. For FKBP5 and SLC6A4, no association of DNAM with mRNA expression could be observed (for details see Supplementary information 5 and Suppl. Fig. 4).
No effects of antenatal sGC treatment on DNAM or mRNA expression in stress-associated genes
First, group differences (PP/GC, PP/non-GC, controls) in mean DNAM (FKBP5, SLC6A4) and mRNA expression levels (NR3C1, FKBP5, SLC6A4) were analyzed by means of separate ANOVAs or Kruskal-Wallis-tests, respectively. In the overall sample, no significant differences in DNAM between the three groups in any of the genes (FKBP5: χ²(2) = 5.61, p = 0.061; BF10 = 0.574; SLC6A4: F(2,104) = 0.05, p = 0.955; BF10 = 0.096; Fig. 1A, B) were observed. Likewise, no significant differences in mRNA expression between the three groups in any of the genes (NR3C1: χ²(2) = 0.33, p = 0.846; BF10 = 0.098; FKBP5: F(2,100) = 1.00, p = 0.370; BF10 = 0.202; SLC6A4: F(2,95) = 2.75, p = 0.069; BF10 = 0.845; Fig. 1C–E) were observed. For completeness, we also tested for group differences in DNAM at specific CpG sites (FKBP5, SLC6A4) and co-methylated factors (SLC6A4) by replacing mean DNAM by DNAM at five individual CpG sites for FKBP5, 84 individual CpG sites and two co-methylated factors for SLC6A4 in the ANOVA. However, the results were largely comparable and showed no significant differences between the three groups for site- as well as factor-specific DNAM after correction for multiple testing (number of CpGs tested per gene; all Padj ≥ 0.190). Together, these findings indicate comparable DNAM and mRNA expression levels in stress-associated genes in individuals of the PP/GC, PP/non-GC, and control group. Results of the Bayes factor analyses largely confirmed the findings of the frequentist analyses. There was anecdotal to strong evidence for the H0 of no group differences of DNAM and mRNA expression levels (for the results of Bayes factors analyses for single and site-specific DNAM see Supplementary Information 6). Comparable results were achieved when controlling for potential confounding effects and after exclusion of identified outliers.

Upper part: There were no significant differences in mean DNA methylation levels between the three groups (pathological pregnancy group with antenatal synthetic glucocorticoid treatment (PP/GC), pathological pregnancy group without antenatal synthetic glucocorticoid treatment (PP/non-GC), untreated control group from physiological pregnancies). Data show mean with 95% confidence interval. A Mean DNA methylation of FKBP5. B Mean DNA methylation of SLC6A4. Lower part: There were no significant differences in mRNA expression levels between the three groups. C mRNA expression of FKBP5. D mRNA expression of SLC6A4. E mRNA expression of NR3C1.
No associations of DNAM and mRNA expression with cortisol stress reactivity
Next, we investigated whether (1) DNAM and (2) mRNA expression were related to differences in cortisol stress reactivity. We set up regression models to test whether (1) mean DNAM and (2) mRNA expression were related to differences in AUCi cortisol stress reactivity. In resulting models, neither mean DNAM (FKBP5: t(93) = 0.43, p = 0.672, β = 0.04; BF10 = 0.234; SLC6A4: t(93) = –0.02, p = 0.983, β = 0.002; BF10 = 0.216; Fig. 2A, B) nor mRNA expression (NR3C1: t(88) = –1.02, p = 0.309, β = –0.11; BF10 = 0.349; FKBP5: t(88) = –0.72, p = 0.476, β = –0.08; BF10 = 0.276; SLC6A4: t(84) = 0.54, p = 0.594, β = .06; BF10 = 0.255; Fig. 2C–E) were significantly related to cortisol stress reactivity. Again, we also tested whether DNAM at specific CpG sites (FKBP5, SLC6A4) and of co-methylated factors (SLC6A4) was associated with cortisol release. However, the results were largely comparable and site- as well as factor-specific DNAM was unrelated to cortisol stress reactivity (all Padj ≥ 0.202). Together these findings do not provide evidence for an association of DNAM and mRNA expression with cortisol stress reactivity. The Bayes factor analyses suggested that there is anecdotal to moderate evidence for the H0 of no effects of DNAM and mRNA expression on cortisol stress reactivity. Comparable results were achieved when controlling for potential confounders.

Upper part: Mean DNA methylation levels were not significantly related to cortisol stress reactivity (indexed by the cortisol AUCi). A Mean DNA methylation of FKBP5. B Mean DNA methylation of SLC6A4. Lower part: mRNA expression levels were not significantly related to cortisol stress reactivity. C mRNA expression of FKBP5. D mRNA expression of SLC6A4. E mRNA expression of NR3C1.
No associations of DNAM and mRNA expression with hairC
Next, we set up regression models to test whether (1) mean DNAM and (2) mRNA expression were related to differences in hairC. In resulting models, neither mean DNAM (FKBP5: t(95) = 0.48, p = 0.630, β = 0.05; BF10 = 0.237; SLC6A4: t(95) = –0.56, p = 0.575, β = –0.06; BF10 = 0.246; Fig. 3A, B) nor mRNA expression (NR3C1: t(91) = –0.15, p = 0.880, β = –0.02; BF10 = 0.220; FKBP5: t(91) = –0.31, p = 0.757, β = –0.03; BF10 = 0.220; SLC6A4: t(86) = –0.34, p = 0.734, β = –0.04; BF10 = 0.235; Fig. 3C–E) were significantly related to hairC. Again, we also tested whether DNAM at specific CpG sites (FKBP5, SLC6A4) and co-methylated factors (SLC6A4) was associated with hairC. However, the results were largely comparable and site- as well as factor-specific DNAM was not significantly related to hairC (all Padj ≥ 0.845). Together these findings do not provide evidence for an association of DNAM and mRNA expression with hairC. The Bayes factor analyses provided moderate evidence for the H0 of no effects of DNAM and mRNA expression on HairC. Comparable results were achieved when controlling for potential confounders.

Upper part: Mean DNA methylation levels were not significantly related to hair cortisol concentration (hairC). A Mean DNA methylation of FKBP5. B Mean DNA methylation of SLC6A4. Lower part: mRNA expression levels were not significantly related to hairC. C mRNA expression of FKBP5. D mRNA expression of SLC6A4. E mRNA expression of NR3C1.
Discussion
The present study investigated whether DNAM and respective mRNA expression profiles of three stress-related candidate genes (NR3C1, FKBP5, SLC6A4) act as mediators of the persistent effects of sGC treatment on cortisol reactivity that have been previously observed by our group [4, 5]. For this, DNAM and mRNA expression profiles of children/adolescents from PP who had received sGC treatment during pregnancy (DEX or BETA) were compared to controls from pregnancies without complications. In order to separate the direct effects of sGC exposure from those related to maternal stress induced by antenatal hospitalization, a second control group was recruited consisting of children/adolescents of mothers who had been admitted to hospital for serious pregnancy complications but did not receive any sGC therapy. Importantly, the current sample included only term-born individuals to exclude the confounding effects of preterm delivery [28] and LBW [29] on DNAM profiles. We found no evidence of group differences in DNAM and mRNA expression levels of three stress-associated genes (NR3C1, FKBP5, SLC6A4). Likewise, no associations of respective DNAM/mRNA expression levels with acute cortisol stress reactivity or long-term cortisol levels in hair were observed.
While the fetal epigenome is particularly responsive to intrauterine stress exposures [8], recent efforts to generate robust associations with DNAM at specific stress-related candidate loci remained challenging. Expanding prominent findings from rodent models [12], a number of human studies tried to provide evidence for a link between childhood (e.g. van der Knaap et al. [47]) and in utero (for a meta-analysis see [11]) adversity and increased NR3C1-1F DNAM, rendering this locus a potential candidate for investigating effects of sGC treatment. However, an increasing number of studies could not demonstrate such an effect (e.g. refs. [48, 49]), and one important message from human studies targeting NR3C1-1F was that overall DNAM levels measured in peripheral and hippocampus cells were generally low with limited sample variance observed for most individual sites [39, 46, 48]. In the present study, the NR3C1-1F promoter region was also found to be largely unmethylated and thus was excluded from further analyses (Suppl. Figure 1A). Although subtle differences in DNAM may theoretically promote changes in HPA-axis functioning, it is important to note that even the most sensitive method like targeted deep bisulfite sequencing is not able to reliably detect DNAM below 1% [38]. Beyond the question of their biological plausibility [50], these technical restrictions should be carefully considered when interpreting previously observed effects of early environmental adversity on NR3C1-1F DNAM levels.
In addition to NR3C1-1F, DNAM changes within the FKBP5 gene were considered as a second promising mediator of altered HPA-axis functioning following antenatal sGC treatment. Several previous studies reported demethylation of a GRE located in intron 7 of the FKBP5 gene following early environmental adversity (e.g. Klengel et al. [14]), but this could not be confirmed by others (e.g. Alexander et al. [15]). In contrast, different forms of maternal stress (e.g., maternal affective disorders and perceived distress) were repeatedly associated with increased FKBP5 DNAM in cord blood and placental samples, although these studies targeted different regions of the FKBP5 gene [17, 18]. Of particular relevance, in vitro dexamethasone treatment has been found to induce an active demethylation of FKBP5 intron 7 in human hippocampal progenitor cell lines, which may impose an increased risk for a chronic state of hypercortisolism [14]. While we observed substantial DNAM variability in our sample, our findings revealed no effects of sGC treatment on FKBP5 DNAM and respective mRNA expression levels. In accordance with an independent study on healthy adults by our group [15], our data provide no evidence for a biological relevance of FKBP5 DNAM levels in terms of changes in mRNA expression or acute and chronic cortisol output. The latter findings thus conflict with studies reporting negative associations of FKBP5 intron 7 DNAM and single measures of early morning cortisol concentrations [51] as well as averaged cortisol awakening levels sampled over one month [52].
Lastly, we targeted a promoter-associated CpG island in the SLC6A4 gene that has already been linked to early environmental adversity and HPA-axis alterations in previous research (for a review see [9]). The few studies that investigated prenatal stress exposures produced overall conflicting results, including a negative correlation between SLC6A4 DNAM levels and maternal depressive mood during pregnancy [21] as well as no association with different maternal prenatal stressors (e.g. Wankerl et al. [22]). In turn, site-specific changes in SLC6A4 DNAM levels have been linked to increased cortisol stress reactivity in studies depending on an individual’s genetic predisposition [19, 53], while this link was not observed by others [54]. Findings of the current study, however, could not provide further evidence that DNAM and respective mRNA expression profiles of SLC6A4 are sensitive to prenatal sGC treatment or play a key mediating role in its persisting effects on HPA-axis functioning.
Several limitations of the present study have to be considered. First, the number of mother/child dyads who agreed to donate blood was relatively small. Second, we did not account for genetic variants that are known to moderate associations of early adversity on DNAM profiles [14] and may also amplify epigenetic effects on HPA-axis functioning [19, 20, 55]. The presence of genetic variants throughout the whole genome affecting DNAM pattern cannot be excluded [56]. Particularly with regard to hairC, which is a highly heritable marker [57], the interplay between genetic and epigenetic effects should be further elucidated in future studies [55]. Third, DNAM of candidate genes was determined in surrogate peripheral tissue. Even though a genome-wide study showed significant correlations between DNAM in the live human brain and whole blood within individuals (for brain-blood correlation see website IMAGE-CpG [58]: FKBP5: r = 0.95; SLC6A4: r = 0.94), the relevance for crucial brain structures involved in HPA-axis regulation (e.g. the hippocampus) is not yet reliably proven. Fourth, the mixture of cell types in whole blood samples introduces a potential bias due to possible significant differences in both the level and variability of DNAM between different sample types (e.g. whole blood vs. homogeneous cell types) [59]. In accordance with previous studies, cell type heterogeneity may constitute a potential confounder of DNAM profiles (e.g. Jones et al. [60]).
In summary, our study provides no evidence for a substantial effect of prenatal sGC treatment on NR3C1, FKBP5, and SLC6A4 DNAM levels in late childhood or adolescence that compares to those observed in the aftermath of prenatal stress in other studies (e.g. NR3C1 [11]; FKBP5 [17]; SLC6A4 [21]). Although cortisol excess reflects a core mediator of maternal stress transfer to the fetal epigenome [24], it is important to note that the effects of maternal GC may differ from those of sCG used in neonatal practice (e.g. sGCs readily cross the placenta whereas maternal GCs are converted into inactive cortisone [61]). Moreover, inconsistent findings might result from differences regarding the choice of the specific tissue used to investigate epigenetic markers as well as different analytical methods used to quantify DNAM. Consequently, the mechanisms underlying the persistent effects of sGC on HPA-axis functioning observed in our sample remain unknown. Future studies should consider other mechanisms to examine these effects on stress reactivity, e.g. by including functional measures of GR sensitivity [49]. Future genome-wide methylation analysis studies might shed light on potentially unexplored epigenetic mediators in this context, however, they would require very large samples to achieve adequate statistical power. A first small-scaled epigenome-wide association study on 29 children at risk for, but not having, congenital adrenal hyperplasia already provided preliminary evidence for 9672 DMPs associated with DEX treatment during the first trimester of pregnancy [26]. Functional enrichment of those DMPs was mainly associated with immune functioning and inflammation, however, a set of DMPs was also implicated in steroidogenesis, thus highlighting the potential relevance for long-term cortisol output. Regarding the use of genome-wide DNAM data, epigenetic scores (ES) might reflect a powerful strategy to aggregate effects of single loci, which can then serve to robustly predict changes in central response systems even in smaller samples. In a landmark study of human HPCs, sGC were recently found to induce widespread changes of DNAM at sites involved in cellular and organ development, transcription, neurogenesis, and neuronal differentiation [25]. To account for the cross-tissue relevance of those GC-induced DNAM changes, the authors calculated a weighted ES of those differentially methylated sites that were found to overlap in HPCs and peripheral human blood cells. Intriguingly, when applying this DEX-sensitive ES to DNAM data of human newborn cord blood samples, it successfully predicted prenatal stress exposure and might thus offer an innovative future strategy to investigate molecular changes following antenatal sGC treatment.
References
McGowan PO, Matthews SG. Prenatal stress, glucocorticoids, and developmental programming of the stress response. Endocrinology 2018;159:69–82.
Braun T, Challis JR, Newnham JP, Sloboda DM. Early-life glucocorticoid exposure: The hypothalamic-pituitary-adrenal axis, placental function, and long-term disease risk. Endocr Rev. 2013;34:885–916.
Bolt RJ, van Weissenbruch MM, Lafeber HN, Delemarre‐van de Waal HA. Glucocorticoids and lung development in the fetus and preterm infant. Pediatr Pulmonol. 2001;32:76–91.
Alexander N, Rosenlöcher F, Stalder T, Linke J, Distler W, Morgner J, et al. Impact of antenatal synthetic glucocorticoid exposure on endocrine stress reactivity in term-born children. J Clin Endocrinol Metab. 2012;97:3538–44.
Ilg L, Kirschbaum C, Li S-C, Rosenlöcher F, Miller R, Alexander N. Persistent effects of antenatal synthetic glucocorticoids on endocrine stress reactivity from childhood to adolescence. J Clin Endocrinol Metab. 2019;104:827–34.
Matthews SG, McGowan PO. Developmental programming of the HPA axis and related behaviours: epigenetic mechanisms. J Endocrinol. 2019;242:T69–T79.
Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 2: mechanisms. Nat Rev Endocrinol. 2014;10:403–11.
Cao-Lei L, de Rooij SR, King S, Matthews SG, Metz GAS, Roseboom TJ, et al. Prenatal stress and epigenetics. Neurosci Biobehav Rev. 2020;117:198–210.
Provenzi L, Giorda R, Beri S, Montirosso R. SLC6A4 methylation as an epigenetic marker of life adversity exposures in humans: a systematic review of literature. Neurosci Biobehav Rev. 2016;71:7–20.
Palma-Gudiel H, Córdova-Palomera A, Leza JC, Fañanás L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci Biobehav Rev. 2015;55:520–35.
Palma-Gudiel H, Córdova-Palomera A, Eixarch E, Deuschle M, Fañanás L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: a meta-analysis. Epigenetics 2015;10:893–902.
Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. Epigenetic programming by maternal behavior. Nat Neurosci. 2004;7:847–54.
Wochnik GM, Rüegg J, Abel GA, Schmidt U, Holsboer F, Rein T. FK506-binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem. 2005;280:4609–16.
Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, et al. Allele-specific FKBP5 DNA demethylation mediates gene–childhood trauma interactions. Nat Neurosci. 2013;16:33–41.
Alexander N, Kirschbaum C, Stalder T, Muehlhan M, Vogel S. No association between FKBP5 gene methylation and acute and long-term cortisol output. Transl Psychiatry. 2020;10:175.
Zannas AS, Binder EB. Gene-environment interactions at the FKBP5 locus: Sensitive periods, mechanisms and pleiotropism. Genes Brain Behav. 2014;13:25–37.
Duis J, Cox OH, Ji Y, Seifuddin F, Lee RS, Wang X. Effect of genotype and maternal affective disorder on intronic methylation of FK506 binding protein 5 in cord blood DNA. Front Genet. 2018;9:648.
Monk C, Feng T, Lee S, Krupska I, Champagne FA, Tycko B. Distress during pregnancy: Epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior. Am J Psychiatry. 2016;173:705–13.
Alexander N, Wankerl M, Hennig J, Miller R, Zänkert S, Steudte-Schmiedgen S, et al. DNA methylation profiles within the serotonin transporter gene moderate the association of 5-HTTLPR and cortisol stress reactivity. Transl Psychiatry. 2014;4:e443.
Alexander N, Illius S, Stalder T, Wankerl M, Muehlhan M, Kirschbaum C. Serotonin transporter gene methylation predicts long-term cortisol concentrations in hair. Psychoneuroendocrinology 2019;106:179–82.
Devlin AM, Brain U, Austin J, Oberlander TF. Prenatal exposure to maternal depressed mood and the MTHFR C677T variant affect SLC6A4 methylation in infants at birth. PLoS ONE. 2010;5:e12201.
Wankerl M, Miller R, Kirschbaum C, Hennig J, Stalder T, Alexander N. Effects of genetic and early environmental risk factors for depression on serotonin transporter expression and methylation profiles. Transl Psychiatry. 2014;4:e402.
Porter RJ, Gallagher P, Watson S, Young AH. Corticosteroid-serotonin interactions in depression: a review of the human evidence. Psychopharmacology 2004;173:1–17.
Provençal N, Binder EB. The effects of early life stress on the epigenome: From the womb to adulthood and even before. Exp Neurol. 2015;268:10–20.
Provençal N, Arloth J, Cattaneo A, Anacker C, Cattane N, Wiechmann T, et al. Glucocorticoid exposure during hippocampal neurogenesis primes future stress response by inducing changes in DNA methylation. Proc Natl Acad Sci USA. 2020;117:23280–5.
Karlsson L, Barbaro M, Ewing E, Gomez-Cabrero D, Lajic S. Epigenetic alterations associated with early prenatal dexamethasone treatment. J Endocr Soc. 2019;3:250–63.
Edelmann MN, Sandman CA, Glynn LM, Wing DA, Davis EP. Antenatal glucocorticoid treatment is associated with diurnal cortisol regulation in term-born children. Psychoneuroendocrinology 2016;72:106–12.
Provenzi L, Guida E, Montirosso R. Preterm behavioral epigenetics: a systematic review. Neurosci Biobehav Rev. 2018;84:262–71.
Küpers LK, Monnereau C, Sharp GC, Yousefi P, Salas LA, Ghantous A, et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat Commun. 2019;10:1893.
Stalder T, Steudte-Schmiedgen S, Alexander N, Klucken T, Vater A, Wichmann S, et al. Stress-related and basic determinants of hair cortisol in humans: a meta-analysis. Psychoneuroendocrinology 2017;77:261–74.
Steudte-Schmiedgen S, Wichmann S, Stalder T, Hilbert K, Muehlhan M, Lueken U, et al. Hair cortisol concentrations and cortisol stress reactivity in generalized anxiety disorder, major depression and their comorbidity. J Psychiatr Res. 2017;84:184–90.
Schalinski I, Elbert T, Steudte-Schmiedgen S, Kirschbaum C. The cortisol paradox of trauma-related disorders: lower phasic responses but higher tonic levels of cortisol are associated with sexual abuse in childhood. PLoS ONE. 2015;10:e0136921.
Ilg L, Kirschbaum C, Li S-C, Wimberger P, Nitzsche K, Rosenlöcher F, et al. No association of antenatal synthetic glucocorticoid exposure and hair steroid levels in children and adolescents. J Clin Endocrinol Metab. 2020;105:e575–e582.
Kirschbaum C, Pirke KM, Hellhammer DH. The ‘Tier Social Stress Test’ – A tool for investigating psychobiological stress responses in a laboratory setting. Neuropsychobiology 1993;28:76–81.
Buske-Kirschbaum A, Jobst S, Wustmans A, Kirschbaum C, Rauh W, Hellhammer D. Attenuated free cortisol response to psychosocial stress in children with atopic dermatitis. Psychosom Med. 1997;59:419–26.
Gao W, Kirschbaum C, Grass J, Stalder T. LC–MS based analysis of endogenous steroid hormones in human hair. J Steroid Biochem Mol Biol. 2016;162:92–99.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215.
Moser DA, Müller S, Hummel EM, Limberg AS, Dieckmann L, Frach L, et al. Targeted bisulfite sequencing: a novel tool for the assessment of DNA methylation with high sensitivity and increased coverage. Psychoneuroendocrinology 2020;120:104784.
McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonté B, Szyf M, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–8.
Vandesompele, J, De Preter, K, Pattyn, F, Poppe, B, Van Roy, N, De Paepe, A et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:research0034.1.
Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300.
Pruessner JC, Kirschbaum C, Meinlschmid G, Hellhammer DH. Two formulas of computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology 2003;28:916–31.
Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc. 1995;90:773–95.
Schönbrodt FD, Wagenmakers E-J. Bayes factor design analysis: Planning for compelling evidence. Psychon Bull Rev. 2018;25:128–42.
Moser DA, Molitor A, Kumsta R, Tatschner T, Riederer P, Meyer J. The glucocorticoid receptor gene exon 1-F promoter is not methylated at the NGFI-A binding site in human hippocampus. World J Biol Psychiatry. 2007;8:262–8.
van der Knaap LJ, Oldehinkel AJ, Verhulst FC, van Oort FVA, Riese H. Glucocorticoid receptor gene methylation and HPA-axis regulation in adolescents. The TRAILS study. Psychoneuroendocrinology 2015;58:46–50.
Alexander N, Kirschbaum C, Wankerl M, Stauch BJ, Stalder T, Steudte-Schmiedgen S, et al. Glucocorticoid receptor gene methylation moderates the association of childhood trauma and cortisol stress reactivity. Psychoneuroendocrinology 2018;90:68–75.
Elwenspoek MMC, Hengesch X, Leenen FAD, Sias K, Fernandes SB, Schaan VK, et al. Glucocorticoid receptor signaling in leukocytes after early life adversity. Dev Psychopathol. 2020;32:853–63.
Leenen FAD, Muller CP, Turner JD. DNA methylation: conducting the orchestra from exposure to phenotype? Clin Epigenetics. 2016;8:92.
Yehuda R, Daskalakis NP, Bierer LM, Bader HN, Klengel T, Holsboer F, et al. Holocaust exposure induced intergenerational effects on FKBP5 methylation. Biol Psychiatry. 2016;80:372–80.
Lee RS, Mahon PB, Zandi PP, McCaul ME, Yang X, Bali U, et al. DNA methylation and sex-specific expression of FKBP5 as correlates of one-month bedtime cortisol levels in healthy individuals. Psychoneuroendocrinology 2018;97:164–73.
Ouellet-Morin I, Wong CCY, Danese A, Pariante CM, Papadopoulos AS, Mill J, et al. Increased serotonin transporter gene (SERT) DNA methylation is associated with bullying victimization and blunted cortisol response to stress in childhood: a longitudinal study of discordant monozygotic twins. Psychol Med. 2013;43:1813–23.
Duman EA, Canli T. Influence of life stress, 5-HTTLPR genotype, and SLC6A4 methylation on gene expression and stress response in healthy Caucasian males. Biol Mood Anxiety Disord. 2015;5:2.
Li-Tempel T, Larra MF, Sandt E, Mériaux SB, Schote AB, Schächinger H, et al. The cardiovascular and hypothalamus-pituitary-adrenal axis response to stress is controlled by glucocorticoid receptor sequence variants and promoter methylation. Clin Epigenet. 2016;8:12.
Gertz J, Varley KE, Reddy TE, Bowling KM, Pauli F, Parker SL, et al. Analysis of DNA methylation in a three-generation family reveals widespread genetic influence on epigenetic regulation. PLoS Genet. 2011;7:e1002228.
Rietschel L, Streit F, Zhu G, McAloney K, Frank J, Couvy-Duchesne B, et al. Hair cortisol in twins: Heritability and genetic overlap with psychological variables and stress-system genes. Sci Rep. 2017;7:15351.
Braun PR, Han S, Hing B, Nagahama Y, Gaul LN, Heinzman JT, et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl Psychiatry. 2019;9:47.
Hannon E, Mansell G, Walker E, Nabais MF, Burrage J, Kepa A, et al. Assessing the co-variability of DNA methylation across peripheral cells and tissues: implications for the interpretation of findings in epigenetic epidemiology. PLoS Genet. 2021;17:e1009443.
Jones MJ, Moore SR, Kobor MS. Principles and challenges of applying epigenetic epidemiology to psychology. Annu Rev Psychol. 2018;69:459–85.
Benediktsson R, Calder AA, Edwards CR, Seckl JR. Placental 11β-hydroxysteroid dehydrogenase: a key regulator of fetal glucocorticoid exposure. Clin Endocrinol 1997;46:161–6.
Acknowledgements
This research was funded by a joint research grant from the German Research Foundation (DFG) to N.A. (AL 1485/5-1), C.K. (KI 537/36-1), and S.C.L (LI 879/15-1). C.K. and S.C.L are also supported by SFB-940. S.C.L. is also supported by EXC 2050/1. We thank Dr. Liesa Ilg for her valuable effort in participant recruitment. We deeply appreciated the children and adolescents who participated in the study as well as their families.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
L.-S.C., C.K., and N.A. designed the study and obtained funding. P.W. and K.N. enabled recruitment of the participants. N.A. supervised the project parts related to DNA methylation and gene expression analysis. D.M. supervised epigenetic analyses and laboratory work packages including DNA methylation and gene expression analyses. S.M. conducted DNA and RNA/cDNA work in the laboratory. S.M. and L.F. performed the statistical analyses and figures. S.M. wrote the first manuscript draft. D.M. and N.A. revised the manuscript critically. All authors commented on the manuscript and approved the final version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Müller, S., Moser, D., Frach, L. et al. No long-term effects of antenatal synthetic glucocorticoid exposure on epigenetic regulation of stress-related genes. Transl Psychiatry 12, 62 (2022). https://doi.org/10.1038/s41398-022-01828-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-01828-x
- Springer Nature Limited