
Abstract
The immune response holds a pivotal role in cardiovascular disease development. As multifunctional cells of the innate immune system, macrophages play an essential role in initial inflammatory response that occurs following cardiovascular injury, thereby inducing subsequent damage while also facilitating recovery. Meanwhile, the diverse phenotypes and phenotypic alterations of macrophages strongly associate with distinct types and severity of cardiovascular diseases, including coronary heart disease, valvular disease, myocarditis, cardiomyopathy, heart failure, atherosclerosis and aneurysm, which underscores the importance of investigating macrophage regulatory mechanisms within the context of specific diseases. Besides, recent strides in single-cell sequencing technologies have revealed macrophage heterogeneity, cell–cell interactions, and downstream mechanisms of therapeutic targets at a higher resolution, which brings new perspectives into macrophage-mediated mechanisms and potential therapeutic targets in cardiovascular diseases. Remarkably, myocardial fibrosis, a prevalent characteristic in most cardiac diseases, remains a formidable clinical challenge, necessitating a profound investigation into the impact of macrophages on myocardial fibrosis within the context of cardiac diseases. In this review, we systematically summarize the diverse phenotypic and functional plasticity of macrophages in regulatory mechanisms of cardiovascular diseases and unprecedented insights introduced by single-cell sequencing technologies, with a focus on different causes and characteristics of diseases, especially the relationship between inflammation and fibrosis in cardiac diseases (myocardial infarction, pressure overload, myocarditis, dilated cardiomyopathy, diabetic cardiomyopathy and cardiac aging) and the relationship between inflammation and vascular injury in vascular diseases (atherosclerosis and aneurysm). Finally, we also highlight the preclinical/clinical macrophage targeting strategies and translational implications.
Similar content being viewed by others

Introduction
The immune response is an important driver of cardiovascular disease (CVD) occurrence and development. Macrophages are key immune cells that exert significant impact on the entire process from inflammation to repair in CVD by expressing specific phenotypes.1,2,3 Generally, upon injury, macrophages are massively recruited to the damaged area by C-C chemokine receptor type 2 (CCR2) and become the dominant immune cells. Macrophages not only play a major role in the inflammatory response by phagocyting tissue debris and releasing a large number of pro-inflammatory cytokines and proteinases, but also secrete a variety of mediators to stimulate extracellular matrix (ECM) production, cell proliferation and angiogenesis.3,4 In addition, macrophages occupy a central position and participate in cross-talk with other cells mainly through the release of different mediators, such as affecting the chemotaxis and functions of other immune cells to regulate immune response, facilitating or suppressing the generation of vascular endothelial cells (ECs) and regulating fibrosis by directly facilitating the activation and proliferation of fibroblasts and promoting their differentiation into myofibroblasts.5 In spite of these common features, there are differences in the phenotype and function of macrophages in specific disease contexts. For example, in the late stage of ischemic injury, resident macrophages tend to proliferate and play a repair role, whereas, in other cardiac diseases, recruited macrophages play a major role, with or without resident macrophage loss. In particular, macrophages in atherosclerosis (AS) phagocytose oxidized low-density lipoprotein (OxLDL) to form foam cells, which are mainly involved in lipid metabolism. Hence, it can be seen that macrophages are indispensable contributors to the development of various CVD.
Myocardial fibrosis, a common pathological outcome of various CVD, is characterized by excessive deposition and abnormal distribution of collagen. Macrophages play an important role in the occurrence, progression and repair of myocardial fibrosis. The structural quality, fibrillary composition and metabolic properties of fibrosis differ under diverse etiologies, resulting in distinct pathophysiological characteristics and clinical manifestations.6,7 Based on histopathological characteristics, fibrosis can primarily be classified into replacement fibrosis and interstitial fibrosis.4 After myocardial ischemic injury, cardiomyocyte death and replacement fibrosis occur, leading to systolic dysfunction. In non-ischemic injury, interstitial fibrosis mainly occurs, contributing to diastolic dysfunction.6 Therefore, it is necessary to consider the diverse disease contexts and types of fibrosis separately when investigating fibrotic pathways. Inflammation is also the main feature of vascular diseases, which can give rise to thrombosis, hardening and narrowing of blood vessel walls and CVD such as myocardial infarction (MI).8 Thus, for effective CVD therapy, identifying and targeting cells along with molecules that regulate fibrosis and inflammation becomes imperative in order to limit or reverse their overdevelopment without disrupting tissue repair. Besides, with the development of emerging technologies such as single-cell RNA sequencing (scRNA-seq), the cellular heterogeneity, microenvironmental signaling, and intracellular regulation during the process of CVD have been elucidated to a greater extent.9,10 For the first time, we comprehensively summarize macrophage classifications and the mechanisms by which macrophages regulate the development of CVD in a range of contexts, including ischemic cardiac injury (acute myocardial infarction (AMI), ischemia-reperfusion injury (IRI), and chronic myocardial infarction (CMI)), non-ischemic cardiac injury (pressure overload (PO), myocarditis, dilated cardiomyopathy (DCM), diabetic cardiomyopathy, and cardiac aging) and vascular diseases (AS and aneurysms), where we concentrate on macrophage-regulated fibrosis formation in cardiac diseases. In addition, we propose the heterogeneity of macrophages from a single-cell perspective and provide new insights into the complex biological processes underlying macrophage-mediated CVD. Finally, aimed at providing new intervention targets and therapeutic strategies for the clinical treatment of CVD, preclinical strategies and published/ongoing clinical trials targeting macrophages are further consolidated.
Origin, phenotype, and function of macrophages in cardiovascular system
The traditional view holds that macrophages are derived from circulating monocytes and are classified into M1/M2 macrophages based on the different stimuli required for in vitro culture. M2 macrophages can be subdivided into four subsets: M2a, M2b, M2c, and M2d.11 M1 macrophages highly express markers such as cluster of differentiation (CD) 80, CD86, and inducible nitric oxide synthase (iNOS), which are primarily associated with the inflammatory response.12 M2 macrophages highly express markers such as CD163, CD206, Arg1, FIZZ1, and YM1. In addition to the M2b subset, which secretes both pro-inflammatory and anti-inflammatory factors to regulate the immune response, other M2 subsets exhibit a repair phenotype mainly through the secretion of anti-inflammatory and pro-fibrotic factors.11 Since there are multiple influencing factors in vivo, the extreme classification of M1/M2 macrophages cannot summarize the complex and diverse functions of macrophages.13,14 In recent years, macrophages have been defined and classified as tissue-resident macrophages and monocyte-derived macrophages according to their different origins, and they have distinct phenotypes and functions. Referring to tissue-specific subsets that differentiate during organogenesis and are capable of establishing stable spatial and functional relationships with specific tissue cells, tissue-resident macrophages are mainly of embryonic origin and equipped with strong self-renewal, anti-inflammatory, and homeostasis maintenance abilities.15 Circulating monocyte-derived macrophages are primarily pro-inflammatory and actively produce high levels of pro-inflammatory cytokines and chemokines.
Origin, phenotype and function of macrophages in heart
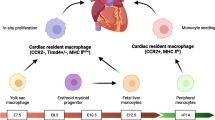
In recent years, CCR2 expression, which can reflect the dynamic changes in macrophage phenotype and the origin of cardiac macrophages, has been utilized for the classification of cardiac macrophages.16,17,18 CCR2- macrophages derived from embryonic yolk sac and fetal liver monocytes are maintained in the absence of monocyte recruitment, whereas CCR2+ macrophages are sustained through monocyte recruitment. Among them, CCR2- subset and a few CCR2+ subsets are resident macrophages. In addition, resident and recruited macrophages in the heart also express major histocompatibility complex class II (MHC-II)/human leukocyte antigen-DR (HLA-DR) to varying degrees, which are associated with antigen presentation and the activation of T cells.19 The introduction of MHC-II markers allows for better differentiation of macrophage subsets with distinct functions. Based on CCR2 and MHC-II/HLA-DR markers, mouse cardiac macrophages can be categorized into three subsets: CCR2-MHCIIlow, CCR2-MHC-IIhigh, and CCR2+MHC-IIhigh (Fig. 1a). Human cardiac macrophages can be categorized into two subsets: CCR2-HLA-DRhigh and CCR2+HLA-DRhigh.16,20 The CCR2+MHC-IIhigh subset replaces CCR2- macrophages during aging and myocardial injury.16,18,20,21 Notably, MHC-II markers in embryonic-derived macrophages are gradually upregulated after birth, which first appear in the CCR2+ subset and then in the CCR2- subset.18 Thus, the majority of cardiac macrophages in neonatal mice are CCR2-MHC-IIlow subset, whereas the adult mouse heart contains three resident macrophage subsets.21

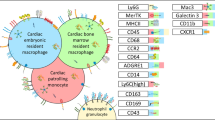
Origin, phenotype and function of macrophages in cardiovascular system under homeostasis, MI and AS. a In cardiac homeostasis, three types of resident macrophages exist in the heart. CCR2-MHClow macrophages and CCR2-MHChigh macrophages are derived from yolk sac cells and fetal liver monocytes and maintain the number of subpopulations through self-renewal, while monocytes also contribute a small amount to the number of subpopulations. CCR2+MHChigh macrophages are derived from fetal liver monocytes and are gradually replaced by circulating monocytes during development. Artery-resident macrophages, predominantly located in the adventitia during homeostasis, are derived from yolk sac cells, fetal liver monocytes and bone marrow (after birth). Main functions and transcriptome signature of each subset are highlighted in the colored corresponding boxes. b When MI occurs, cardiac TLF+ macrophages undergo self-renewal. In addition, a large number of Ly6Chigh monocytes infiltrate into the heart and mainly differentiate into three types of macrophages, including MHC+ macrophages, ISG+ macrophages and Trem2+ macrophages. In AS, macrophages can be classified into four main subsets, including proliferating macrophages, inflammatory macrophages, IFNIC and foamy/TREM2+ macrophages. Proliferating macrophages maintain the number of subpopulations through completely self-renewal and other subsets are derived from Ly6Chigh monocytes. Main location, functions and transcriptome signature of each subset are highlighted in the colored corresponding boxes. (Created with BioRender.com)
Different subsets of macrophages focus on specific functions.3 The functions of resident macrophages include secreting anti-inflammatory mediators, promoting tissue repair, clearing apoptotic cells and damaged mitochondria, regulating myocardial fibrosis and inhibiting hypertrophy. In cardiac diseases, monocytes are recruited to lesion sites via C-C motif chemokine ligand (CCL) 2/ C-X3-C motif chemokine ligand 1 (CX3CL1) and predominantly differentiate into the CCR2+MHC-IIhigh macrophage subset.22 The effects of recruited macrophages on cardiac function and cardiac remodeling would be deeply discussed in the following context given diverse functions in relation to specific pathological states. In particular, MHC-IIhigh subset macrophages pivotally involve in immunodetection by scavenging the environment, recognizing and clearing pathogens, and presenting antigens.3,22 The distinct roles of specific macrophage subsets in myocardial fibrosis vary across different diseases, resulting in bidirectional regulatory effects on myocardial fibrosis23 (Table 1). When it comes to promoting fibrosis, firstly, macrophages secrete a variety of pro-fibrotic mediators, such as transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), interleukin (IL)-10, vascular endothelial growth factor (VEGF), and amphiregulin (AREG), which directly induce the proliferation and activation of fibroblasts through the fibroblast receptors PDGFR, TGF-βR, and epidermal growth factor receptor (EGFR), thereby facilitating collagen synthesis.24,25,26 Furthermore, macrophages secrete substances that inhibit the degradation of the ECM, such as tissue inhibitor of matrix metalloproteinases (TIMPs), facilitating cardiac scar formation and myocardial remodeling.27 Additionally, macrophages have the potential to differentiate into fibroblasts and secrete collagen fibers, but the specific subset of macrophages with this capability remains unidentified.28,29,30 When referring to anti-fibrosis, some macrophages invovled can not only express a large number of matrix metalloproteinases (MMPs) but also stimulate other cells to produce MMPs, effectively degrading ECM components, which is crucial for the regression of fibrosis.31 Additionally, with a constant number of macrophages, it is generally believed that the polarization of macrophages towards the M2 phenotype can suppress inflammation over time, leading to a reduction in fibrosis.32,33 Macrophages can also regulate fibrosis through phagocytosis and modulation of inflammatory responses. While suppressing fibrosis by removing necrotic tissue and temporary matrix through phagocytosis,31 some pro-inflammatory or anti-inflammatory factors secreted by macrophages can directly act on interleukin 1 receptor (IL-1R), IL-6R complex, and angiotensin-II type 1 receptor (AT1R) on fibroblasts, or induce the increase of pro-fibrotic factors, thereby promoting fibrosis.25,26,34,35,36 In the regulation of both pro-repair fibroblasts and pro-fibrotic myofibroblasts, macrophages play a crucial role in maintaining a delicate balance, primarily through the secretion of inflammatory mediators. A majority of these pro-inflammatory mediators, including IL-1β, IL-6, and IL-23, which are released by macrophages, induce fibrosis and unfavorable cardiac remodeling in both ischemic and non-ischemic cardiac diseases.7,37 However, in the context of ischemic cardiac disease, which necessitates the production of replacement scar, early inflammation is advantageous for preserving cardiac repair.38 Conversely, macrophages aid in resolving chronic inflammation in cardiac disease through the process of phagocytosis and the secretion of anti-inflammatory mediators such as TGF-β and IL-10, thereby promoting cardiac repair.39 It is worth noting that different macrophage subsets may partially exhibit similar functions under M1/M2 classification and CCR2 classification. CCR2+ macrophages primarily display pro-inflammatory characteristics akin to those of M1 macrophages, but they can transition to a phenotype resembling the M2 subset during later stages of injury. The CCR2- macrophages have comparable anti-inflammatory and reparative functions to M2 macrophages.
Origin, phenotype and function of macrophages in vascular system
Artery-resident macrophages are predominantly distributed in the adventitia during homeostasis and have been found to originate from two main sources in mice. In the embryo, macrophages mainly develop from yolk sac-derived C-X3-C motif chemokine receptor 1 (CX3CR1)+ endothelial microparticles (EMPs), with a smaller contribution from fetal liver monocytes.40 After birth, these macrophages are immediately colonized and replaced by monocyte-differentiated macrophages. In adulthood, only about 20% of arterial-resident macrophages are still yolk sac-derived.40 Artery-resident macrophages express the CD206 marker.41,42,43 However, unlike cardiac macrophages, the CCR2+ subset also exists in artery-resident macrophages. In common with cardiac macrophages, arterial macrophages in neonatal mice are MHC-IIlow and develop MHC-IIhigh macrophages after a period of birth.40 Independent of the replenishment of circulating monocytes, adult mouse artery-resident macrophages are sustained primarily through self-renewal. In addition, embryonic and monocyte-derived arterial macrophages have comparable self-renewal abilities3,40 (Fig. 1a). Although mouse vascular macrophages have been extensively studied, there is still limited data available on the origin and phenotype of human vascular macrophages. Nowadays, numerous studies are dedicated to mapping the development and differentiation of human vascular macrophages using high-precision single-cell transcriptome sequencing technology. It is found that the categorization of arterial macrophages at the single-cell level is cross-correlated with the traditional M1/M2 categorization.
Generally speaking, macrophages play an essential role in regulating phagocytosis, immune surveillance, inflammation and remodeling in blood vessel3 (Table 2). The phagocytosis and immune surveillance functions of macrophages are primarily performed by resident macrophages. Inflammatory response and vascular remodeling occur in vascular diseases. Macrophage proliferation in the early stages of vascular disease mainly depends on monocyte recruitment and differentiation.40,44,45 Among them, lymphocyte antigen 6 complex, locus C (Ly6C)high monocytes chiefly differentiate into M1 macrophages, while it remains unclear which macrophage subpopulation Ly6Clow monocytes preferentially differentiate into. In terms of inflammation, macrophages facilitate chronic vascular inflammation by releasing pro-inflammatory cytokines such as IL-1, IL-6, and tumor necrosis factor (TNF). Mainly, inflammatory macrophages act similarly to the M1 phenotype.8,46 With respect to anti-inflammation, macrophages secrete anti-inflammatory factors, such as IL-10 and TGF-β, to suppress inflammation, similar to the M2 phenotype.46,47,48,49 A distinct population of foam cells in AS serves as an early marker of atherosclerotic plaques in mice and humans, which exhibit lipid phagocytosis and metabolic functions.50,51 However, dead foam cells release lipids and tissue factors to form the necrotic core, a crucial component of unstable plaques, which facilitates plaque rupture and subsequent intravascular clot formation, ultimately leading to MI.52 Macrophages also exhibit a high degree of matrix-degrading activity by releasing MMPs, which leads to the degradation of collagen.53 This process damages the vessel wall and results in adverse remodeling of the vessel wall.53 Furthermore, in addition to M1/M2 macrophages, recent studies have identified several novel macrophage subtypes in atherosclerotic plaques: Mox, M4, Mhem, and M(Hb) macrophages, which exhibit unique gene expression profiles and functional properties.54 Mox macrophages are bone marrow-derived cells with decreased expression of M1-M2 related genes, which can facilitate heme detoxification, reduce oxidative stress, and inhibit foam cell formation.55 M4 macrophages, mostly found in unstable plaques, highly express chemokines, such as CCL2 and CXCL4, and proteases, such as MMP-12, which recruit monocytes and neutrophils to degrade ECM proteins.56 In ruptured hemorrhage sites, M(Hb) and hemin-induced Mhem macrophages exist. Mhem, with a high expression of CD163 and heme oxygenase-1 (HO-1),57,58,59 promotes erythrocyte turnover by phagocytosis of senescent and damaged erythrocytes, thereby recycling iron and heme. M (Hb) highly expresses CD206 and CD163, which can remove free hemoglobin and inhibit its pro-oxidation effects.60
Heterogeneity and regulatory mechanisms of cardiac macrophages
Ischemic injury
Ischemic injury directly leads to myocardial death, and since the regenerative capacity of cardiomyocytes is limited, repair of the infarcted heart mainly relies on scar tissue formation. There are three types of ischemic injury: acute ischemia, ischemia-reperfusion and chronic ischemia. In all three types of ischemia, the damaged cardiomyocytes and ECM release damage associated molecular patterns (DAMPs) to activate pattern recognition receptors (PRRs) on the surviving parenchymal cells, which secrete inflammatory cytokines and chemokines to recruit monocytes and other inflammatory cells.38 However, the types of cardiac remodeling and fibrosis induced by the three ischemic injuries differ. Acute ischemia mainly causes inflammation and replacement fibrosis at the infarct zone. Ischemia-reperfusion restores blood supply on the basis of acute ischemia, resulting in smaller infarct area and scar. The most important pathological process of chronic ischemia is interstitial fibrosis in the remote zone, which is induced by persistent chronic inflammation and altered cardiac structure.
Acute myocardial infarction
AMI is defined as the extensive death of cardiomyocytes and acute injury to the myocardium resulting from acute myocardial ischemia. Currently, the paradigm of MI in animal models is primarily divided into inflammatory, anti-inflammatory, and reparative stages.24 After MI, macrophages and other inflammatory cells are recruited to the infarct zone, leading to the production of pro-inflammatory cytokines to intensify inflammation and remove necrotic tissue.38 With the removal of necrotic tissue, macrophages switch phenotypes to produce anti-inflammatory cytokines that mediate the termination of inflammation and transition into the anti-inflammatory phase.24 Anti-inflammatory cytokines facilitate the differentiation of fibroblasts into myofibroblasts, which produce replacement fibrosis during the reparative phase.38 It should be noted that the high concentration of pro-inflammatory mediators prevents the pro-fibrotic mediators from exerting pro-fibrotic effects during the inflammatory phase,61 which may inhibit the premature emergence of collagen-producing cells, as the inflammatory phase is dominated by the clearance of infarct cells and matrix debris rather than collagen deposition.61 If inflammatory conduction is excessively blocked during the inflammatory phase, the risk of cardiac rupture leading to death and wall thinning leading to cardiac dilation increases, despite subsequent reductions in myofibroblast infiltration and collagen deposition.62,63,64 Unlike early intervention in inflammation to block the inflammatory cascade, late intervention in inflammation may primarily eliminate the direct effects of pro-inflammatory mediators on fibroblasts.65 In this review, we categorize the MI paradigm into two phases: the inflammatory phase and the reparative phase, discussing the fundamental principle that macrophages tend to secrete inflammatory cytokines during the inflammatory phase but anti-inflammatory cytokines to participate in scar formation during the reparative phase.
Inflammatory phase
The inflammatory phase is the period distinguished by recruitment of inflammatory cells and clearance of necrotic tissue, usually between 0 and 4 days after ischemia. Ly6Chigh monocytes are recruited to the infarct zone through CCR2/CCL2 signaling and differentiate into CCR2+MHC-IIhigh macrophages, replacing the lost resident macrophages,66,67 so recruited CCR2+ macrophages play a dominant role in the inflammatory phase.68 When compared with tissue-resident macrophages, recruited CCR2+ macrophages express higher levels of inflammatory chemokines (monocyte chemoattractant protein-1 (MCP-1)), cytokines (IL-1β, IL-6, TNF-α), and genes implicated in adverse cardiac remodeling (MMP-9, TIMP-1).20,21 Different subsets of surviving resident macrophages play distinct roles in the process of recruiting monocytes. The tissue-resident CCR2- macrophages can inhibit monocyte recruitment, playing an important role in preventing myocardial fibrosis after cardiac injury.13,21 The tissue-resident CCR2+ macrophages contribute to the recruitment of neutrophils and monocytes. Thereby, the depletion of this subset attenuates inflammation and myocardial fibrosis following MI.13,69
Recruited macrophages clear necrotic tissue and create an environment conducive to scar repair through three mechanisms, including the synthesis of pro-inflammatory mediators, the synthesis of MMPs, and phagocytosis (Fig. 2a). These three mechanisms interact with each other, which is reflected in the fact that inflammation promotes the recruitment of macrophages to perform phagocytosis, phagocytosis promotes the normal progress of inflammation, and MMP is also involved in the regulation of substances related to inflammation and phagocytosis. Most of the pro-inflammatory mediators synthesized during the inflammatory phase play a pro-fibrotic role, including IL-1,65,70 NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome,16,71 IL-6,72,73 and angiotensin-II (Ang-II),74 among which IL-1 plays a dominant role. IL-1 can be divided into IL-1α and IL-1β. IL-1α enhances the release of pro-inflammatory mediators such as IL-6 and MCP-1 and the expression of fibrosis genes such as connective tissue growth factor (CTGF), ultimately promoting myocardial fibrosis.75 Compared to IL-1α, IL-1β has contradictory effects. On the one hand, IL-1β secreted by recruited macrophages inhibits the expression of α-smooth muscle actin (α-SMA) in cardiac fibroblasts (CFs) and delays the transformation of myofibroblasts.61 On the other hand, IL-1β increases the fibrotic mediator TGF-β1 in the infarct zone and collaborates with TNF-α to increase the AT1R density on CFs, which prompts collagen deposition during the reparative phase.36,65 As an effector mediating pro-inflammatory signaling cascades in innate immunity, the caspase-recruitment domain family member 9 (CARD9) can upregulate the macrophages to express lipocalin 2 (Lcn2) and MMP-9, which consequently contributes to myocardial apoptosis, the deterioration of cardiac function and adverse remodeling after MI.76

Regulations of myocardial fibrosis by macrophages after ischemic injury. a In the inflammatory phase of AMI, DAMP activates retained cells in the heart to release pro-inflammatory mediators, thereby promoting monocyte infiltration and differentiation into CCR2+ macrophages. CCR2+ macrophages secrete mediators (such as MMPs, miR-15, and VEGFA) to regulate inflammation and fibrosis in order to clear necrotic tissue and prepare for subsequent cardiac repair. b In the reparative phase of AMI, restorative Ly6ClowCD206+ macrophages become the main macrophage subset in the heart. They secrete anti-inflammatory and pro-fibrotic mediators such as TGF-β, IL-10, galectin-3, and IL-10 to promote the conversion of fibroblasts into myofibroblasts which secrete collagen to form scars. c When CMI occurs, CCR2+ macrophages continue to infiltrate into the heart, interact with T cells, and secrete a large amount of pro-inflammatory and pro-fibrotic factors, causing interstitial fibrosis in remote zone. d When IRI occurs in the heart, a large number of CCR2+ macrophages accumulate in the early stage. They upregulate LGR4, Dectin-1 and CCL17 to promote inflammation and myocardial fibrosis, or upregulate the expression of CD44 and receive small extracellular vesicles secreted by M2 macrophages to convert to a reparative phenotype and attenuate the inflammatory response caused by ROS. (Created with BioRender.com)
MMP can affect fibrosis by regulating inflammatory signal transduction and degrading substrates dominated by ECM, but the former is the main function in the inflammatory phase.77 During the inflammatory phase, MMP-9, MMP-12, and MMP-28 are important MMPs secreted by macrophages, among which MMP-9 is more widely studied. MMP-9 exerts impacts on fibrosis mainly by affecting the expression of other MMP isoforms, including MMP-2, MMP-8, MMP-12, and MMP-13, to regulate the infiltration of macrophages and neutrophils.78,79 At present, it is not yet clear whether MMP-9 promotes or inhibits fibrosis, which may be due to the presence of multiple MMP subtypes involved. Unlike MMP-9, the fibrosis induced by MMP-12 and MMP-28 is beneficial for maintaining cardiac function in the later stage. Not only does MMP-12 stimulate the synthesis of cluster of differentiation (CD) 44 on the surface of neutrophils and enhance the interaction between CD44 and hyaluronan in the ECM, promoting the expression of apoptotic genes in neutrophils and the timely resolution of inflammation,80 but also downregulate the expression of C-X-C Motif Chemokine Ligand (CXCL) 1, CXCL2, and CXCL5 in the heart to prevent neutrophil infiltration and significantly reduce the secretion of MMP-9.81 MMP-28 can improve post-MI remodeling and dysfunction by inhibiting M2 macrophage activation, ECM synthesis, and collagen cross-linking.82
Phagocytosis consists of four processes: recognition, binding, internalization, and degradation of dying cells.39 After MI, apoptotic cells express “Find-me” and “Eat-me” signals (e.g., lipid mediators and nucleotides), which can attract macrophages and bind to phagocytosis-associated receptors on the cells, including myeloid-epithelial-reproductive receptor tyrosine kinase (Mertk), milk fat globule epidermal growth factor 8 (Mfge8), CD36, and legumain. Binding to these receptors initiates the phagocytosis process to remove neutrophils and myocardial debris,83 and inhibition of this process will lead to the obstruction of inflammatory program and greater fibrosis.84 The externalization of phosphatidylserine on the injured cell membrane is one of the earliest signals sufficient to activate the phagocytotic process, while Mertk- and Mfge8-expressing monocyte/macrophages play nonredundant roles in the recognition of phosphatidylserine,84 which mediates the clearance of damaged cardiomyocytes and favors the secretion of VEGFA to locally repair the dysfunctional heart.84 CD36, a scavenger receptor, is important for macrophage phagocytosis of apoptotic neutrophils. MI triggers macrophage infiltration into the infarct area to release abundant CXCL4, which decreases CD36 expression in a direct or MMP-9 dependent manner to inhibit macrophage phagocytosis of dead myocytes and neutrophils, eventually resulting in adverse remodeling post-MI.83 Resident macrophage-derived legumain promotes the efferocytosis of apoptotic cardiomyocytes, bringing about the recruitment of CCR2+ MHC-IIhigh macrophages and the inhibition of pro-inflammatory cytokine secretion, thereby improving cardiac repair.85 In addition, recent studies have revealed that mitochondrial metabolism has the potential to affect macrophage efferocytosis. Macrophage mitochondrial complex I deficiency can promote glycolysis and increase mitochondrial reactive oxygen species (ROS) production, which aggravates the early inflammatory response and impairs efferocytosis, thereby hindering the proliferation and activation of fibroblasts and scar formation after MI.86 Apart from collagen present in the interstitium, vascular rupture will lead to the deposition of coagulation-related fibrin, whose clearance is mainly related to CCR2+ macrophages.87
Reparative phase
The reparative phase refers to the process of gradual resolution of inflammation, proliferation of myofibroblasts, and scar formation after the inflammatory phase, usually between 4 and 14 days after the onset of MI. After 3 days of MI, anti-inflammatory mediators are gradually generated to suppress neutrophil infiltration, enhance macrophage phagocytosis of apoptotic neutrophils, and transform the CCR2+Ly6Chigh recruited macrophages into reparative phenotypes.84,88 Except for the remaining macrophages in the inflammatory phase, Ly6Clow monocytes are recruited through CX3CR1/CX3CL1 signaling and differentiate into CCR2-Ly6Clow macrophages.89,90 It is generally accepted that reparative macrophages are characterized by low expression of Ly6C, CCR2, MHC-II, and high expression of CD206 and MerTK, as well as high expression of a series of anti-inflammatory and fibrosis-related genes, including Il10, hypoxia-inducible factor 1 α (Hif1a), Vegfa, insulin-like growth factor 1 (Igf1), secreted phosphoprotein 1 (Spp1), and Tgfb.22,91,92 Macrophages are capable of facilitating endothelial-to-mesenchymal transition,93 and certain macrophages undergo mesenchymal transition to adopt a fibroblast-like phenotype, directly contributing to collagen production.28,29,30 However, the specific macrophage subset equipped with the potential for fibroblast differentiation remains unidentified. Fibroblast-like macrophages express Acta2, type I collagen, fibroblast specific protein-1, prolyl-4-hydroxylase, and fibroblast activation protein and other markers, thereby secreting collagen and promoting fibrosis progression.28,29,30
Reparative macrophages regulate fibrosis mainly through fibrosis mediators and anti-inflammatory factors, of which TGF-β and IL-10 are garnering significant interest (Fig. 2b). Macrophages are an important source of TGF-β during the reparative phase, which can be induced by a variety of substances, such as hypoxia-induced V-set and Ig domain-containing 4 (VSIG4),94 tissue factor (TF)-protease-activated receptor 2 (PAR2) signaling,95 renin-angiotensin-aldosterone system (RAAS),96 MMP-14,97 and galectin-3.98,99 Expressed primarily in the peri-infarct zone,7 TGF-β predominantly transmits downstream signals through the small mothers against decapentaplegic (Smad) family,100 the most important of which targets fibroblasts through the TGF-β/Smad3 axis to motivate their migration, transdifferentiation and synthesis of collagen and fibronectin.101,102,103,104 Compared to Smad3, Smad2 plays a partial but limited role in conducting TGF-β signaling.95,97,104,105 The process of TGF-β-driven myofibroblast activation is also modulated by negative feedback from Smad7 through inhibition of Smad2/3, extracellular signal-regulated kinase (ERK), protein kinase B (Akt), and EGFR signaling.106,107 It is worth mentioning that Smad3 in macrophages contributes to the acquisition of an anti-inflammatory phenotype, yet it does not exert a marked impact on subsequent collagen deposition, demonstrating that fibrosis is not always in connection with inflammation.108 In view of anti-inflammatory factors, IL-10 is a pleiotropic cytokine and plays a differential role in the regulation of fibrosis. Hypoxia-induced VSIG4 promotes IL-10 expression in M2 macrophages, which ultimately accounts for the transformation of CFs into myofibroblasts.94 IL-10 also synergizes with macrophage colony-stimulating factor (M-CSF) to activate signal transducers and activators of transduction 3 (STAT3) and ERK in cardiac macrophages, which in turn elevates the expression of galectin-3 and MerTK, driving cardiac macrophage polarization and osteopontin (OPN) production.109,110 As a consequence, this process contributes to fibrosis.111 However, several studies have also found that IL-10 may play a role in inhibiting fibrosis. On the one hand, IL-10 can inhibit human antigen R (HuR)/MMP-9 signaling and activate the STAT3 to suppress collagen deposition.112,113 On the other hand, IL-10 stimulates myofibroblasts to enter a hyper-activated state represented by enriched hyaluronan levels and reduced collagen through the regulation of macrophage M2 polarization.114 In this state, myofibroblasts exhibit heightened proliferation, but collagen I secretion and collagen I–III ratio are reduced, thereby significantly attenuating myocardial fibrosis,114 which may imply that fibroblast activation does not necessarily represent increased collagen deposition.
Regardless of some progress, the heterogeneity of macrophages during the reparative phase has not been thoroughly explored,13,67 which impedes the further comprehension of the mechanisms by which reparative macrophages regulate fibrosis. In recent years, some studies on subsets have shed new light on the mechanism of fibrosis. During the reparative phase, the number of CCR2- resident macrophages gradually increases, but the ratio of resident macrophages to recruited macrophages does not return to the pre-infarction level.67 Moreover, genes that confer critical repair functions on resident macrophages (T-cell immunoglobulin- and mucin-domain-containing molecule-4 (Timd4), lymphatic vessel endothelial receptor 1 (Lyve1), Igf1, etc.) are not adopted by recruited macrophages, suggesting that recruited macrophages cannot compensate for the depletion of resident macrophages.67 Even if monocytes can be transformed into peripheral-derived resident macrophages, the time window for effective protection of cardiac function may have been missed,67 which put emphasis on the significance of understanding the functions of resident macrophages and the heterogeneity of recruited macrophages for fibrosis and cardiac repair.
Chronic myocardial infarction
CMI refers to the persistent ischemic injury of the myocardium, which can be regarded as a subsequent stage of AMI and can deteriorate into heart failure (HF). The commonly used model for constructing CMI in mice is the ligation of the coronary artery for several weeks. Interstitial fibrosis in the remote zone stands out as a pivotal characteristic of CMI, resulting in elevated cardiac stiffness and impaired heart function.115 Prolonged ischemia and heightened mechanical stress on the non-infarcted myocardium induce infiltration of inflammatory cells and activation of pro-fibrotic cytokines in the remote zone.115 In addition, continuously activated cells in old scars secrete pro-fibrotic factors that might traverse the interstitial gaps to the remote zone, triggering the activation and proliferation of local fibroblasts and collagen deposition.74
Although cardiac macrophages in CMI shares the origin from recruited monocytes and local macrophage proliferation as in AMI to some extent, the prolonged ischemic and stressful environment adds fuel to the proliferation of macrophages. Exposed to chronic stress in CMI, the heart elevates the release of norepinephrine (NE) from sympathetic nerves.116,117 On the one hand, NE controls the release of hematopoietic stem and progenitor cell (HSPC) through β3-adrenergic signaling. On the other hand, it downregulates the expression of CXCL12 to decrease HSPC homing.116,117 The strong cardiosplenic axis has also been found in CMI, with an increase in the proliferation of HSPC and innate immune cells in the spleen.117,118 Expanded intramedullary and extramedullary hematopoiesis causes circulating monocytes to continuously proliferate and mobilize to the heart, resulting in macrophage infiltration. Besides recruited monocytes, the activation of the mitogen-activated protein kinase (MAPK) pathway induces local cardiac macrophage proliferation in response to increased ventricular wall tension caused by thinning left ventricular wall and ventricular dilatation.117
The pro-inflammatory phenotype macrophages exhibit in CMI is attributed to the reduced mitochondrial oxidative phosphorylation in myocardial tissues, which in turn promotes the anti-inflammatory functions of macrophages38,119,120 (Fig. 2c). Besides, the interactions between cardiac macrophages and increased T cells which are exerted via cytokines in CMI are of vital importance for regulating fibrosis. Increased release of the inflammatory factors IL-1β and TNF-α by macrophages accounts for the inflammation and fibrosis in the myocardium.121,122 IL-1β and TNF-α continuously stimulate the upregulation of AT1R on fibroblasts within the peri-infarct zone, thereby intensifying the pro-fibrotic effect.36 TNF-α induces distinct effects specific to the tumor necrosis factor receptor (TNFR), with TNFR1 exacerbating fibrosis in the remote zone while TNFR2 mitigating it, which may also offer insights into the negative results seen in clinical trials of TNF antagonists.123 In dealing with the effects of T cells on macrophage activation, T helper (Th) 2 cells and regulatory T cells (Tregs) are the main phenotypes involved in CMI.124 Th2 cells secrete IL-4 and IL-13, whereas Tregs secrete IL-10, IL-13 and TGF-β, all of which can stimulate macrophage M2 polarization, leading to the production of pro-fibrotic cytokines such as TGF-β, galectin-3, and MMP-9.125,126,127,128 Galectin-3, an emerging biomarker associated with fibrosis, has been found to correlate with the development and severity of HF. It promotes fibrosis by inducing fibroblast proliferation and differentiation into myofibroblasts, as well as inducing macrophage M2 polarization.128,129 In terms of the effects of macrophages on T cells activation, IL-1β and IL-23 primarily produced by M1 macrophages synergize with toll-like receptor (TLR) signaling to promote the expansion of γδ T cell and the production of IL-17A.130 Regardless of the fact that IL-17A is not involved in the early inflammatory response, it plays a role in the later stage of remodeling, by means of enhancing the infiltration of macrophages, the secretion of pro-inflammatory cytokines and MMPs, as well as fibroblast proliferation and pro-fibrotic gene expression, which facilitates fibrosis as a consequence.130 In addition, the selective endogenous expression of thrombospondin (TSP)-1, a TGF-β activator and angiogenesis inhibitor, may serve as a “barrier” in the peri-infarct zone. TSP-1 locally inhibits the synthesis of inflammatory cytokines and chemokines by activating TGF-β, which limits the infiltration of macrophages and myofibroblasts, as well as the extension of inflammatory response to the non-infarcted area.131
Ischemia–reperfusion injury
Owing to the exposure of the myocardium to oxidative stress, which exacerbates myocardial dysfunction and causes structural damage during the reperfusion phase, reperfusion following acute ischemia sometimes fails to restore myocardial function and instead results in IRI.132 IRI can also induce MI, but it typically causes a non-transmural infarction with fewer necrotic cells and a smaller infarct area, leading to a smaller scar. Ischemia and reperfusion collectively induce cardiac remodeling, encompassing replacement fibrosis and interstitial fibrosis.133 In IRI, the precise demarcation between inflammatory and reparative phases remains elusive, probably due to the rapid maturation of the fibrous scar. Studies tend to focus on CCR2+ macrophages infiltrating in the early stage of the injury, while paying less attention to reparative macrophages in the later stage.133 Although numerous findings suggest that there are shared mediators and pathways that regulate inflammation and fibrosis akin to the non-reperfused infarction,7,38 unique mechanisms also make a vast influence on reperfused infarction (Fig. 2d).
In the early stage of IRI, phagocytosis is primarily dominated by CCR2-MHC-IIlow macrophages through MerTK.91 However, the hydrolysis of MerTK by ROS after IRI results in decreased levels of the anti-inflammatory mediators IL-10 and TGF-β, along with an increase in the pro-inflammatory mediators IL-1β and TNF-α, which eventually hinder the resolution of inflammation and cardiac repair.91 In accord with MerTK, AXL also mediates the phagocytosis of macrophages, but it is mainly expressed in MHC-IIhigh macrophages.134 AXL and TLR4 co-stimulate STAT1 signaling to direct a HIF-1α-dependent shift towards glycolytic metabolism in cardiac macrophages, thereby polarizing macrophages into inflammatory phenotypes and facilitating IL-1β secretion.134 While CCR2+ macrophages, recruited mainly through MCP-1, dominate the inflammatory and fibrotic responses in the early stage of IRI.135 Subsequently recruited macrophages can regulate inflammation and fibrosis through the expression of leucine-rich repeat-containing G protein-coupled receptor (LGR) 4,136 dendritic cell-associated C-type lectin-1 (Dectin-1),137 CCL17,138 and CD44.139 LGR4 orchestrates a pro-inflammatory phenotype in macrophages by enhancing activator protein-1 (AP-1) transcriptional activity via the protein kinase A (PKA) / cyclic AMP-responsive element binding protein (CREB) pathway mediated c-Fos, Fosl1, and Fosb transactivation, thereby aggravating the local myocardial inflammatory response.136 Dectin-1 is a PRR chiefly expressed on macrophages.137 On the one hand, Dectin-1 induces macrophage M1 polarization, giving rise to the release of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-23. On the other hand, it upregulates CXCL1 and granulocyte colony-stimulating factor (G-CSF) in macrophages, which mediate neutrophil infiltration.137 Early augmented inflammatory responses contribute to the aggravation of myocardial injury and ultimately culminate in the development of more severe fibrosis. Notably, the long-term effect of G-CSF may aid in preventing fibrosis. In the early stage, G-CSF accelerates the uptake of necrotic tissue by expanding neutrophil and macrophage populations, and promotes the dissolution of collagen by upregulating the expression of myocardial MMPs.140 In the later stage, G-CSF decreases the population of macrophages to inhibit the ongoing inflammatory response.140 CCL17, a chemokine selectively expressed in CCR2+ macrophages, promotes inflammation and fibrosis by inhibiting Tregs chemotaxis, thereby relieving the suppressive effect of Tregs on pro-inflammatory macrophages.138 CD44 is a widely distributed glycoprotein that mediates various cell-to-cell and cell-matrix interactions. It inhibits post-infarction inflammatory responses through interactions with hyaluronic acid, stimulates the TGF-β signaling pathway, promotes fibroblast infiltration and proliferation, and ultimately enhances collagen deposition.139 In the late stage of IRI, CCL2 stimulates the transformation of CCR2+ macrophages into a reparative phenotype and releases TGF-β to promote fibrosis.141 When it comes to pro-repair CCR2- macrophages, Li et al. unveiled their ability to produce small extracellular vesicles (sEVs). When taken up by CCR2+ macrophages, the sEVs microRNA (miR)-181b-5p regulates glucose uptake and glycolysis in macrophages while mitigating mitochondrial ROS generation, which promotes left ventricular remodeling and fibrosis by polarizing macrophages towards a reparative phenotype.142 As opposed to what are mentioned above, M2b macrophages are anti-fibrotic macrophages that inhibit fibroblast activation by regulating the MAPK signaling pathway.143
Non-ischemic injury
The occurrence and development of fibrosis are similarly observed in non-ischemic injuries. Multiple stimuli can trigger fibrosis in the absence of ischemia through fibrotic signaling pathways in macrophages, including mechanical stress and RAAS activation in PO, ROS in DCM and cardiac aging, and metabolic impairments associated with hyperglycemia in diabetic cardiomyopathy, etc. In ischemic injury, inflammation usually precedes fibrosis in a sequential manner, while in non-ischemic injury, inflammation and fibrosis commonly coexist. In non-ischemic injury, interstitial fibrosis is a chronic and progressive epiphenomenon of the sustained repression of non-circumscribed, self-perpetuating inflammation and the concomitant chronic activation of pro-fibrotic stimuli.
Pressure overload
PO is a mechanical disorder that causes cardiac hypertrophy and myocardial fibrosis, with hypertension and valvular heart disease being its primary causes. Contrary to MI where cell death releases antigens, immune responses in PO may be initiated by DAMPs and endogenous cardiac neoantigens, and compensatory mechanisms such as myocardial fibrosis and hypertrophy are adopted in response to the increased load.144,145 PO can be divided into the compensation period and the decompensated period.133 CCR2- macrophages play a dominant role in the compensation period, inhibiting fibrosis and myocardial hypertrophy.146,147,148 However, with the continuous infiltration of monocytes, monocyte-derived CCR2+ macrophages replace CCR2- macrophages to play a dominant role in the decompensation period, promoting myocardial fibrosis and hypertrophy.148,149,150 Depleting CCR2+ macrophages as early as possible in the compensation period can mitigate myocardial fibrosis, while depletion of CCR2+ macrophages or splenectomy in the decompensation period fails to halt the development of fibrosis,149 which attaches significant importance to early regulation of CCR2+ macrophages.148,149,150 Nevertheless, most of the current research on PO focuses on the mechanism by which recruited macrophages regulate myocardial fibrosis and hypertrophy, while the mechanism of tissue-resident macrophages has not been thoroughly investigated.
PO can be simulated by transverse aortic constriction (TAC) or Ang-II infusion. In both models, the onset of fibrosis and myocardial hypertrophy is mainly initiated by the neurohumoral system (mainly RAAS) and mechanical stress, while macrophage-mediated inflammation plays an important role in the subsequent progression of cardiac remodeling (Fig. 3a). Ang-II and aldosterone, which belong to RAAS, play a dominant role in PO-induced macrophage recruitment. Ang-II activates calcium/calmodulin-dependent protein kinase IIδ (CaMKIIδ) and initiates the nuclear factor-κB (NF-κB) pathway and inflammasome activation in cardiomyocytes, leading to CCR2+ macrophage recruitment.151,152,153 This process represents a potential initiating factor for PO. Ang-II also mediates macrophage recruitment through direct activation or up-regulation of chemokines to activate macrophage surface receptors TLR2,154 C-X-C motif chemokine receptor (CXCR) 2,155 CXCR4,156 Dectin-1,157 lymphocyte function-associated antigen 1 (LFA-1).158 Aldosterone promotes macrophage infiltration by activating mineralocorticoid receptor (MR), which may be mainly mediated by MR/IL-6/ cyclooxygenase-2 (COX 2) and MMP-1 and MMP-9 signaling pathways.159 Gamma-aminobutyric acid subtype A (GABAA) receptors, recognized as major neurotransmitter receptors in the central nervous system, have also been implicated to increase the number of Ly6Clow macrophages in the heart during PO and the number of circulating Ly6Chigh monocytes during late PO, thereby favoring myocardial fibrosis and hypertrophy.160 In addition to the above common recruitment pathways, myocardial fibrosis and hypertrophy may each have some distinct recruitment pathways. Under sustained PO stimulation, sympathetic activation and subsequent intrarenal cell-to-cell interactions contribute to the expression and secretion of colony-stimulating factor 2 (CSF2). Nephrogenic CSF2 stimulates Ly6Clow macrophages in the heart to produce AREG and activate the cardiac hypertrophy program.161

Regulations of myocardial fibrosis by macrophages after non-ischemic injury. a When PO occurs in the heart, Ang-II can recruit CCR2+ macrophages and cause them to secrete pro-inflammatory cytokines and chemokines such as IL-6, CCL2, and CXCL1. Mechanical stress directly or indirectly activates macrophages to exert pro-fibrotic functions. Interstitial fibrosis eventually develops in the heart. b When myocarditis occurs, viruses cause cardiomyocytes necrosis, and the necrotic cardiomyocytes produce DAMP which then recruits macrophages to the heart. Macrophages secrete mediators (such as OPN, NO, and IL-1β) that act on fibroblasts to promote the occurrence of myocardial interstitial fibrosis. c When DCM occurs, ROS activates CCR2+ macrophages to secrete pro-inflammatory and pro-fibrotic substances that act on fibroblasts, leading to interstitial fibrosis in the heart, while resident macrophages reduce myocardial fibrosis by improving cardiac metabolism or secreting the anti-fibrotic substance IGF-1. d When diabetic cardiomyopathy occurs, macrophages induce the production of pro-inflammatory substances through Dectin-1, Glectin-3, and SGLT-1, and release the fibrotic substances such as OPN and Human antigen R. Pro-inflammatory and pro-fibrotic substances jointly act on fibroblasts to promote the occurrence of interstitial fibrosis. e As cardiac aging occurs, increased ROS continues to promote monocytes to infiltrate into the heart and differentiate into CCR2+ macrophages. Macrophage-derived MMP-9 induces a series of pro-inflammatory and pro-fibrotic factors to act on fibroblasts, leading to interstitial fibrosis in the aging heart (Created with BioRender.com)
Under the stimulation of the neurohumoral system, inhibition of peroxisome proliferator activated receptor γ (PPARγ) signaling and activation of NF-κB signaling in macrophages initiate downstream inflammasome activation and expression of inflammatory genes such as IL-1β, which eventually cause fibrosis154,155,156,157 and cardiac hypertrophy.155,157,162,163 Clonal hematopoiesis mediated by Tet2 mutations also accelerates cardiac hypertrophy and HF through the NLRP3/IL-1β pathway.164,165 In the context of Ang-II, CARD9 in macrophage cytoplasm also activates the NF-κB/MAPK signaling pathway and the expression of pro-inflammatory cytokines, thereby boosting fibrosis.166 Recently, Chen et al. have also revealed that WW domain-containing protein 2 (WWP2) in macrophages interacts with the transcription factor interferon regulatory factor (IRF)-7 to drive downstream CCL5 and interferon (IFN) signaling, which facilitates Ly6Chigh monocyte infiltration and myofibroblast activation.167 However, some substances such as C terminus of Hsp70-interacting protein (CHIP), NOD-like receptor family caspase recruitment domain family domain containing 5 (NLRC5), and heat shock protein family A member 8 (HSPA8) in macrophages can inhibit macrophage recruitment and inflammatory gene expression to suppress the progression of fibrosis.168,169 Following the activation of MR on the surface of macrophages by aldosterone, macrophages secrete IL-10,170 galectin-3,171 CTGF,172 MMP-1, MMP-9,159 and other mediators that regulate fibrosis. It is worth noting that IL-10 showed opposite effects on fibrosis in different studies, which may be related to diverse sources of IL-10. Macrophage-derived IL-10 stimulates macrophage autocrine secretion of OPN and TGF-β, which activates fibroblasts.170,173 Conversely, systemically derived IL-10 may be beneficial for the inhibition of fibrosis, which is achieved by suppressing activation of bone marrow-derived fibroblasts mediated by the TGF-β-Smad-miRNA-21 pathway174 and blocking the NF-κB pathway,175 among other pathways.
Mechanical stress activates macrophages in both direct and indirect ways. Under the stimulation of mechanical stress, CCR2+ macrophages activate CD4+ T cells through antigen presentation,144,150 which produce TGF-β through integrin adhesion to CFs and induce myofibroblast transformation.144,176 The switch of CD4+ T cells to Th2 cells fosters M2 macrophage polarization, which promotes CF activation and myocardial fibrosis through TGF-β signaling.177 Mechanical stress activates fibroblasts to produce serum- and glucocorticoid-inducible kinase 1 (SGK1), chemokines, and other substances, which can induce macrophages to migrate and secrete pro-fibrotic mediators.178 Apart from indirect activation, mechanical stress can also directly induce M2 macrophage polarization by modifying macrophage morphology and actin cytoskeleton contractility,179 which may be associated with pro-fibrotic effects.177 Macrophages can also release secreted protein acidic and rich in cysteine (SPARC) to facilitate the processing of procollagen into insoluble fibrillar collagen, contributing to the development of fibrosis.180 In addition, macrophage migration inhibitory factor (MIF) antagonizes stress-induced cardiac hypertrophy and fibrosis responses by activating autophagy181 as well as maintaining a redox homeostasis phenotype.182 It is notable that the cardioprotective effect of CCR2- macrophages is activated by mechanical stress in DCM.183
Inflammatory factors play a crucial role in adverse cardiac remodeling in PO. Among them, IL-6 related to fibrosis, IFN-γ and GATA3 related to myocardial hypertrophy have received more attention. Multiple studies based on PO models such as Ang-II infusion, aldosterone infusion, and TAC have found that IL-6 can recruit macrophages and directly activate fibroblasts, underscoring its pivotal role regulating fibrosis.159,169,184,185 Aldosterone and mechanical stress stimulate IL-6 synthesis by ECs, facilitating the recruitment of CCR2+ macrophages.159,185,186,187 In fibrosis regulation, CFs synthesize IL-6 in a macrophage-dependent manner,184 which induces the activation of TGF-β/Smad in CFs through IL-6 trans-signaling,185 thereby promoting the proliferation and differentiation of CFs.153,154,184,188 Among them, Smad3 signaling can also contribute to fibrosis by downregulating miR-25 and miR-29a.189 However, oncostatin M (OSM), a member of the IL-6 superfamily, plays a role in inhibiting fibrosis by directly preventing TGF-β-mediated CF from activation under hypoxic conditions.190 In terms of cardiac hypertrophy, IFN-γ is a common pro-inflammatory but anti-hypertrophic cytokine expressed in CD68+ macrophages, with the IFN-γ/Stat5 axis potentially mitigating PO-induced cardiac hypertrophy by activating the phosphatidylinositol 3-kinase (PI3K)/Akt pathway.191 Macrophage-derived GATA3 appears to facilitate PO-induced cardiac hypertrophy, possibly by regulating Th2 cell polarization and increasing the number of Ly6Clow macrophages.192
Myocarditis
Myocarditis is a pathological condition distinguished by the infiltration of inflammatory cells into the myocardium and the occurrence of non-ischemic necrosis in cardiomyocytes.193 Viruses are the primary inducing factors, among which coxsackievirus B3 (CVB3) is the most prevalent.194 Upon infection with CVB3, cardiomyocytes exhibit aberrant antigens that are subsequently identified by T cells and B cells, ultimately resulting in the necrosis of cardiomyocytes.195 Released by impaired cells, DAMPs are able to bind to PRRs on monocytes, stimulating the secretion of chemokines, such as CCL2 and MIF-α,196 which in turn initiate the recruitment of monocytes and the activation of macrophages, thus accelerating early inflammatory responses and later myocardial remodeling.195,197,198,199 (Fig. 3b) Ly6Chigh M1 macrophages are dominant in the early inflammatory response and contribute to the removal of viruses and necrotic cells. In contrast, Ly6Clow M2 macrophages predominate during later myocardial remodeling, attenuating the inflammatory response and promoting myocardial fibrosis.200,201 After viral invasion into the myocardium, cardiac infiltrating macrophages release significant amounts of cytokines and MMPs in response to the infection, primarily including IL-1β, IL-6, TNF-α, and MMP-9.202,203 In this process, by activating macrophage NLRP3 inflammasome, CVB3 induces the production of IL-1β, thereby facilitating myocardial injury.204 Meanwhile, CVB3 upregulates miR-223205 and miR-19b-3p206 in cardiac infiltrating macrophages, which activate the NF-κB pathway and trigger the release of the inflammatory factor TNF-α, leading to myocardial injury. Regarding MMP-9 secreted by macrophages, not only does it contribute to ECM hydrolysis and the blockade of viral transmission, but it also lowers the chemotactic activity and diminishes the invasion of inflammatory cells by influencing the expression of IFN-β, IFN-γ, IL-6, and MIP-1, subsequently decreasing the inflammatory response and fibrosis in viral-induced myocarditis.207 In terms of promoting later fibrosis, the virus induces macrophages to secrete IL-1, which may bring about an elevation in circulating levels of IL-6, thereby facilitating myocardial fibrosis.208 Concurrently, macrophages are also prompted by virus to express iNOS and synthesize excess nitric oxide (NO), amplifying the activation of p44/42 MAPK in CFs and augmenting the expression of CTGF, whose functions include stimulating the proliferation of CFs and enhancing collagen secretion.209 Furthermore, the initiation of vitamin D signaling in macrophages results in the upregulation of pERK and the secretion of OPN, which then acts on fibroblasts to enhance the expression of type I collagen through the OPN-ERK-Elk1 pathway and the PI3K cascade, ultimately resulting in fibrosis.210
Dilated cardiomyopathy
DCM is a primary cardiomyopathy characterized by left ventricular or biventricular dilation, accompanied by decreased ventricular systolic function.211 The possible causes of DCM include heredity, poisoning, infection, endocrine, metabolic disorders, and other factors. These factors can induce DNA damage and ROS production, resulting in mitochondrial dysfunction, cellular vacuolation, myocardial apoptosis, and interstitial fibrosis.211 Infusing doxorubicin (DOX) and truncating titin variants are the most commonly used models for constructing DCM. Under inflammation induced by damage factors such as DOX, pro-inflammatory macrophages derived from peripheral blood monocytes are the main subset of macrophages in DCM.212 ROS is a critical factor in causing damage in DCM (Fig. 3c), and its production partly depends on the activation of TLR4 pathway.213 Furthermore, TLR4 has been reported to be associated with fibrosis.214 Shimazu et al. discovered that myeloid differentiation factor 2 (MD-2), synthesized by monocytes, was essential for TLR4 activation in DCM.215 MD-2 directly acts on monocytes and ECs through TLR4/NF-κB pathway to stimulate the synthesis of chemokines and pro-inflammatory cytokines, which could facilitate monocyte recruitment and macrophage activation.216,217 Moreover, the NLRP3 inflammasome, synthesized by recruited macrophages in DCM, facilitates the cleavage of apoptosis-associated speck-like protein containing a CARD (ASC), caspase-1, IL-1β, IL-18, and gasdermin-D (GSDMD) into active states, which promote inflammation, cardiomyocyte pyroptosis and myocardial fibrosis.218,219 In genetic DCM, recruited macrophages are also the main source of OPN.220 Infiltrating macrophages may promote the secretion of galactin-3 via OPN, which will facilitate fibrosis.220
Despite not being the dominant subpopulation in DCM, resident macrophages are beneficial to mitigate fibrosis and adverse cardiac remodeling.212,221 As for the proliferation of resident macrophages, cardiomyocytes activate resident macrophages by transient receptor potential vanilloid 4-dependent pathways.183 Additionally, DOX can induce the production of lipid peroxidation products, which produce class A1 scavenger receptor (SR-A1) ligands. These ligands act on SR-A1 on the surface of macrophages and activate the downstream c-Myc signaling pathway to promote resident macrophage proliferation.212 In terms of regulating fibrosis, resident macrophages are capable of actively ingesting dysfunctional mitochondria and other cellular debris released from cardiomyocytes through the phagocytic receptor Mertk, thereby improving myocardial metabolism and inhibiting fibrosis.222 In addition, CTGF, which serves as a downstream mediator of the TGF-β pathway as well as boosts the proliferation of fibroblasts and the production of ECM, is upregulated in DCM.223 The secretion of insulin-like growth factor 1 (IGF-1) by resident macrophages can effectively suppress fibrosis and enhance cardiac function by inhibiting the production of CTGF.146,223 Under the M1/M2 paradigm, M2-like macrophages can transfer mitochondria to injured cardiomyocytes via exosome or extracellular vesicle dependent pathways, thereby inhibiting oxidative stress. This transfer of mitochondria may explain why the adoptive transfer of M2 macrophages can alleviate myocardial fibrosis.32,33
Diabetic cardiomyopathy
Diabetic cardiomyopathy is defined as myocardial structural and functional abnormalities in diabetics, with metabolic disorders and myocardial fibrosis being prominent features.224 In general, pathophysiological processes related to diabetic cardiomyopathy, such as glucose abnormality, deposition of advanced glycation end products (AGEs), release of adipokines, activation of RAAS, microvascular dysfunction, and oxidative stress, collectively contribute to the infiltration of macrophages into the cardiac interstitial space. Efferocytosis of macrophages, as well as the secreted bioactive mediators TNF-α and resistin, play crucial roles in the metabolic disorders of diabetic cardiomyopathy, especially the hyperglycemic state and the accumulation of harmful substances. Macrophages exposed to high glucose reduce the expression of miR-126, resulting in a corresponding increase in the expression of A distegrinin and metalloprotease 9 (ADAM9). ADAM9 can enhance high glucose-induced cleavage of MerTK, leading to shedding of soluble Mer (sMER) and loss of MerTK function,225 which brings about adverse consequences such as defective elimination of abnormal mitochondria in myocardial tissue, obstruction of clearance of apoptotic cardiomyocytes, extracellular accumulation of metabolic wastes, ultimately causing imbalance of cardiometabolic balance and ventricular dysfunction.222,225 In diabetic cardiomyopathy, macrophages secrete large amounts of TNF-α, which can significantly reduce the content of cellular glucose transporter 4 (GLUT4) and the tyrosine phosphorylation level of insulin receptor substrate 1 (IRS1), causing impairment of glucose uptake by heart tissue cells.226,227,228 Furthermore, pro-inflammatory cytokines represented by TNF significantly increase the expression of resistin (an adipokine that contributes to insulin resistance) in macrophages, which further helps maintain a high glucose state.229,230 It is worth noting that resistin can also promote the expression of inflammatory cytokines, which means that they promote the production of each other, thus forming a vicious loop.231
Macrophages further interact with fibroblasts, ultimately leading to interstitial and perivascular fibrosis232,233 (Fig. 3d). Hyperglycemia, one of the main characteristics of diabetic cardiomyopathy, triggers an inflammatory response in macrophages, contributing to the development of myocardial fibrosis.234,235 Dectin-1, a PRR primarily expressed on macrophages, plays a crucial role in mediating inflammatory responses in innate immunity and is significantly upregulated in the heart tissue of diabetic mice.236 Under the influence of high glucose, Dectin-1 favors the transformation of macrophages into an inflammatory phenotype by stimulating the activation of the spleen tyrosine kinase (Syk)/NF-κB pathway.236 High glucose levels stimulate macrophage expression of galectin-3, leading to increased NF-κB p65 activation. This activation, in turn, induces macrophage infiltration into the heart and promotes M1 macrophage polarization.237 Meanwhile, abnormal glycemic variability (changes in blood glucose over time) in diabetics promotes M1 macrophage polarization through sodium-glucose cotransporter 1 (SGLT1).238 These inflammatory macrophages secrete inflammatory cytokines, including TNF-α, IL-1β, IL-6, etc., which act on CFs and promote the occurrence of myocardial fibrosis. Notably, substance P (SP) can promote the transformation of macrophages into M2 phenotype, playing an important role in regulating ECM remodeling. However, SP is significantly decreased in diabetic hearts, resulting in a greatly elevated proportion of M1 macrophages under high glucose conditions.239 In addition to inducing M1 macrophage polarization, hyperglycemia can also facilitate the secretion of pro-fibrotic factors by macrophages, which directly target CFs. Macrophage-derived exosome-associated HuR, an RNA-binding protein, is secreted more under the induction of hyperglycemia and can directly act on fibroblasts to upregulate the expression of fibrosis-related genes.240 The development of diabetic cardiomyopathy is often accompanied by the activation of RAAS,241 which promotes macrophage to infiltrate into the myocardium and secrete OPN. As an important pro-fibrotic substance, OPN promotes CF attachment to the ECM, and CF growth and ECM production.242 Adiponectin (APN) is an adipokine with anti-inflammatory function that can inhibit the Ang-II-induced inflammatory response by activating macrophage autophagy, thereby reducing the degree of myocardial fibrosis.243 However, its levels are significantly reduced in diabetic hearts, increasing myocardial fibrosis.243,244
Cardiac aging
Cardiac aging is characterized by pathological changes in the heart, including hypertrophy, systolic and diastolic dysfunction, lipid deposition, and fibrosis, which culminates in HF. These changes are influenced by factors that occur with age, such as telomere shortening, oxidative stress, metabolic dysfunction, and epigenetic changes245,246 (Fig. 3e). As an individual ages, cardiac resident macrophages are gradually replaced by monocyte-derived CCR2+ macrophages.247,248 This transition is primarily attributed to the accumulation of ROS resulting from dysfunctional mitochondria caused by impaired autophagy function in the aging heart.249 ROS activates the Ras-Erk pathway in fibroblasts to promote the high expression of MCP-1, which in turn prompts monocytes infiltration and polarization into M2a macrophages.250,251,252 Additionally, ROS induces vascular ECs to express high levels of the adhesion molecule vascular cell adhesion molecule 1 (VCAM-1), which further facilitates monocyte infiltration into the heart.253 The accumulation of macrophages in the aging heart gives rise to a significant secretion of MMP-9, which plays a crucial role in the progression of aging-related interstitial fibrosis. MMP-9 can directly cleave and activate latent TGF-β in the ECM, leading to the expression of pro-fibrotic periostin (POSTN) and CTGF.254 Excessive MMP-9 levels also diminish the expression of angiogenesis-related genes, such as integrin β3 and platelet/endothelial cell adhesion molecule 1, resulting in insufficient angiogenesis and an imbalanced oxygen supply to cardiac tissue.255 This imbalance sets off inflammatory responses that are critical to subsequent fibrosis.256,257 Furthermore, MMP-9 plays a role in regulating macrophage subtypes by promoting their conversion to an inflammatory M1 phenotype.258 It is worth mentioning that SPARC produced by fibroblasts, which increases alongside MMP-9, also fosters M1 macrophage polarization.259 These factors contribute to the chronic inflammatory state of the aging heart, leading to the release of fibrotic cytokines and growth factors and ultimately triggering the accumulation of collagen in the ECM.260
Heterogeneity and regulatory mechanisms of vascular macrophages
Atherosclerosis
AS is a chronic inflammatory response driven by lipids, and the pathological basis is the accumulation of OxLDL in the arterial intima261 (Fig. 4). As a key mediator of inflammatory response, macrophages are involved in all stages of AS development, including plaque germination, calcification, rupture, and regression.8

Regulatory mechanisms of macrophages in AS. a Foam cells not only bind and uptake circulating lipids to promote plaque progression, but also facilitate cholesterol efflux to prevent plaque progression. M1 macrophages promote foam cell formation mainly by releasing pro-inflammatory cytokines (e.g., TNF, IL-6, IL-1). M2 macrophages secrete anti-inflammatory cytokines (e.g., IL-10 and TGF-β), which inhibit pro-inflammatory cytokines release and foam cell formation, plaque formation, and promote cholesterol efflux. However, M2 macrophages can also promote plaque formation by releasing GDF15 and VEGF-A. Besides, M(Hb) and Mhem macrophages can also mediate cholesterol efflux. Apoptotic cells, which promote plaque formation, release a “Find-me” signal to attract macrophages, and then the “Eat-me” signal on the surface of apoptotic cells combines with the “bridging molecules” signal on the surface of macrophages to initiate the phagocytosis process, while normal cells avoid being phagocytosed by macrophages through the “Don’t eat-me” signal. b Macrophage-derived MMPs thin the fibrous cap by directly degrading ECM proteins such as collagen and elastin, causing plaque rupture. M2 macrophages can inhibit the production of MMPs by M1 macrophages through the secretion of IL-10. Clinical PPARα agonists have been found to suppress the production of MMP-12 induced by IL-1β. c. During plague regression, the Wnt signaling pathway in plaque macrophages is activated to promote macrophage migration, and Sema3E and netrin-1 inhibit this process. Statins and LRP-1 deficiency promote the regression by activating the CCR7-dependent migration pathway in macrophages. However, scavenger receptors Msr1 and CD36 inhibit plaque regression by promoting macrophage proliferation. M2 macrophages are the main promoters of plaque regression. Tregs and HDL could promote M2 polarization, while miR-33 inhibits M2 polarization. (Created with BioRender.com)
Plaque progression
Plaque progression refers to pathological processes such as leukocyte infiltration, lipid accumulation, necrotic core expansion, and fibrous cap formation in AS plaques.262,263 AS plaques locally produce chemokines, such as CCL2, CCL5, CX3CL1 and CXCL12, which bind to receptors on monocytes to promote their migration from the blood into tissues. CCR2, CCR5, and CX3CR1 are important receptors on the surface of monocytes that can affect the outcome of AS plaques. After migration to the corresponding site, monocytes also need to enter the vessel wall through transepithelial migration, which is mainly achieved by the adhesion molecules on monocytes (e.g., LFA1, PSGL1) acting on ECs.264 The increased number of plaque macrophages depends on increased myelopoiesis of monocytes,265,266 induction of chemokines (like CCR2),267,268 increased expression of intra-plaque migration inhibitory molecules (like netrin-1)269 and macrophage self-proliferation.270
Macrophages affect the progression and complication of AS and the formation of rupture-prone plaques by mediating inflammation, lipid metabolism, and efferocytosis (Fig. 4a). M1 macrophages dominate in progressive plaques, mainly by releasing pro-inflammatory cytokines, including IL-1, IL-6, TNF, etc., promoting chronic inflammation of plaques and causing plaque progression and instability.8,263,271,272,273,274 The pro-inflammatory NLRP3 inflammasome/IL-1 axis has been most extensively studied, which promotes thrombosis and plaque progression through mechanisms such as myeloid cell recruitment, EC activation and angiogenesis.275,276,277,278,279 IL-6 promotes the development of AS by inducing vascular smooth muscle cells (VSMC) proliferation, activating ECs, promoting thrombosis, and promoting lipid accumulation in macrophages.280,281 TNF is associated with endothelial dysfunction and can promote ROS production, reduce NO bioavailability and increase endothelial permeability.274,282 Absence of TNF can attenuate the development of AS disease,283,284,285 but some studies have also produced conflicting results.286 Other pro-inflammatory cytokines produced by plaque macrophages are generally considered to promote plaque progression and increase the death risk in patients, such as IL-8,287,288 IL-12,289,290 and IL-18.291,292,293 M2 macrophages can secrete anti-inflammatory cytokines, including IL-10 and TGF-β, which help to terminate inflammation and inhibit the formation of necrotic core.47,48,49 IL-10 has anti-inflammatory properties and has a protective effect on AS,294,295 which may be achieved by inhibiting the release of pro-inflammatory factors,296 MMP-9 and apoptosis-inducing substances like caspase-3.297 In addition, by upregulating the transporters ATP-binding cassette transporter A1 (ABCA1) and ABCG1, IL-10 also increases cholesterol efflux and disposal of harmful lipoproteins by macrophages.297,298 Another anti-inflammatory cytokine, TGF-β, is generally believed to prevent AS and stabilize plaques by inhibiting inflammation, promoting cholesterol efflux from macrophages, and promoting collagen secretion.299,300,301 However, the growth differentiation factor (GDF) 15 of the TGF-β family seems to have a role in promoting the progression of AS.302,303 It is worth noting that not all M2 macrophages will contribute to the regression of AS. CD163+ M2 macrophages promote angiogenesis, vascular permeability, and leukocyte infiltration through the CD163/HIF1α/VEGF-A pathway, thereby promoting AS progression.304,305 OxLDL inhibits the expression of Krüppel-like factor (KLF) 2 in M2 macrophages, thus enhancing the production of pro-inflammatory cytokines such as IL-6 and MCP-1.306 This suggests that we need to distinguish M2 macrophages formed by different stimuli, or use other more sophisticated classifications, and be aware of the transformation of macrophage functional phenotypes under different stimuli and environments.
Macrophages in atherosclerotic plaques uptake apolipoprotein B-containing lipoproteins (apoB-LPs) to form lipid-dense cells called foam cells.52 After formation, foam cells activate the endoplasmic reticulum stress and apoptosis pathways and release MMPs, which are involved in the process of plaque necrotic core expansion and have pro-atherosclerotic function.307,308 Compared with non-foamy macrophages, foamy macrophages express few inflammatory genes but more lipid processing genes.308 Cholesterol metabolism in macrophages includes three stages: uptake, esterification, and efflux, of which the uptake and efflux stages have received more attention. The uptake process is the main step of the intracellular accumulation of modified LDL and the formation of fatty streaks. However, if the function of cholesterol efflux is effective, the formation of foam cells and the development of foam cells into apoptotic cells will be inhibited.297 SR is a class of receptors on the cell membrane of macrophages and other cell types, which is involved in the removal of many foreign substances and wastes through extensive ligand specificity. Macrophages can bind and uptake circulating lipids through several SRs, such as SR-A,309,310 CD36309,310,311,312 and lectin-like oxidized LDL receptor-1(LOX-1).313,314,315 Sustained activation of SR-mediated uptake processes leads to lipid accumulation and cell necrosis, which facilitates the progression of plaques to more advanced necrotic lesions. Ox-LDL binds to CD36 and triggers the TLR4/TLR6 complex, which initiates sterile inflammation.316 The combined elimination of SR-A and CD36 results in the downregulation of inflammatory genes such as Il-1α and Ccl2, and a significant reduction in macrophage apoptosis and plaque necrosis.309 LOX-1 promotes inflammatory response and AS progression by activating the NF-κB and MAPK pathways.314 After cellular uptake, the modified lipoproteins are carried to intracellular lysosomes for hydrolysis and esterification. Macrophages facilitate cholesterol and phospholipid efflux through multiple transporters, such as ABCA1, ABCG1 and SR-B1, which prevent excessive accumulation of intracellular cholesterol and formation of foam cells.317,318,319,320,321 The effects of ABCG1322 and SR-B1323 on AS may depend on the stage of AS development, related to the functional diversity of these molecules. Deficiency of ABCG1 leads to the accumulation of cholesterol in the early stage of AS, resulting in an enlarged plaque lesion area.322 However, in the late stages of AS, cholesterol accumulation caused by ABCG1 deficiency leads to increased macrophage apoptosis, which reduces the susceptibility to AS and delays the progression of lesions.322 The dual role of SR-B1 in cholesterol homeostasis may be due to the fact that SR-B1 mediates both the uptake of cholesterol-rich lipoproteins and the efflux of cholesterol to high-density lipoprotein (HDL).323 Except for foam cells, M(Hb) macrophages and Mhem macrophages are also involved in lipid metabolism. Compared with foam cells, M(Hb) and Mhem macrophages express high levels of liver X receptor (LXR)-α involved in cholesterol efflux and low levels of SR involved in lipid uptake, thereby promoting cholesterol efflux and preventing foam cell formation.60,324 Besides, since iron levels in macrophages may drive cholesterol efflux, manipulating iron levels and iron metabolism-related substances like hepcidin in macrophages can inhibit the generation of foam cells and the development of AS.325,326
Efferocytosis is the process by which macrophages eliminate apoptotic cells, thereby limiting secondary necrosis caused by apoptotic cells327,328,329 and terminating the inflammatory response,271,330 which is conducive to preventing the progression of AS. Apoptotic cells release a “Find-me” signal to attract macrophages, and then the “Eat-me” signal (such as phosphatidylserine and intercellular adhesion molecule 3 (ICAM-3)) on the surface of apoptotic cells combines with the “bridging molecules” signal (like mammary-derived growth factor 8 (MFGE8)) on the surface of macrophages to initiate the phagocytosis process. Living cells avoid being phagocytosed by macrophages through the “Don’t eat-me” signal, such as CD47 and CD31. SR-B1 on the surface of macrophages mediates efferocytosis and reduces atherosclerotic lesion necrosis through intracellular Src/PI3K/Rac1 signaling.331 M2 macrophages in plaques show higher phagocytosis than M1 macrophages, which is due to the involvement of highly expressed opsonins and receptors involved in phagocytosis, such as PPARγ332 and Mertk.333,334 In the early stage of AS, macrophages exhibit a capacity to respond to apoptosis, thereby mitigating the expansion of the necrotic core within atherosclerotic plaques328 As plague progresses, macrophage efferocytosis within plaques is impaired, leading to chronic and unresolved inflammation and enhanced macrophage apoptosis in advanced plaques, ultimately promoting the formation of a necrotic core.335,336,337 Impaired efferocytosis in advanced plaques is mainly caused by lipid competition for recognition receptors,338,339 downregulation of “bridging molecule” signals, upregulation of “Don’t eat me” signals,327,340 and impairment to mitochondrial fission.341 CD47 binds to inhibitory signal regulatory protein α (SIRPα) on macrophages to induce the “Don’t eat-me” signal. CD47 blocking antibodies or SIRPα deletion improve efferocytosis in plaques, attenuate oxidized LDL-induced inflammation and induce M2 macrophage polarization, thereby reducing the formation of necrotic core.342,343,344
Arterial calcification is caused by the crystallization of calcium and phosphate in the form of hydroxyapatite, which can accumulate in the ECM of the artery wall. The degree of plaque calcification is also a measure of plaque stability.345 The inability of microcalcification formed by M1 macrophages to form stable structures is associated with an increased risk of plaque rupture.346,347 However, macrocalcification formed by M2 macrophages can stabilize AS plaques.348 M1 macrophages induce osteogenic transdifferentiation of VSMCs and further mineralization of plaque lesions mainly by secreting pro-inflammatory cytokines (such as IL-1β and TNF-α).349,350,351 Anti-inflammatory cytokines (like IL-10) secreted by M2 macrophages may be beneficial to osteoblastic differentiation of VSMCs and plaque macrocalcification.348 In addition, OSM secreted by plaque macrophages induces osteoblastic differentiation of VSMCs and M2 macrophage polarization through the Janus Kinase 3 (JAK3)/STAT3 pathway, thereby promoting plaque macrocalcification and stability.352
Plaque rupture
Rupture-prone plaques contain a large necrotic core and a thin fibrous cap, and are also characterized by high MMP activity, ECM proteolysis, VSMC dedifferentiation, impaired exocytosis and chronic inflammation353 (Fig. 4b). Among them, macrophage-derived MMPs thin the fibrous cap by directly degrading ECM proteins such as collagen and elastin,53,354,355,356 so MMP-1, MMP-8, and MMP-12, which belong to collagenase, have a greater impact on plaque stability.357 Newly recruited monocytes may upregulate a broad spectrum of MMPs through a prostaglandin (PG)-dependent pathway.358 Different macrophages secrete different MMPs to participate in plaque rupture. M1 macrophages mainly release MMP-1, MMP-3, MMP-10 and other MMPs, while M2 macrophages reduce MMP-2 and increase MMP-11, MMP-12, MMP-25 and other MMPs.359 And M4 can participate in fibrous cap degradation and plaque rupture by producing MMP-7.360 Clinically, PPARα agonists are used to lower lipids for the treatment of AS. PPARα agonists have also been found to inhibit IL-1β-induced MMP-12 production, thereby preventing inflammation and plaque rupture.361
Plague regression
As LDL-cholesterol in circulating blood continues to decrease, plaque regression may occur. During the regression process, plaque composition can change significantly from that of progressive plaques, with increased fibrotic cap thickness,362 decreased macrophage content, and M2 macrophage polarization47,363,364,365 (Fig. 4c). At present, the mechanisms underlying plaque regression are relatively less studied than those driving plaque progression, and mainly rely on a cholesterol-free diet or the use of cholesterol-metabolizing drugs (e.g., statins and ezetimibe).366,367,368 The reduction in the number of plaque macrophages mainly depends on the inhibition of local proliferation369,370 and the efflux of macrophages from the site of inflammation.371 One study showed that Msr1 and CD36,involved in the uptake of modified lipoproteins, are mediators of plaque macrophage proliferation.369 Statins and low-density lipoprotein receptor–related protein 1 (LRP-1) deficiency promote the regression of AS by activating the CCR7-dependent migration pathway in macrophages.372,373,374 During regression, the Wnt signaling pathway in plaque macrophages is activated to promote macrophage migration.375 At the same time, the classical Wnt/β-catenin signaling regulates the STAT pathway in macrophages to terminate the elevated inflammatory response and prevent AS.376 Sema3E377 and netrin-1269 are upregulated in macrophages in advanced plaques, which serve as negative regulators of macrophage migration, promoting macrophage retention and chronic inflammation, and targeted inhibition of negative regulators facilitates plaque regression. Notably, inhibition of monocyte recruitment was found to be critical for plaque macrophage regression in a model of plaque regression.378 In regression plaques, macrophages exhibit downregulation of adhesion-related genes (e.g., cadherin, vinculin) and upregulation of movement-related genes (e.g., actin and myosin) and M2 phenotype-related genes (e.g., arginase I and CD163).379 Tregs are essential for macrophage efflux, M2 polarization and pro-catabolic functions in regressing plaques, including clearance of apoptotic cells and cellular debris and production of specialized pro-lipolytic mediators.380 The antagonism of miR-33, a microRNA that is elevated in macrophages in progressive lesions, promotes macrophages tilt toward the M2 state and causes plaque regression.381,382 During plaque regression, the increase in the concentration of functional HDL particles is an important contributor to plaque regression. HDL can mediate cholesterol efflux and induce M2 polarization,383,384 which is dependent on the STAT6 pathway385 and the expression of activating transcription factor 3 (ATF3).386
Aneurysm
Aneurysms generally occur in the aorta, and the main pathological characteristics of aorta aneurysms (AA) are smooth muscle cell (SMC) apoptosis, inflammatory response and matrix degradation.387 Macrophages play an important role in all stages of AA development, and are affected by the microenvironment such as hemodynamics, changes in circumferential stress, perivascular adipose tissue (PVAT) and intraluminal thrombus (ILT) (Fig. 5). M1 macrophages are involved in the development of AA mainly by secreting inflammatory factors and MMPs, promoting ECM destruction and VSMCs apoptosis.46,388 However, M2 macrophages are involved in vascular repair mainly by inhibiting inflammation.46,388,389 Since it is crucial to inhibit further development and rupture of AA, studies have mostly focused on early M1 macrophages.

Regulatory mechanisms of macrophages in aneurysm. Macrophages mainly regulate inflammatory response, ECM remodeling and VSMC apoptosis in AA. Macrophages clear apoptotic VSMCs through phagocytosis and produce large amounts of ROS, which further activate macrophages. Macrophages can also secrete pro-inflammatory cytokines (such as IL-6, TNF, IL-1β), chemokines (such as CXCL1 and CCL2) and ANGPTL2 to promote the development of inflammation. On the contrary, macrophages also secrete anti-inflammatory factors such as IL-10 and TGF-β by upregulating the transcription factor KLF6 or downregulating the activation of PPARδ. In addition, macrophages release proteinases such as MMP-9, MMP-2, and MMP-3 by activating the STING, TERT, JNK and p38 pathways, leading to aortic wall bleeding and rupture. In turn, macrophage recruitment, accumulation, proliferation, and activation are modulated by microenvironmental conditions (such as hemodynamics, circumferential stress, PVAT and ILT). (Created with BioRender.com)
When an artery is injured, monocytes are recruited to the injury site by chemokines such as CCR2 and CX3CR1, and differentiate into macrophages.390,391,392 Inflammatory response is one of the main characteristics of AA, and macrophages play an important role in regulating inflammation. M1 macrophages promote inflammation by releasing ROS, pro-inflammatory cytokines, and chemokines. When M1 macrophages clear early cell debris through phagocytosis, they produce large amounts of ROS, which together with ROS derived from ECs, VSMC and other immune cells in the aortic wall further activate macrophages, thus continuously enhancing this cycle.393,394,395,396,397 M1 macrophages can also secrete pro-inflammatory cytokines (such as IL-6, TNF, IL-1β, etc.) to promote the development of inflammation.388,398,399,400 Recent studies have found that S-Nitrosylation of Septin2 and adenosine deaminase acting on RNA (ADAR1) in macrophages in AA can promote the activation of the NF-κB signaling pathway, which in turn activates NLRP3 inflammasome, resulting in the release of IL-1 and enhanced degradation of ECM.401,402,403 Activation of NLRP3-caspase-1 inflammasome is also associated with the degradation of contractile proteins.404 Infiltrating macrophages can also highly express angiopoietin-related protein 2 (ANGPTL2), which induces macrophages to further release pro-inflammatory factors such as TNF-α, IL-1β, and IL-6 and MMPs in an autocrine manner.405 In addition to inflammatory factors, M1 macrophages promote the recruitment of inflammatory cells by producing chemokines such as CXCL1 and CCL2, forming a positive feedback that continuously promotes chronic inflammation.406,407,408 Among them, CXCL1 recruits neutrophils which secrete IL-6, and the increase in IL-6 levels in turn promotes the differentiation of monocytes into macrophages which secrete CCL2, thus recruiting more monocytes into the aneurysmal artery wall.395,409,410 In contrast to M1 macrophages, M2 macrophages promote vascular reconstruction and repair by secreting anti-inflammatory factors such as IL-10 and TGF-β, inhibiting the production of inflammatory factors and MMPs, clearing hemoglobin, and regulating oxidative stress,46,406,411,412 which may be achieved by upregulating the transcription factor KLF6 or downregulating the activation of PPARδ.413,414 Nevertheless, it has been found that the deficiency of IL-12p40 promotes the development of abdominal aortic aneurysms by promoting the recruitment of M2 macrophages.415 Therefore, M2 macrophages are not exclusively beneficial to aneurysms.
In addition to inflammation, ECM degradation is also one of the main features of aneurysms. Macrophages release proteinases such as MMP-9, MMP-2, and MMP-3 to degrade the ECM, leading to aortic wall bleeding and rupture.395 Among them, MMP-9 may play a more important role in AA due to its highest content.416,417 Luo et al. found that SMC damage and subsequent DNA release into the cytoplasm activated the STING-TBK1-IRF3 pathway, promoting SMC apoptosis and necrosis.418 Macrophages phagocytose DNA released by damaged SMCs and activate stimulator of interferon genes (STING) and its target protein IRF3, which enters the nucleus and binds to the MMP-9 promoter to induce MMP-9 expression.418 MMP-2 is also the primary MMPs during the early stages of AA formation, leading to the initial breakdown of elastic tissue.419,420 Telomerase reverse transcriptase (TERT) in bone marrow-derived macrophages promotes MMP-2 expression.421 Besides, there are a large number of exosomes in the adventitia of aneurysmal arteries, mainly from macrophages, which can induce the expression of MMP-2 in VSMCs by activating the JNK and p38 pathways.422
The microenvironment of AA, including hemodynamics, changes in circumferential stress, PVAT and ILT, can also influence macrophage action. Increased aortic blood flow and wall shear stress can promote macrophage apoptosis, induce the expression of antioxidant genes such as HO-1 in macrophages, and reduce ROS production.423,424 However, lower shear stress can induce inflammatory responses by promoting macrophage infiltration.425 PVAT induces endothelial dysfunction and macrophage infiltration by secreting RAS components, adipocytokines, cytokines, and chemokines like CCL2, promoting inflammation and aortic dilation.426,427,428,429 Adverse remodeling following ECM degradation can lead to the formation of ILT.395 ILT can form an inflammatory microenvironment containing cytokines, proteinases and ROS. At the same time, its growth competes for oxygen and nutrients, causing local hypoxia in the aortic wall, which is related to the activation of inflammatory macrophages, increased inflammation levels, degradation of elastin in the arterial wall and the decrease of SMCs, thereby damaging the arterial wall.388,426,430
New perspectives brought by single-cell technologies
Single-cell technologies, particularly scRNA-seq, allow resolution of gene expression at the single-cell level to reveal cellular heterogeneity. Compared with lower resolution sequencing technologies such as RNA-seq and bulk RNA-seq, scRNA-seq can perform accurate and unbiased cell clustering, discover rare cell subsets, and provide transcriptome profiles of cell subsets rather than just several markers through a series of algorithms.431,432 In addition, scRNA-seq can also perform multi-dimensional data mining, including revealing the differentiation trajectory of key cell populations, the interaction between cell populations in specific physiological and pathological states, and the identification of key transcription factors.431 Thus, scRNA-seq can provide new insights into the complex biological process of macrophage-mediated CVD pathogenesis, which includes uncovering into the diversity of macrophages as well as searching for new mechanisms and potential therapeutic targets.431Moreover, spatial transcriptomics (ST) can supplement the positional information at the spatial level lost by scRNA-seq, revealing the spatial distribution of macrophages for exploring the real cell interaction mechanism.433,434
Cardiac macrophages
Previously, recruitment macrophages and resident macrophages could be roughly distinguished by CCR2 expression. However, recent scRNA-seq studies have shown that monocyte-derived macrophages can acquire a variety of cell fates, and some of these subsets have low CCR2 expression, suggesting that the use of CCR2 expression to distinguish the origin of macrophages is not precise enough.13,67,435,436,437 Meanwhile, scRNA-seq reveals that the recruited macrophages have low expression of reparative genes such as TIMD4, LYVE1 and folate receptor 2 (FOLR2), and combined with this feature, macrophages of different origins could be better distinguished.67 The use of scRNA-seq defines the most dominant resident macrophage subset, namely TLF+ (expressing TIMD4 and/or LYVE1 and/or FOLR2) macrophages18,67,147,435,436 (Fig. 1b). The renewal of TLF+ macrophages is independent of circulating monocytes,18 and their transcriptome signatures are mainly functions of maintaining homeostasis, such as endocytosis, cell transport, and angiogenesis.18,67 TLF+ macrophages have also been found to inhibit fibrosis in MI67 and PO.19,147 In the context of scRNA-seq applied to AMI, different subsets of recruited macrophages were further divided, and it was found that interferon-stimulated gene (ISG+) macrophages and MHC-II+ macrophages are important pro-inflammatory subsets in the inflammatory phase, while triggering receptor expressed on myeloid cells 2 (Trem2+) macrophages are the major anti-inflammatory subset in the reparative phase (Fig. 1b). ISG+ macrophages activate the IRF3-IFN axis by uptake of DNA from infarcted myocardium, which facilitates the production of pro-inflammatory cytokines and chemokines, ultimately worsening cardiac function.67,435,438,439 MHC-II+ macrophages, another major pro-inflammatory subset, enrich transcripts associated with a pro-inflammatory and pathogenic profile, such as Il1b, Nlrp3, and Tlr2.67,435,439,440,441 Previously, these two pro-inflammatory subsets may have been broadly described as M1 macrophages. Trem2+ macrophages predominate in late-infarcted hearts and highly express tissue repair, exocytosis, and anti-inflammatory genes.434,435,437,440,442 Injection of soluble Trem2 in mice can inhibit fibrosis and improves infarcted heart function,434 and cardioprotective effects of Trem2 have also been found in PO442 and sepsis-induced cardiomyopathy.440
In terms of exploring new potential mechanisms, recent scRNA-seq studies have found that miR-21, ALKBH5, SPP1, Runx1 and NLRP3 can become new targets for the regulation of macrophage inflammation. MiR-21, an important microRNA driving fibrosis, was found to be essential for the elevation of M1 subsets in PO. Ligand-receptor interaction analysis based on scRNA-seq and in vitro model validation confirmed that M1 macrophages secrete miR-21 in a paracrine manner, which activated the differentiation of CFs into myofibroblasts.441 Also in PO, scRNA-seq and lineage tracing revealed that cardiac macrophages derived from circulating monocytes preferentially undergo macrophage-to-myofibroblast transition through the ALKBH5/IL-11/IL-11RA1 axis, resulting in hypertensive myocardial fibrosis and dysfunction in mice.30 In the Hulsmans et al. atrial fibrillation mouse model that integrates hypertension, obesity, and mitral valve regurgitation, scRNA-seq suggested that CCR2+Trem2+ macrophages promoted the progression of fibrosis and atrial fibrillation by secreting SPP1, which was verified in the bone marrow transplantation model.443 Ligand-receptor interaction analysis also showed that SPP1 may act on integrins, CD44, and the prostaglandin E2 (PGE2) receptor on fibroblasts to activate the TGF-β pathway.443 CCL3+ pro-inflammatory macrophages and TNMD+ fibroblasts are enriched in the right ventricle of patients with arrhythmogenic right ventricular cardiomyopathy (ARVC). CCL3+ pro-inflammatory macrophages strongly interact with fibroblasts via NLRP3, and pharmacological inhibition of CCL3+ pro-inflammatory macrophages significantly alleviated RV dilatation and dysfunction in a mouse model of ARVC.444 In addition, a single-nucleus RNA sequencing study predicted that downregulation of runt-related transcription factor 1 (RUNX1) transcriptional activity in cardiac macrophages and fibroblasts may promote cardiac recovery in patients with HF by gene regulatory network construction. This possibility was confirmed in subsequent animal experiments.445 Utilizing ST, it was found that macrophages were dispersed across the whole heart on day 1 after MI and began to penetrate deep into the infarct area from day 3, and their numbers peaked on days 5 and 7.434 Other immune cells, such as B cell and T cell, were always dispersed across the entire mouse heart (not clustered in the infarcted area), which confirmed the importance of macrophages for scar formation.434 One study using ST in MI patients observed crosstalk between SPP1+ macrophages and neighboring fibroblasts in the infract zone. Moreover, ligand-receptor interaction analysis found that SPP1+ macrophages may act on fibroblasts through PDGF-C, PDGF-D, and thrombospondin-1 (THBS1) signaling to affect the progression of fibrosis.26 Another study using ST identified monocyte-derived basic helix-loop-helix family member e41 (Bhlhe41+) macrophages in the developing infarct zone of MI. By analyzing spatial ligand-receptor interaction and in combination with animal model experiment, the results suggested that Bhlhe41+ macrophages could increase the secretion of granulin (GRN) to antagonize the effect of TNF-α on TNFR1, thereby inhibiting myofibroblast activation and limiting the expansion of the infarct zone.446 This mechanism was verified by co-culture systems and depletion of Bhlhe41+ macrophages in mice. Meanwhile, Bhlhe41+ macrophages were found to help limit the expansion of developing infarct area and improve cardiac function.446 The combination of ST with single-cell data allows us to have further insight into fibro-myeloid spatial relations in different histomorphological regions (infarcted, border, and remote zones). These results also support the application of ST to explore the spatial distribution patterns and roles of macrophages and other cells in heart diseases.
Vascular macrophages
scRNA-seq analysis of the diversity of macrophages in mouse and human blood vessels identifies resident macrophages involved in the maintenance of homeostasis, inflammatory macrophages, anti-inflammatory macrophages, and proliferative macrophages in vascular diseases308,407,447,448,449,450,451,452,453,454 (Fig. 1b). Resident macrophages are mainly found in the adventitia of healthy and diseased blood vessels.455 Resident macrophages are capable of proliferating and resemble an M2-like phenotype, and their transcriptome is characterized by the expression of Lyve-1, FOLR2, F13a1, and Wfdc17, which are involved in signaling pathways related to phagocytosis, intercellular adhesion, chemotaxis, and vascular calcification.447,449,453,454,456,457,458,459 Inflammatory macrophages in AS and AA are mainly present in the intima and adventitia of the vascular walls, respectively.455 Inflammatory macrophages are enriched in M1-related genes, expressing genes encoding pro-inflammatory mediators (including IL-1β, Nlrp3, Tlr2), chemokines (e.g., Cxcl2, Ccl3, Ccl4), and transcription factors (e.g., Cebpb, Egr1).407,447,448,453,454,457,459 A special group of IFN-induced macrophages related to inflammation is also found in AS, mainly present in the intima, expressing genes such as Isg15, Irf7, and Ifit1, which promote macrophage recruitment and foam cell formation.460,461,462 Anti-inflammatory macrophages mainly highly express genes related to anti-inflammation, phagocytosis, and proteinase, such as Pf4, Mrc1, Arg1, and Ctsa, promoting anti-inflammation and vascular remodeling.407,454 Trem2+ macrophages are important anti-inflammatory macrophages that infiltrate diseased blood vessels but are not present in healthy mice. They are lipid-rich and resemble an M2-like phenotype, characterized by the expression of Lgals3, Cd9, Ctsd, and Spp1, and enriched with signaling pathways related to cholesterol metabolism, oxidative phosphorylation, the lysosome, and the proteasome.447,448,461 While Trem2+ macrophages can regulate LDL levels by removing apoptotic cells and lipids to prevent lipid metabolism disorders and also play an anti-atherosclerotic role by inhibiting inflammation and advanced calcification, they also express some molecules that exacerbate plaque rupture (e.g, Lgals3 and Ctsb).407,459,463 Proliferating macrophages represent macrophages that are expanding or renewing, whose transcriptome is characterized by the expression of Mki67, Stmn1, Top2a, and Tuba1b and the enrichment of signaling pathways for cell proliferation.407,447,454,459,463
With regard to the comprehension of disease mechanisms, analysis of intercellular interaction based on scRNA-seq shows that macrophages primarily interact with ECs, T cells, and VSMCs.452 ECs and macrophages mainly exert adhesion through ICAM1-VCAM1/ITGB2 and ACKR1-CCL8/CXCL1, perform transendothelial migration through SELL–CD44, and participate in angiogenesis through PDGFBR-PDGFB. These functions may be related to the initiation of AS.448,457 T cells and macrophages activate each other through VCAN-TLR1/2, CCL5-CCR1/5, and ITGAL-ICAM1 to induce cytotoxicity and antigen presentation and regulate lipid accumulation and foam cell formation by regulating LRP1 ligands on macrophages.448,451 For VSMC, CCL5, which is highly expressed by macrophages, interacts with CCR5 on VSMC to drive VSMC proliferation and conversion to the synthetic phenotype, thereby causing vascular remodeling and plaque progression.451,464 In addition, scRNA-seq is used to explore the downstream mechanisms of intervention targets for vascular diseases, including netrin-1, miR-33, and CD47/SIRPα, among which netrin-1 is a common target of AS and AA. In AS, silencing of myeloid netrin-1 in mice resulted in downregulation of genes involved in pro-inflammatory responses (S100a8/9) and upregulation of genes involved in lipid metabolism, anti-inflammatory (Il10, Tgfb), and cell migration (Ccr7) in macrophages, thereby promoting resolution of inflammation and reducing plaque burden in the aorta.465 In AA, netrin-1 activates the release of MMP-3 in VSMCs, leading to matrix degradation, which promotes the formation of AA. Therefore, a deficiency of netrin-1 can prevent AS and AA.466 Anti-miR-33 reduces the proliferation and retention of MHC-IIhigh inflammatory and Trem2+ macrophages, decreases the accumulation of vascular lipid, promotes macrophage apoptosis and cytotoxicity clearance, and increases collagen content, thus playing a role in tissue repair and the resolution of inflammation.467,468 Interruption of CD47/SIRPα signaling by precision-engineered nanoparticles causes macrophages to downregulate the expression of pro-inflammatory transcription factors (CCL2, CCL7, CCL8, and PF4), upregulate the expression of genes related to inflammation resolution (SOCS3 and Zfp36), and also enrich genes related to phagocytosis and antigen presentation, resulting in the reduction of plaque burden.469 Up to now, few studies have been published on ST in vascular diseases.470,471 A spatial transcriptional map study found that macrophage-derived MMP-9 was more prominent in the narrowest areas of plaques (unstable) than in the distal areas (stable),472 which may help us better understand the characteristics of ruptured plaques.
Current status of preclinical macrophage targeting strategies
Inhibition of macrophage recruitment
In cases of inflammation or injury in cardiovascular tissue, recruited macrophages act as the primary inflammatory cells that mediate the balanced regulation of inflammatory immunity and play a central role in the interaction between various cells. Therefore, inhibiting the recruitment of macrophages is a promising therapeutic strategy for CVD.3 The most studied CVD is AS. In AS, monocytes aggregate into plaques through chemokine-mediated recruitment,473 adhesion molecule-mediated adhesion, and junction adhesion molecule-mediated exudation.474 Inhibiting these targets not only prevents the subsequent accumulation and proliferation of macrophages in the plaque but also prevents the instability and rupture of atherosclerotic plaques.475 Recruitment of monocytes is primarily mediated by the CCL2-CCR2 axis.476 When CCL2 or CCR2 is deficient, macrophage recruitment to the blood vessel wall is reduced in mice, and consequently, atherosclerotic lesion size is also reduced.477,478,479 Conversely, if CCL2 is overexpressed, the number of macrophages and the accumulation of oxidized lipids in mice atherosclerotic plaques are significantly increased, thereby promoting the progression of AS.480 One study shows that the combined deletion of CCL2, CX3CR1, and CCR5 significantly reduces macrophage invasion and plaque lesion size compared to deletion alone.481 Similarly, the combined loss of CCR2 and CX3CL1 significantly reduces the accumulation of macrophages in the lesions and decreases the instability of atherosclerotic plaques.482 These results suggest that targeting multiple chemokines or receptors simultaneously is a potential therapeutic strategy.476 For monocyte adhesion, this process is mainly mediated by the binding of VCAM-1 on vascular ECs and very late antigen 4 (VLA-4) integrin on circulating monocytes.483 Direct inhibition of VCAM-1 has been shown to prevent monocytes from infiltrating into the subcutaneous space, thereby effectively preventing macrophage maturation and foam cell transformation required for the formation of atherosclerotic lesions.484 However, highly specific peptide and antibody therapeutics that selectively inhibit VCAM-1/VLA-4 interactions have recently emerged as a promising adherence-based anti-AS therapy.485 During the exudation process, inhibiting the junctional adhesion molecule A (JAM-A) can effectively reduce inflammation and monocyte recruitment to atherosclerotic endothelium, thus decreasing the formation of the AS.486 In addition, as an inflammatory cytokine with chemokine-like characteristics, MIF also plays a critical role in the overall macrophage recruitment process.487,488 Treatment with MIF antibody in atherosclerotic mice significantly reduces the content of macrophages in the lesion as well as the levels of circulating and local aortic inflammatory mediators, thereby inhibiting the area of plaque development.489 In MI, many recent preclinical studies have also focused on targeting the CCL2-CCR2 axis.473 Studies have shown that reducing CCR2 expression through CCR2 inhibitors can significantly inhibit monocyte recruitment in the heart, thereby easing the inflammatory cascade and reducing MI size.490
Inhibition of foam cell formation and macrophage survival
Foam cells are prototype cells in atherosclerotic plaques, formed by the excessive accumulation of cholesterol esters by macrophages.491 Therefore, inhibiting foam cell formation by targeting critical proteins involved in macrophages cholesterol uptake,492 esterification,493 and efflux494 is one of the important strategies for treating AS. Studies have shown that by silencing SR-A alone, foam cell formation can be significantly reduced, thereby decreasing the occurrence of AS.310 However, the role of acetyl coenzyme A acetyltransferase 1 (ACAT-1) in cholesterol esterification in AS is still controversial. Pharmacological inhibition of ACAT-1 has been found to lead to increased foam cell formation in atherosclerotic mouse and rabbit models, which, in turn, facilitates plaque formation.495 The overexpression of ACAT-1 also facilitates the accumulation of cholesterol ester and the formation of macrophage-derived foam cells, which increase the occurrence of AS.496 Finally, in cholesterol efflux, a related study has found that treatment with PPARα and PPARγ agonists in LDL-receptor deficient mice induces LXRα and LXR-mediated ABCA1 expression, which promotes cholesterol efflux and reduces foam cell formation, thereby inhibiting the development of AS.497
The role of macrophage death in CVD is complex. It may either promote tissue repair and remodeling or exacerbate tissue damage and inflammation, depending on the mode and extent of its death.498,499 The death modes of macrophages are predominantly apoptosis, regulated necrosis (including necroptosis, pyroptosis, and ferroptosis), and autophagy.490,499,500,501,502,503 Apoptosis is an orderly process of cell death that eliminates excess or damaged cells and prevents an inflammatory response.504 In contrast to apoptosis, regulated necrosis induces an inflammatory response.505 Autophagy is a non-apoptotic form of cell death that prevents inflammation.499 Current therapeutic strategies targeting these modes of cell death to regulate macrophage survival have predominantly focused on atherosclerotic disease, with less emphasis on macrophage death modes in the cardiac field. For AS, liposomes containing drugs, such as clodronate, are widely used to induce apoptosis of macrophages because they can be delivered to macrophages through phagocytosis without causing cytotoxicity to non-phagocytes.506,507 Studies have shown that the administration of clodronate liposomes (Clo-Lip) inhibits mitochondrial oxygen consumption, leading to macrophage apoptosis and preventing the progression of AS.508 However, systemic administration of clodronate-containing liposomes also reduces blood monocytes, which increases the risk of immunosuppression and infection. Notably, when recombinant tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is administered systemically to mice with diabetes-induced AS, no adverse effects are observed. It is found that TRAIL induces apoptosis of infiltrating macrophages in atherosclerotic plaques but does not induce apoptosis of circulating macrophages, significantly weakening the development of AS.509 In terms of targeting macrophage necroptosis, intervention in mice with atherosclerotic lesions using the pharmacological necroptosis inhibitor necrostatin-1 (Nec-1) has been found to prevent further progression of the lesions and reduce markers of plaque instability, known as necrotic core and necrotic cell death.510 In addition, the activation of the NLRP3 inflammasome during pyroptosis is required for the formation of AS. Therefore, targeted destruction of the NLRP3 inflammasome significantly protects atherosclerotic mice from the disease and reduces lesion size.275 In terms of targeting ferroptosis in macrophages, studies have found that the use of micheliolide (MCL)511 or IL-37512 can activate the nuclear factor erythroid 2-related factor 2 (NRF2) pathway, thereby inhibiting ferroptosis in macrophages and reducing the progression of AS. Finally, by targeting mechanistic target of rapamycin (mTOR), a critical protein activated by the autophagy pathway, such as a mTOR inhibitor513 or silencing mTOR with small interfering RNA,514,515 the activation of macrophage autophagy genes can be induced, leading to the clearance of macrophages in plaques and facilitating a stable plaque phenotype. However, in the heart, the immune microenvironment in which macrophages reside is more complex than that in blood vessels. It requires both M1 macrophages to clear dead cells and M2 macrophages to facilitate infarction repair and angiogenesis promptly. Therefore, uniformly targeted strategies for macrophage depletion are often ineffective, hindering wound healing and left ventricular remodeling after myocardial injury.516,517 However, it has been found that the absence of the apoptosis inhibitor of macrophage (AIM) selectively reduces the level of M1 macrophages in MI, which decreases the incidence of heart rupture and improves the survival rate.518 At present, there are few studies on targeting specific macrophage subsets for cell death in MI, which may be a potential therapeutic strategy to promote post-MI repair.
Regulation of macrophage function
Macrophages have many functions in the cardiovascular system, such as regulating inflammation and fibrosis, lipid metabolism, efferocytosis, etc. Regulating the function of macrophages is a feasible idea for the treatment of CVD. For the cardiac system, current research is mainly based on ischemic injury models to explore the regulation of macrophage function by cytokines and cell therapy. IL-1, IL-4 and IL-10 are widely studied cytokines that regulate macrophage function. Anti-IL-1β antibody or anakinra (an exogenous recombinant human IL-1Ra) treatment reduces the intensity of inflammation, prevents excessive accumulation of white blood cells, and inhibits cardiomyocyte apoptosis by inhibiting IL-1,277,519,520 while systemic infusion or targeted delivery of IL-4 and IL-10 during the inflammatory phase can induce macrophages to produce repair-phase bioactive mediators with anti-inflammatory, pro-angiogenesis, and collagenesis functions.114,521,522,523 Based on this, the infarct size is reduced, the pumping function of the heart is improved, and the degree of adverse fibrosis is reduced after MI.114,277,519,520,521,522,523 For cell therapy, the current focus is on the infusion of mesenchymal stem cells (MSCs) and ex vivo reprogrammed macrophages. MSCs regulate the function of macrophages from pro-inflammatory to anti-inflammatory by means of direct intercellular communication or paracrine. They have the advantages of strong immunomodulatory ability, low antigenicity, easy acquisition and easy expansion in vitro, etc., but there are problems such as low survival rate and implantation rate.524,525 Reprogramming macrophages in vitro prompts them to produce specific functions such as anti-inflammation, promoting angiogenesis, and preventing myocardial cell apoptosis, which not only allows personalized treatment for patients, but also avoids off-target effects that are prone to occur when regulating macrophage function in vivo.506 For example, after infusion of Cardiac Nestin+ MSCs, in vitro M-CSF and IL-4 combined treated macrophages or hypoxia-induced in vitro reprogrammed macrophages into MI animal models, it is observed that pathological fibrosis of the heart infarction area or distal end is reduced, microangiogenesis is enhanced, and cardiomyocyte hypertrophy is weakened.526,527,528
For the vascular system, current research is mainly based on AS models to explore the regulation of macrophage function by epigenetics and cytokines. Epigenetics plays a significant role in regulating the inflammatory response and lipid metabolism of macrophages,529 and currently focusing on microRNAs, such as miR-21, miR-155, miR-33 and miR-144-3p. In the advanced stage of AS, local delivery of miR-21 to carotid plaque or extensive inhibition of miR-155 expression can inhibit the secretion of inflammatory mediators such as TNF-α, MCP1, IL-6 and IL-1β by macrophages, and promote the expression of IL-10;530,531,532 however, anti-miR-33 and anti-miR-144-3p therapies promote ABCA1 and ABCG1 mediated cholesterol efflux in macrophages, alleviating lipid accumulation and inflammatory response.533,534 Based on this, AS plaque burden can be reduced, plaque rupture can be prevented, and the progression of AS can be delayed.531,532,533,534 It is worth noting that miR-21 can also regulate the function of macrophages in the heart, and the delivery of miR-21 significantly inhibits the macrophage-mediated inflammatory response in the infarcted myocardium, effectively reducing the infarct size and myocardial fibrosis.535,536 For cytokines, cytokines such as IL-1, IL-19, and IL-13 play an important role in AS lesions. The administration of IL-19 and IL-13 can activate pathways such as STAT3, STAT6, and KLF4 to promote macrophages to perform anti-inflammatory, lipid efflux, efferocytosis and other functions, and anti-IL-1β antibody can inhibit IL-1-mediated chronic inflammation and lipid metabolism disorders, thereby improving the stability of atherosclerotic plaques.363,537,538,539
In addition, many antihyperglycemic and lipid-lowering drugs that have been clinically applied have also been found to regulate macrophage function, such as Dapagliflozin, Pioglitazone, Sitagliptin and Rosuvastatin. Dapagliflozin is a highly potent and selective sodium-glucose co-transporter 2 (SGLT2) inhibitor that has been shown to reduce fibrosis and AS formation. In terms of regulating fibrosis, Dapagliflozin effectively alleviates myocardial fibrosis after MI by inhibiting macrophage inflammatory pathways (especially NF-κB) and promoting repair function mediated by the RONS/STAT3 pathway.540,541 Besides, it can also promote the transformation of M1 macrophages into M2 phenotype by inhibiting LPS-induced TLR-4 overexpression and NF-κB activation in macrophages, reducing the rate of atherosclerotic plaque formation and increasing plaque stability.542,543 Pioglitazone is a PPARγ agonist, and intravenous administration after MI reduces infarct and border zone fibrosis by skewing macrophages toward a pro-healing M2 phenotype through inhibition of NF-κB.544 Sitagliptin promotes the deflection of macrophages toward the M2 phenotype through SDF-1/CXCR1 signaling, and Rosuvastatin promotes cholesterol efflux and the secretion of anti-inflammatory mediators by increasing the expression of ABCA1, ABCG1, Arg-1 and CD206 in macrophages, so they can reduce the formation of early lesions, alleviate plaque load and prevent further development of AS.545,546 Notably, most current studies on the regulation of macrophage function in CVD lack comparisons between the results of intervention at different time points, so it is necessary to strengthen the exploration of the optimal time window for intervention (Table 3).
Clinical progress and translational implications
Most current clinical trials on CVD involve anti-inflammatory therapies based on inflammatory cytokines and chemokines (such as IL-1, IL-6, TNF-α, and CCL2) and anti-lipid therapies that inhibit foam cell formation (Table 4). The CANTOS trial is the first successful immunotherapy trial in CVD. A neutralizing antibody canakinumab against IL-1β, an inflammatory cytokine primarily produced by macrophages, was the first drug shown in a clinical trial to specifically and successfully reduce inflammation and the recurrence rate of cardiovascular events in patients after MI.547 Colchicine, a broadly anti-inflammatory drug, can not only inhibit the production of IL-1β activated by the NLRP3 inflammasome in macrophages548 but also interfere with the TNF-α-induced NF-κΒ pathway549 to reduce inflammation. The COLCOT (Colchicine Cardiovascular Outcomes Trial) demonstrated that colchicine treatment in patients with MI significantly reduced the risk of ischemic cardiovascular events such as resuscitative cardiac arrest, MI, stroke, and angina.550 The LoDoCo2 (Low-Dose Colchicine for Secondary Prevention of Cardiovascular Disease) trial also demonstrated that 0.5 mg of colchicine once a day significantly reduced the risk of cardiovascular events in patients with chronic coronary artery disease.551 As a downstream inflammatory signaling of IL-1, IL-6 also participates in the inflammatory response and immunomodulation, thereby affecting the occurrence and development of CVD.552 The ASSAIL-MI trial showed that early treatment with tocilizumab, an IL-6 antibody, enhanced myocardial salvage in patients with STEMI, and there was a tendency to reduce the size of MI.553 For anti-lipid therapies, systemic ACAT inhibition has been shown to reduce circulating TNF-α levels and improve vascular endothelial function in hypercholesterolemic subjects.554 However, several subsequent trials in patients with hypercholesterolemia555 and coronary AS556,557 showed that ACAT inhibition did not improve the disease but actually promoted AS. This may be attributed to the fact that inhibition of ACAT-1 causes free cholesterol to accumulate to toxic levels in macrophages, leading to cell death.555 Therefore, targeting ACAT as a therapeutic strategy for CVD may need to be considered carefully. Finally, two phase II clinical trials targeting the inhibition of the CCL2-CCR2 axis (NCT01269242, NCT00715169) have been successfully conducted. Treatment with bindarit, a CCL2 inhibitor, could prevent restenosis in patients after percutaneous coronary intervention.558 Furthermore, in patients with CVD risk factors, treatment with the anti-CCR2 monoclonal antibody MLN1202 significantly reduced the level of C-reactive protein (CRP), an established biomarker of inflammation associated with coronary artery disease.268
For the ongoing clinical trials, anti-inflammatory therapies, including broad immunosuppression and those targeting specific cytokines, are primarily utilized. With regard to broad immunosuppression, colchicine is the most widely used and is being tested in patients with acute coronary syndrome (NCT01906749, NCT00754819), coronary heart disease (NCT05130892), MI (NCT03048825), and high-risk patients with ischemic stroke or transient ischemic attack (NCT02898610). The incidence of major adverse cardiovascular events (MACE), such as MI, death, hospitalization for unstable angina, and HF, is evaluated after treatment. Hydroxychloroquine and methotrexate, originally used as broad anti-inflammatory drugs for rheumatism, have both been found to significantly reduce the risk of CVD in patients with rheumatoid arthritis.559,560 There are currently two clinical trials ongoing using hydroxychloroquine in patients with MI (NCT02648464) and coronary heart disease (NCT02874287), with the incidence of MACE as the primary endpoint. Additionally, a clinical trial is underway that uses LDL-like nanoparticles to deliver methotrexate to patients with coronary heart disease (NCT04616872). In terms of targeting cytokines, for IL-6, trials of the anti-IL-6 receptor monoclonal antibody sarilumab in patients with rheumatoid arthritis (NCT04350216) and high CRP levels, and of the anti-IL-6 monoclonal antibody ziltivekimab in patients with chronic kidney disease and high CRP levels (NCT05021835) are ongoing, with the changes in atherosclerotic plaques and the incidence of MACE as the primary endpoints, respectively. Targeting the pro-inflammatory cytokine TNF-α, the inhibitor etanercept is also being clinically tested in patients with acute ST-segment elevation myocardial infarction (STEMI) (NCT01372930). Regarding the use of anti-lipid therapy, there is an ongoing clinical trial of the anti-LOX1 receptor antibody MEDI6570 in patients with previous MI, with noncalcified plaque volume as the primary endpoint (NCT04610892). It is expected that the publication of these clinical trial results will bring new insights into the understanding of CVD treatment.
Nanomaterials and cell therapy are two promising strategies for the further translation of preclinical treatment modalities for CVD into clinical practice.475,561,562,563 The spatial structures of biomolecules such as cytokines, chemokines, and microRNA are affected by biological, physical and chemical factors such as biological enzymes, temperature, pH, and ionic strength of the surrounding environment in vivo, and also face problems such as off-target and difficulty in breaking through the biofilm barriers, which to a certain extent hinders the efficacy of drugs.564,565 Based on the characteristics such as the loading capacity and modifiability,564,566,567 nanomaterials can achieve the encapsulation and delivery of biomolecules to isolate the environment in vivo,568 assemble themselves with biomolecules or residues to mitigate off-target effects,536,569,570 and use material sources with lipid-soluble or positive surface potential properties to help therapeutic drugs cross cell membranes,571 which provides a solution to obstacles in the clinical translation of drugs. Cell therapy has the advantages of individualization, durability, and low drug resistance, and can solve refractory CVD that cannot be solved by traditional drugs.563 When preparing in vitro reprogrammed macrophages for adoptive transplantation therapy, autologous macrophages are not only less efficient in the collection and processing process, but more importantly, the weak proliferation and difficult genetic manipulation characteristics of macrophages themselves increase the difficulty of modification and expansion in vitro.506,572 With induced pluripotent stem cells (iPSCs) from healthy donors as the source, repair macrophages can be prepared in large quantities by utilizing their good plasticity and proliferation, which will greatly improve the efficiency of macrophage-based cell therapy.572 MSCs transplantation can contribute to the treatment of CVD, however, MSCs-based cell therapy may cause many adverse reactions in organisms, such as immune response, embolism, graft-versus-host disease, and risk of malignant tumors.573,574,575,576 The main way for MSCs to exert function is secretion of exosomes, and the infusion of exosomes or further isolation of effector substances in exosomes can minimize safety issues of live cell management, showing reduced immunogenicity and tumor development risk.577 It is worth noting that many nanomaterials and cell therapies have been used in various clinical fields, which provides a precedent for clinical translation in the cardiovascular field563,564 (Fig. 6).

Promising targets for preclinical strategies and clinical trials. This chart outlines promising targets for preclinical strategies and clinical trials aimed at macrophage intervention. These targets primarily focus on three essential mechanisms: inhibition of macrophage recruitment, inhibition of foam cell formation and macrophage survival, and regulation of macrophage function. The blue section underscores targets and therapeutic agents for preclinical strategies on the basis of subdivided macrophage regulatory mechanisms. The red section pertains to drugs currently undergoing clinical trials. (Created with BioRender.com)
Conclusion and perspective
This article comprehensively reviews the regulatory mechanisms of macrophages in ischemic and non-ischemic cardiac injuries, as well as vascular diseases, which involve inflammation responses and their impact on fundamental pathological processes such as myocardial fibrosis, myocardial hypertrophy, myocardial metabolic disorders, and vascular injury. Additionally, the advancements in targeted macrophage therapy have garnered considerable attention in both preclinical strategies and clinical trials. From macrophage recruitment to its role in mediating CVD progression, three characteristics are captured. First, monocyte-derived CCR2+ macrophages are considered to be the main macrophage subset that plays a pivotal role in CVD. Multiple factors in different CVD backgrounds can catalyze macrophage recruitment, such as cell death caused by ischemia and viral infection, mechanical stress and neurohumoral systems in PO, ROS in DCM and cardiac aging, the abnormal metabolic state in diabetic cardiomyopathy, endothelial damage and plaque formation in AS, as well as hemodynamic changes in AA.6 Second, macrophage-secreted mediators may exhibit diverse roles contingent upon different etiologies or different stages of the same etiology. For example, MMP-9 exerts pro-inflammatory and pro-fibrotic effects in AMI78,79 and cardiac aging,254 while in viral myocarditis, it mitigates myocardial damage and fibrosis by impeding viral proliferation.207 IL-1β is involved in both pro-inflammatory and pro-fibrotic processes in AMI,70 IRI,65 and viral myocarditis.208 Nonetheless, it should be noted that early inhibition of IL-1β in AMI leads to insufficient scar formation and cardiac rupture,578 while its early suppression in IRI can diminish infarct size and ameliorate ventricular remodeling.65 Third, there may exist some shared pathways that play a crucial role across various diseases, and these crucial pathways may intricately intertwine within the same disease, collectively driving disease progression. For instance, the NLRP3/IL-1 axis lays a solid foundation for initiating inflammation, amplifying inflammation, and promoting myocardial apoptosis in a variety of diseases, such as AMI, PO, and AS. OPN plays a pro-fibrotic role in myocarditis,210 diabetic cardiomyopathy,242 and especially the MI reparative phase.109,110 Galectin-3 exhibits an important pro-fibrotic function in CMI128,129 and is also significantly upregulated in AMI,98,99 PO,171 and diabetic cardiomyopathy.237 Therefore, based on the complexity of macrophage-mediated signaling pathways in cardiovascular pathogenesis, simplistic applications of cellular mediators like IL-1, IL-10, and MMP-9 often fail to yield expected benefits, elucidating the challenges encountered in most cardiovascular clinical trials when in pursuit of efficacy.579,580 In the future, it is imperative to investigate more critical pro-inflammatory and pro-fibrotic mediators that underlie pathological cardiac remodeling and ensure these molecules do not induce severe adverse reactions. Notably, heart failure with preserved or reduced ejection fraction is an increasingly intriguing topic.581 As HF signifies the advanced stage of both ischemic and non-ischemic myocardial injury, the macrophage-mediated pathophysiological mechanisms exhibit a degree of convergence.23
In the future, optimizing the following aspects may help to further enhance the conversion and success rates of targeted macrophage therapy to cardiovascular clinical practice, including the refinement of macrophage typing to achieve greater precision and granularity, the exploration of novel research directions, the development of accurate disease models, and the implementation of specific treatment approaches. Advancements in single-cell sequencing offer opportunities for further subdivision of macrophage subsets. Currently, there is a lack of precision in targeting specific macrophage types, with most therapeutic strategies tending to concentrate on promoting the polarization of M2 macrophages and related anti-inflammatory mediators. Such a description may account for the fact that the overall functional shift of macrophages is advantageous for disease recovery. However, certain sub-subtypes of the M2 phenotype are not favorable for disease prognosis, and exclusion of these types of macrophages, such as foam cells in AS, may potentially achieve a better therapeutic effect. Meanwhile, the promising therapeutic value of some newly discovered mechanisms in the treatment of CVD, such as macrophage extracellular traps (MET), warrants further investigation. Although MET has garnered significant interest in fields encompassing pathogen infection, acute kidney injury and cystic fibrosis, its potential role in the cardiovascular field has received limited attention.582,583 In light of preclinical tests, the problems existing in animal models are gradually revealed and improved. For instance, while permanent coronary artery ligation is widely used to simulate AMI, clinical patients have universal access to reperfusion therapy instead. Traditional MI models generally entail pericardial destruction to access the coronary arteries, which may interfere with cardiac repair.584,585 The necessity for more accurate and precise models is highlighted by the fact that fibrosis typically manifests in rodent models over weeks or months, whereas it takes years or decades to develop in humans. Several methods, including the utilization of organoids, heart-on-a-chip, and humanized mice, have been established to investigate disease mechanisms, elucidate cell-to-cell interactions, and conduct drug screening.586 In the meantime, the employment of single-cell resolution analyses is aiding in refining in vivo and in vitro models that recapitulate the phenotypes and functions of immune cells, including macrophages. In the management of CVD, there is a notable absence of exploration into the intervention time window, which may be due to the difficulty in controlling the specific stage of disease progression during the experimental procedures. The lack of exploration of the time window for intervention also makes the delicate balance between pro-and anti-inflammatory cells in vivo elusive. It is widely recognized that managing inflammation early or facilitating M2 macrophage polarization during the transition to an anti-inflammatory environment is beneficial for cardiac remodeling, taking the crossover point between the inflammatory and reparative phases of AMI (e.g., 3–4 days after AMI) and early PO as examples. What’s more, by employing small molecules such as miRNAs and antisense oligonucleotides (ASOs) or novel delivery systems such as nanoparticles and hydrogels, compounds are likely to be more effective and target specific without jeopardizing their critical roles in other physiological functions and avoiding catastrophic side effects, which paves the road for clinical translation of preclinical strategies and immunomodulation of CVD.587
References
Fernández-Velasco, M., González-Ramos, S. & Boscá, L. Involvement of monocytes/macrophages as key factors in the development and progression of cardiovascular diseases. Biochem. J. 458, 187–193 (2014).
Bhattacharya, M. & Ramachandran, P. Immunology of human fibrosis. Nat. Immunol. 24, 1423–1433 (2023).
Frodermann, V. & Nahrendorf, M. Macrophages and cardiovascular health. Physiol. Rev. 98, 2523–2569 (2018).
Frangogiannis, N. G. Cardiac fibrosis. Cardiovasc. Res. 117, 1450–1488 (2021).
Jian, Y. et al. Crosstalk between macrophages and cardiac cells after myocardial infarction. Cell Commun. Signal. 21, 109 (2023).
López, B. et al. Diffuse myocardial fibrosis: mechanisms, diagnosis and therapeutic approaches. Nat. Rev. Cardiol. 18, 479–498 (2021).
Frangogiannis, N. G. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 65, 70–99 (2019).
Libby, P. Inflammation during the life cycle of the atherosclerotic plaque. Cardiovasc. Res. 117, 2525–2536 (2021).
Claridge, B., Drack, A., Pinto, A. R. & Greening, D. W. Defining cardiac fibrosis complexity and regulation towards therapeutic development. Clin. Transl. Discov. 3, e163 (2023).
Vallejo, J., Cochain, C., Zernecke, A. & Ley, K. Heterogeneity of immune cells in human atherosclerosis revealed by scRNA-Seq. Cardiovasc. Res. 117, 2537–2543 (2021).
Witherel, C. E., Abebayehu, D., Barker, T. H. & Spiller, K. L. Macrophage and fibroblast interactions in biomaterial-mediated fibrosis. Adv. Healthc. Mater. 8, e1801451 (2019).
Orecchioni, M., Ghosheh, Y., Pramod, A. B. & Ley, K. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front. Immunol. 10, 1084 (2019).
Bajpai, G. et al. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 124, 263–278 (2019).
Murray, P. J. et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20 (2014).
Epelman, S., Lavine, K. J. & Randolph, G. J. Origin and functions of tissue macrophages. Immunity 41, 21–35 (2014).
Epelman, S. et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104 (2014).
Ginhoux, F. & Guilliams, M. Tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449 (2016).
Dick, S. A. et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci. Immunol. 7, eabf7777 (2022).
Chakarov, S. et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science 363, eaau0964 (2019).
Bajpai, G. et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24, 1234–1245 (2018).
Lavine, K. J. et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl Acad. Sci. USA 111, 16029–16034 (2014).
Lafuse, W. P., Wozniak, D. J. & Rajaram, M. V. S. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells 10, 51 (2020).
DeBerge, M., Shah, S. J., Wilsbacher, L. & Thorp, E. B. Macrophages in heart failure with reduced versus preserved ejection fraction. Trends Mol. Med. 25, 328–340 (2019).
Yap, J. et al. Macrophages in cardiac remodelling after myocardial infarction. Nat. Rev. Cardiol. 20, 373–385 (2023).
Buechler, M. B., Fu, W. & Turley, S. J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 54, 903–915 (2021).
Kuppe, C. et al. Spatial multi-omic map of human myocardial infarction. Nature 608, 766–777 (2022).
Maruyama, K. & Imanaka-Yoshida, K. The pathogenesis of cardiac fibrosis: a review of recent progress. Int. J. Mol. Sci. 23, 2617 (2022).
Haider, N. et al. Transition of macrophages to fibroblast-like cells in healing myocardial infarction. J. Am. Coll. Cardiol. 74, 3124–3135 (2019).
Simões, F. C. et al. Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 11, 600 (2020).
Zhuang, T. et al. ALKBH5-mediated m6A modification of IL-11 drives macrophage-to-myofibroblast transition and pathological cardiac fibrosis in mice. Nat. Commun. 15, 1995 (2024).
Hu, S. et al. Different roles of resident and non-resident macrophages in cardiac fibrosis. Front. Cardiovasc. Med. 9, 818188 (2022).
Liu, Y., Wu, M., Zhong, C., Xu, B. & Kang, L. M2-like macrophages transplantation protects against the doxorubicin-induced heart failure via mitochondrial transfer. Biomater. Res. 26, 14 (2022).
Cao, Y. et al. M2b macrophages protect against doxorubicin-induced cardiotoxicity via alternating autophagy in cardiomyocytes. PLoS One 18, e0288422 (2023).
Hara, A. & Tallquist, M. D. Fibroblast and immune cell cross-talk in cardiac fibrosis. Curr. Cardiol. Rep. 25, 485–493 (2023).
Amrute, J. M. et al. Targeting immune-fibroblast crosstalk in myocardial infarction and cardiac fibrosis. Preprint at https://doi.org/10.21203/rs.3.rs-2402606/v1 (2023).
Gurantz, D. et al. IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J. Mol. Cell. Cardiol. 38, 505–515 (2005).
Smolgovsky, S., Ibeh, U., Tamayo, T. P. & Alcaide, P. Adding insult to injury—inflammation at the heart of cardiac fibrosis. Cell. Signal. 77, 109828 (2021).
Prabhu, S. D. & Frangogiannis, N. G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 119, 91–112 (2016).
Li, J., Chen, Q., Zhang, R., Liu, Z. & Cheng, Y. The phagocytic role of macrophage following myocardial infarction. Heart Fail. Rev. 28, 993–1007 (2023).
Ensan, S. et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat. Immunol. 17, 159–168 (2016).
Hernandez, G. E. et al. Aortic intimal resident macrophages are essential for maintenance of the non-thrombogenic intravascular state. Nat. Cardiovasc. Res. 1, 67–84 (2022).
Williams, J. W. et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 21, 1194–1204 (2020).
Weinberger, T. et al. Ontogeny of arterial macrophages defines their functions in homeostasis and inflammation. Nat. Commun. 11, 4549 (2020).
Stoneman, V. et al. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ. Res. 100, 884–893 (2007).
Bjornson, Z. B., Nolan, G. P. & Fantl, W. J. Single-cell mass cytometry for analysis of immune system functional states. Curr. Opin. Immunol. 25, 484–494 (2013).
Cheng, Z. et al. Diverse roles of macrophage polarization in aortic aneurysm: destruction and repair. J. Transl. Med. 16, 354 (2018).
Rahman, K. et al. Inflammatory Ly6Chi monocytes and their conversion to M2 macrophages drive atherosclerosis regression. J. Clin. Investig. 127, 2904–2915 (2017).
Ramji, D. P. & Davies, T. S. Cytokines in atherosclerosis: key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev. 26, 673–685 (2015).
Chinetti-Gbaguidi, G., Colin, S. & Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 12, 10–17 (2015).
Chistiakov, D. A., Melnichenko, A. A., Myasoedova, V. A., Grechko, A. V. & Orekhov, A. N. Mechanisms of foam cell formation in atherosclerosis. J. Mol. Med. 95, 1153–1165 (2017).
Bobryshev, Y. V., Ivanova, E. A., Chistiakov, D. A., Nikiforov, N. G. & Orekhov, A. N. Macrophages and their role in atherosclerosis: pathophysiology and transcriptome analysis. Biomed. Res. Int. 2016, 9582430 (2016).
Moore, K. J. & Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 145, 341–355 (2011).
Newby, A. C. Metalloproteinase production from macrophages—a perfect storm leading to atherosclerotic plaque rupture and myocardial infarction. Exp. Physiol. 101, 1327–1337 (2016).
Theofilis, P., Oikonomou, E., Tsioufis, K. & Tousoulis, D. The role of macrophages in atherosclerosis: pathophysiologic mechanisms and treatment considerations. Int. J. Mol. Sci. 24, 9568 (2023).
Kadl, A. et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 107, 737–746 (2010).
Gleissner, C. A. et al. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ. Res. 106, 203–211 (2010).
Boyle, J. J. Heme and haemoglobin direct macrophage Mhem phenotype and counter foam cell formation in areas of intraplaque haemorrhage. Curr. Opin. Lipidol. 23, 453–461 (2012).
Boyle, J. J. et al. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 174, 1097–1108 (2009).
Boyle, J. J. et al. Heme induces heme oxygenase 1 via Nrf2: role in the homeostatic macrophage response to intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol. 31, 2685–2691 (2011).
Finn, A. V. et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 59, 166–177 (2012).
Saxena, A. et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J. Immunol. 191, 4838–4848 (2013).
Huang, S. & Frangogiannis, N. G. Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br. J. Pharmacol. 175, 1377–1400 (2018).
Bevan, L. et al. Specific macrophage populations promote both cardiac scar deposition and subsequent resolution in adult zebrafish. Cardiovasc. Res. 116, 1357–1371 (2020).
Dewald, O. et al. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 96, 881–889 (2005).
Bujak, M. et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 173, 57–67 (2008).
Heidt, T. et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 115, 284–295 (2014).
Dick, S. A. et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39 (2019).
Frangogiannis, N. G. et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation 115, 584–592 (2007).
Li, W. et al. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1, e87315 (2016).
Bageghni, S. A. et al. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight 5, e125074 (2019).
Liu, W. et al. Activation in M1 but not M2 macrophages contributes to cardiac remodeling after myocardial infarction in rats: a critical role of the calcium sensing receptor/NRLP3 inflammasome. Cell. Physiol. Biochem. 35, 2483–2500 (2015).
Kobara, M. et al. Antibody against interleukin-6 receptor attenuates left ventricular remodelling after myocardial infarction in mice. Cardiovasc. Res. 87, 424–430 (2010).
Jing, R., Long, T. Y., Pan, W., Li, F. & Xie, Q. Y. IL-6 knockout ameliorates myocardial remodeling after myocardial infarction by regulating activation of M2 macrophages and fibroblast cells. Eur. Rev. Med. Pharmacol. Sci. 23, 6283–6291 (2019).
Weber, K. T., Sun, Y., Bhattacharya, S. K., Ahokas, R. A. & Gerling, I. C. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat. Rev. Cardiol. 10, 15–26 (2013).
Lugrin, J. et al. The systemic deletion of interleukin-1α reduces myocardial inflammation and attenuates ventricular remodeling in murine myocardial infarction. Sci. Rep. 13, 4006 (2023).
Liu, Y. et al. Macrophage CARD9 mediates cardiac injury following myocardial infarction through regulation of lipocalin 2 expression. Signal Transduct. Target. Ther. 8, 394 (2023).
Lindsey, M. L. Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat. Rev. Cardiol. 15, 471–479 (2018).
Ducharme, A. et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J. Clin. Investig. 106, 55–62 (2000).
Iyer, R. P. et al. Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J. Mol. Cell. Cardiol. 100, 109–117 (2016).
Iyer, R. P. et al. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int. J. Cardiol. 185, 198–208 (2015).
Kubota, A., Suto, A., Suzuki, K., Kobayashi, Y. & Nakajima, H. Matrix metalloproteinase-12 produced by Ly6C(low) macrophages prolongs the survival after myocardial infarction by preventing neutrophil influx. J. Mol. Cell. Cardiol. 131, 41–52 (2019).
Ma, Y. et al. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circ. Res. 112, 675–688 (2013).
Lindsey, M. L. et al. Exogenous CXCL4 infusion inhibits macrophage phagocytosis by limiting CD36 signalling to enhance post-myocardial infarction cardiac dilation and mortality. Cardiovasc. Res. 115, 395–408 (2019).
Howangyin, K. Y. et al. Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation 133, 826–839 (2016).
Jia, D. et al. Cardiac resident macrophage-derived legumain improves cardiac repair by promoting clearance and degradation of apoptotic cardiomyocytes after myocardial infarction. Circulation 145, 1542–1556 (2022).
Cai, S. et al. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J. Clin. Investig. 133, e159498 (2023).
Motley, M. P. et al. A CCR2 macrophage endocytic pathway mediates extravascular fibrin clearance in vivo. Blood 127, 1085–1096 (2016).
Chen, W. et al. Endogenous IRAK-M attenuates postinfarction remodeling through effects on macrophages and fibroblasts. Arterioscler. Thromb. Vasc. Biol. 32, 2598–2608 (2012).
Nahrendorf, M. et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204, 3037 (2007).
Hilgendorf, I. et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 114, 1611–1622 (2014).
DeBerge, M. et al. MerTK cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ. Res. 121, 930–940 (2017).
Shiraishi, M. et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J. Clin. Investig. 126, 2151–2166 (2016).
Alonso-Herranz, L. et al. Macrophages promote endothelial-to-mesenchymal transition via MT1-MMP/TGFβ1 after myocardial infarction. Elife 9, e57920 (2020).
Wang, Y. et al. Hypoxia induces M2 macrophages to express VSIG4 and mediate cardiac fibrosis after myocardial infarction. Theranostics 13, 2192–2209 (2023).
Garlapati, V. et al. Targeting myeloid cell coagulation signaling blocks MAP kinase/TGF-β1-driven fibrotic remodeling in ischemic heart failure. J. Clin. Investig. 133, e156436 (2023).
Yu, C. M., Tipoe, G. L., Wing-Hon Lai, K. & Lau, C. P. Effects of combination of angiotensin-converting enzyme inhibitor and angiotensin receptor antagonist on inflammatory cellular infiltration and myocardial interstitial fibrosis after acute myocardial infarction. J. Am. Coll. Cardiol. 38, 1207–1215 (2001).
Alonso-Herranz, L. et al. Macrophages promote endothelial-to-mesenchymal transition via MT1-MMP/TGFbeta1 after myocardial infarction. Elife 9, e57920 (2020).
Cassaglia, P. et al. Genetic deletion of galectin-3 alters the temporal evolution of macrophage infiltration and healing affecting the cardiac remodeling and function after myocardial infarction in mice. Am. J. Pathol. 190, 1789–1800 (2020).
Liu, Y. H. et al. N-acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am. J. Physiol. Heart Circ. Physiol. 296, H404–H412 (2009).
Frangogiannis, N. G. Transforming growth factor-beta in myocardial disease. Nat. Rev. Cardiol. 19, 435–455 (2022).
Dobaczewski, M. et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 107, 418–428 (2010).
Kong, P. et al. Opposing actions of fibroblast and cardiomyocyte smad3 signaling in the infarcted myocardium. Circulation 137, 707–724 (2018).
Bujak, M. et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 116, 2127–2138 (2007).
Huang, S. et al. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J. Mol. Cell. Cardiol. 132, 84–97 (2019).
Chen, B. et al. Differential effects of Smad2 and Smad3 in regulation of macrophage phenotype and function in the infarcted myocardium. J. Mol. Cell. Cardiol. 171, 1–15 (2022).
Humeres, C. et al. Smad7 effects on TGF-beta and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J. Clin. Investig. 132, e146926 (2022).
Wang, B. et al. Decreased Smad 7 expression contributes to cardiac fibrosis in the infarcted rat heart. Am. J. Physiol. Heart Circ. Physiol. 282, H1685–H1696 (2002).
Chen, B. et al. Macrophage Smad3 protects the infarcted heart, stimulating phagocytosis and regulating inflammation. Circ. Res. 125, 55–70 (2019).
Shirakawa, K. et al. MerTK expression and ERK activation are essential for the functional maturation of osteopontin-producing reparative macrophages after myocardial infarction. J. Am. Heart Assoc. 9, e017071 (2020).
Shirakawa, K. et al. IL (Interleukin)-10-STAT3-galectin-3 axis is essential for osteopontin-producing reparative macrophage polarization after myocardial infarction. Circulation 138, 2021–2035 (2018).
Trueblood, N. A. et al. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ. Res. 88, 1080–1087 (2001).
Krishnamurthy, P. et al. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 104, e9–e18 (2009).
Burchfield, J. S. et al. Interleukin-10 from transplanted bone marrow mononuclear cells contributes to cardiac protection after myocardial infarction. Circ. Res. 103, 203–211 (2008).
Jung, M. et al. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res. Cardiol. 112, 33 (2017).
Talman, V. & Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res 365, 563–581 (2016).
Méndez-Ferrer, S., Lucas, D., Battista, M. & Frenette, P. S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 452, 442–447 (2008).
Sager, H. B. et al. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res. 119, 853–864 (2016).
Ismahil, M. A. et al. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ. Res. 114, 266–282 (2014).
Stride, N. et al. Decreased mitochondrial oxidative phosphorylation capacity in the human heart with left ventricular systolic dysfunction. Eur. J. Heart Fail. 15, 150–157 (2013).
Zhang, S. et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. 29, 443–456.e445 (2019).
Testa, M. et al. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J. Am. Coll. Cardiol. 28, 964–971 (1996).
Deswal, A. et al. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103, 2055–2059 (2001).
Hamid, T. et al. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation 119, 1386–1397 (2009).
Zaidi, Y., Aguilar, E. G., Troncoso, M., Ilatovskaya, D. V. & DeLeon-Pennell, K. Y. Immune regulation of cardiac fibrosis post myocardial infarction. Cell. Signal. 77, 109837 (2021).
Shintani, Y. et al. IL-4 as a repurposed biological drug for myocardial infarction through augmentation of reparative cardiac macrophages: proof-of-concept data in mice. Sci. Rep. 7, 6877 (2017).
Hofmann, U. et al. Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circ. Heart Fail. 7, 822–830 (2014).
Weirather, J. et al. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 115, 55–67 (2014).
Suthahar, N. et al. Galectin-3 activation and inhibition in heart failure and cardiovascular disease: an update. Theranostics 8, 593–609 (2018).
Filipe, M. D., Meijers, W. C., Rogier van der Velde, A. & de Boer, R. A. Galectin-3 and heart failure: prognosis, prediction & clinical utility. Clin. Chim. Acta 443, 48–56 (2015).
Yan, X. et al. Deleterious effect of the IL-23/IL-17A axis and γδT cells on left ventricular remodeling after myocardial infarction. J. Am. Heart Assoc. 1, e004408 (2012).
Frangogiannis, N. G. et al. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 111, 2935–2942 (2005).
Algoet, M. et al. Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc. Med. 33, 357–366 (2023).
Bacmeister, L. et al. Inflammation and fibrosis in murine models of heart failure. Basic Res. Cardiol. 114, 19 (2019).
DeBerge, M. et al. Macrophage AXL receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J. Clin. Investig. 131, e139576 (2021).
Hishikari, K. et al. Pharmacological activation of the prostaglandin E2 receptor EP4 improves cardiac function after myocardial ischaemia/reperfusion injury. Cardiovasc. Res. 81, 123–132 (2009).
Huang, C. K. et al. Lgr4 governs a pro-inflammatory program in macrophages to antagonize post-infarction cardiac repair. Circ. Res. 127, 953–973 (2020).
Fan, Q. et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation 139, 663–678 (2019).
Feng, G. et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T cells. Circulation 145, 765–782 (2022).
Huebener, P. et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J. Immunol. 180, 2625–2633 (2008).
Minatoguchi, S. et al. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation 109, 2572–2580 (2004).
Shen, S. C. et al. Macrophages promote the transition from myocardial ischemia reperfusion injury to cardiac fibrosis in mice through GMCSF/CCL2/CCR2 and phenotype switching. Acta Pharmacol. Sin. https://doi.org/10.1038/s41401-023-01222-3 (2024).
Li, L. et al. M2 macrophage-derived sEV regulate pro-inflammatory CCR2(+) macrophage subpopulations to favor post-AMI cardiac repair. Adv. Sci. 10, e2202964 (2023).
Yue, Y. et al. M2b macrophages regulate cardiac fibroblast activation and alleviate cardiac fibrosis after reperfusion injury. Circ. J. 84, 626–635 (2020).
Laroumanie, F. et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 129, 2111–2124 (2014).
Ngwenyama, N. et al. Isolevuglandin-modified cardiac proteins drive CD4+ T-cell activation in the heart and promote cardiac dysfunction. Circulation 143, 1242–1255 (2021).
Zaman, R. et al. Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity 54, 2057–2071.e2056 (2021).
Revelo, X. S. et al. Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circ. Res. 129, 1086–1101 (2021).
Liao, X. et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc. Natl Acad. Sci. USA 115, E4661–E4669 (2018).
Patel, B., Ismahil, M. A., Hamid, T., Bansal, S. S. & Prabhu, S. D. Mononuclear phagocytes are dispensable for cardiac remodeling in established pressure-overload heart failure. PLoS One 12, e0170781 (2017).
Patel, B. et al. CCR2(+) monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC: Basic Transl. Sci. 3, 230–244 (2018).
Willeford, A. et al. CaMKIIdelta-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 3, e97054 (2018).
Suetomi, T. et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca(2+)/calmodulin-dependent protein kinase ii delta signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 138, 2530–2544 (2018).
Zhao, M. et al. Selective Inhibition of NLRP3 inflammasome reverses pressure overload-induced pathological cardiac remodeling by attenuating hypertrophy, fibrosis, and inflammation. Int. Immunopharmacol. 99, 108046 (2021).
Wang, L. et al. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc. Res. 101, 383–392 (2014).
Wang, L. et al. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur. Heart J. 39, 1818–1831 (2018).
Zhang, N. et al. CXCR4-dependent macrophage-to-fibroblast signaling contributes to cardiac diastolic dysfunction in heart failure with preserved ejection fraction. Int. J. Biol. Sci. 18, 1271–1287 (2022).
Ye, S. et al. Dectin-1 acts as a non-classical receptor of ang II to induce cardiac remodeling. Circ. Res. 132, 707–722 (2023).
Lin, Q. Y. et al. Pharmacological blockage of ICAM-1 improves angiotensin II-induced cardiac remodeling by inhibiting adhesion of LFA-1(+) monocytes. Am. J. Physiol. Heart Circ. Physiol. 317, H1301–H1311 (2019).
Liao, C.-W. et al. Interleukin-6 plays a critical role in aldosterone-induced macrophage recruitment and infiltration in the myocardium. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165627 (2020).
Bu, J. et al. The GABA(A) receptor influences pressure overload-induced heart failure by modulating macrophages in mice. Front. Immunol. 12, 670153 (2021).
Fujiu, K. et al. A heart-brain-kidney network controls adaptation to cardiac stress through tissue macrophage activation. Nat. Med. 23, 611–622 (2017).
Li, X. et al. TAK1 activation by NLRP3 deficiency confers cardioprotection against pressure overload-induced cardiomyocyte pyroptosis and hypertrophy. JACC: Basic Transl. Sci. 8, 1555–1573 (2023).
Horio, T. et al. Production and secretion of adrenomedullin in cultured rat cardiac myocytes and nonmyocytes: stimulation by interleukin-1beta and tumor necrosis factor-alpha. Endocrinology 139, 4576–4580 (1998).
Sano, S. et al. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ. Res. 123, 335–341 (2018).
Sano, S. et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1β/NLRP3 inflammasome. J. Am. Coll. Cardiol. 71, 875–886 (2018).
Ren, J. et al. Proinflammatory protein CARD9 is essential for infiltration of monocytic fibroblast precursors and cardiac fibrosis caused by angiotensin II infusion. Am. J. Hypertens. 24, 701–707 (2011).
Chen, H. et al. The E3 ubiquitin ligase WWP2 regulates pro-fibrogenic monocyte infiltration and activity in heart fibrosis. Nat. Commun. 13, 7375 (2022).
Yang, K. et al. Carboxyl terminus of heat shock protein 70-interacting protein inhibits angiotensin II-induced cardiac remodeling. Am. J. Hypertens. 25, 994–1001 (2012).
Yu, Q. et al. Macrophage-specific NLRC5 protects from cardiac remodeling through interaction with HSPA8. JACC: Basic Transl. Sci. 8, 479–496 (2023).
Hulsmans, M. et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 215, 423–440 (2018).
Lin, Y. H. et al. Aldosterone induced galectin-3 secretion in vitro and in vivo: from cells to humans. PLoS One 9, e95254 (2014).
Young, M. J. et al. Macrophage mineralocorticoid receptor signaling plays a key role in aldosterone-independent cardiac fibrosis. Endocrinology 153, 3416–3425 (2012).
Li, J., Yousefi, K., Ding, W., Singh, J. & Shehadeh, L. A. Osteopontin RNA aptamer can prevent and reverse pressure overload-induced heart failure. Cardiovasc. Res. 113, 633–643 (2017).
Verma, S. K. et al. Interleukin-10 inhibits bone marrow fibroblast progenitor cell-mediated cardiac fibrosis in pressure-overloaded myocardium. Circulation 136, 940–953 (2017).
Verma, S. K. et al. Interleukin-10 treatment attenuates pressure overload–induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3–dependent inhibition of nuclear factor-κB. Circulation 126, 418–429 (2012).
Nevers, T. et al. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J. Exp. Med. 214, 3311–3329 (2017).
Li, Y. et al. Interleukin-12p35 deletion promotes CD4 T-cell–dependent macrophage differentiation and enhances angiotensin II–induced cardiac fibrosis. Arterioscler. Thromb. Vasc. Biol. 32, 1662–1674 (2012).
Gan, W. et al. Serum–glucocorticoid-regulated kinase 1 contributes to mechanical stretch-induced inflammatory responses in cardiac fibroblasts. Mol. Cell. Biochem. 445, 67–78 (2017).
McWhorter, F. Y., Wang, T., Nguyen, P., Chung, T. & Liu, W. F. Modulation of macrophage phenotype by cell shape. Proc. Natl Acad. Sci. USA 110, 17253–17258 (2013).
McDonald, L. T. et al. Increased macrophage-derived SPARC precedes collagen deposition in myocardial fibrosis. Am. J. Physiol. Heart Circ. Physiol. 315, H92–h100 (2018).
Xu, X., Hua, Y., Nair, S., Bucala, R. & Ren, J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension 63, 490–499 (2014).
Koga, K., Kenessey, A. & Ojamaa, K. Macrophage migration inhibitory factor antagonizes pressure overload-induced cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 304, H282–H293 (2013).
Wong, N. R. et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity 54, 2072–2088.e2077 (2021).
Ma, F. et al. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS One 7, e35144 (2012).
Chou, C.-H. et al. IL-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc. Res. 114, 690–702 (2018).
Shimojo, N. et al. Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin alphaVbeta3/nuclear factor-kappaB/interleukin-6 axis. Hypertension 66, 757–766 (2015).
Loperena, R. et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc. Res. 114, 1547–1563 (2018).
Khalil, H. et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 127, 3770–3783 (2017).
Divakaran, V. et al. Adaptive and maladptive effects of SMAD3 signaling in the adult heart after hemodynamic pressure overloading. Circ. Heart Fail. 2, 633–642 (2009).
Abe, H. et al. Macrophage hypoxia signaling regulates cardiac fibrosis via Oncostatin M. Nat. Commun. 10, 2824 (2019).
Kimura, A. et al. Protective roles of interferon-γ in cardiac hypertrophy induced by sustained pressure overload. J. Am. Heart Assoc. 7, e008145 (2018).
Yang, M. et al. Deficiency of GATA3-positive macrophages improves cardiac function following myocardial infarction or pressure overload hypertrophy. J. Am. Coll. Cardiol. 72, 885–904 (2018).
Luyt, C. E., Hékimian, G. & Ginsberg, F. What’s new in myocarditis? Intensive Care Med. 42, 1055–1057 (2016).
Cooper, L. T. Jr Myocarditis. N. Engl. J. Med. 360, 1526–1538 (2009).
Tschope, C. et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat. Rev. Cardiol. 18, 169–193 (2021).
Göser, S. et al. Critical role for monocyte chemoattractant protein-1 and macrophage inflammatory protein-1alpha in induction of experimental autoimmune myocarditis and effective anti-monocyte chemoattractant protein-1 gene therapy. Circulation 112, 3400–3407 (2005).
Muller, I. et al. Pathogenic role of the damage-associated molecular patterns S100A8 and S100A9 in coxsackievirus B3-induced myocarditis. Circ. Heart Fail. 10, e004125 (2017).
Leuschner, F. et al. Silencing of CCR2 in myocarditis. Eur. Heart J. 36, 1478–1488 (2015).
Tsou, C. L. et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 117, 902–909 (2007).
Heymans, S., Eriksson, U., Lehtonen, J. & Cooper, L. T. Jr The quest for new approaches in myocarditis and inflammatory cardiomyopathy. J. Am. Coll. Cardiol. 68, 2348–2364 (2016).
Khawaja, A. & Bromage, D. I. The innate immune response in myocarditis. Int. J. Biochem. Cell Biol. 134, 105973 (2021).
Murray, P. J. & Wynn, T. A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737 (2011).
Ahn, J. & Kim, J. Mechanisms and consequences of inflammatory signaling in the myocardium. Curr. Hypertens. Rep. 14, 510–516 (2012).
Bao, J., Sun, T., Yue, Y. & Xiong, S. Macrophage NLRP3 inflammasome activated by CVB3 capsid proteins contributes to the development of viral myocarditis. Mol. Immunol. 114, 41–48 (2019).
Gou, W., Zhang, Z., Yang, C. & Li, Y. MiR-223/Pknox1 axis protects mice from CVB3-induced viral myocarditis by modulating macrophage polarization. Exp. Cell Res. 366, 41–48 (2018).
Jiahui, C. et al. miR-19b-3p/PKNOX1 regulates viral myocarditis by regulating macrophage polarization. Front. Genet. 13, 902453 (2022).
Cheung, C. et al. Ablation of matrix metalloproteinase-9 increases severity of viral myocarditis in mice. Circulation 117, 1574–1582 (2008).
Kraft, L., Erdenesukh, T., Sauter, M., Tschope, C. & Klingel, K. Blocking the IL-1beta signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res. Cardiol. 114, 11 (2019).
Gruhle, S. et al. The prostacyclin agonist iloprost aggravates fibrosis and enhances viral replication in enteroviral myocarditis by modulation of ERK signaling and increase of iNOS expression. Basic Res. Cardiol. 107, 287 (2012).
Szalay, G. et al. Osteopontin: a fibrosis-related marker molecule in cardiac remodeling of enterovirus myocarditis in the susceptible host. Circ. Res. 104, 851–859 (2009).
Schultheiss, H.-P. et al. Dilated cardiomyopathy. Nat. Rev. Dis. Prim. 5, 32 (2019).
Zhang, H. et al. Self-maintenance of cardiac resident reparative macrophages attenuates doxorubicin-induced cardiomyopathy through the SR-A1-c-Myc axis. Circ. Res. 127, 610–627 (2020).
Riad, A. et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur. J. Heart Fail. 10, 233–243 (2008).
Ma, Y. et al. Toll-like receptor (TLR) 2 and TLR4 differentially regulate doxorubicin induced cardiomyopathy in mice. PLoS One 7, e40763 (2012).
Shimazu, R. et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on toll-like receptor 4. J. Exp. Med. 189, 1777–1782 (1999).
Feldtmann, R. et al. Myeloid differentiation factor‐2 activates monocytes in patients with dilated cardiomyopathy. Immunology 167, 40–53 (2022).
Kobayashi, M. et al. Expression of monocyte chemoattractant protein-1 in idiopathic dilated cardiomyopathy. Int. J. Cardiol. 126, 427–429 (2008).
Zhang, L. et al. MCC950 attenuates doxorubicin-induced myocardial injury in vivo and in vitro by inhibiting NLRP3-mediated pyroptosis. Biomed. Pharmacother. 143, 112133 (2021).
Marchetti, C. et al. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and nonischemic injury in the mouse. J. Cardiovasc. Pharmacol. 66, 1–8 (2015).
Psarras, S. et al. Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur. Heart J. 33, 1954–1963 (2012).
Chaffin, M. et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature 608, 174–180 (2022).
Nicolás-Ávila, J. A. et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183, 94–109.e123 (2020).
Touvron, M. et al. Locally expressed IGF1 propeptide improves mouse heart function in induced dilated cardiomyopathy by blocking myocardial fibrosis and SRF-dependent CTGF induction. Dis. Models Mech. 5, 481–491 (2012).
Dillmann, W. H. Diabetic cardiomyopathy. Circ. Res. 124, 1160–1162 (2019).
Suresh Babu, S. et al. MicroRNA-126 overexpression rescues diabetes-induced impairment in efferocytosis of apoptotic cardiomyocytes. Sci. Rep. 6, 36207 (2016).
Bajpai, A. & Tilley, D. G. The role of leukocytes in diabetic cardiomyopathy. Front. Physiol. 9, 1547 (2018).
Lumeng, C. N., Bodzin, J. L. & Saltiel, A. R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 117, 175–184 (2007).
Rao, X., Zhong, J. & Sun, Q. The heterogenic properties of monocytes/macrophages and neutrophils in inflammatory response in diabetes. Life Sci. 116, 59–66 (2014).
Qatanani, M., Szwergold, N. R., Greaves, D. R., Ahima, R. S. & Lazar, M. A. Macrophage-derived human resistin exacerbates adipose tissue inflammation and insulin resistance in mice. J. Clin. Investig. 119, 531–539 (2009).
Steppan, C. M. et al. The hormone resistin links obesity to diabetes. Nature 409, 307–312 (2001).
Tilg, H. & Moschen, A. R. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 6, 772–783 (2006).
Tan, Y. et al. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat. Rev. Cardiol. 17, 585–607 (2020).
Jia, G., Hill, M. A. & Sowers, J. R. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ. Res. 122, 624–638 (2018).
Tuleta, I. & Frangogiannis, N. G. Fibrosis of the diabetic heart: clinical significance, molecular mechanisms, and therapeutic opportunities. Adv. Drug Deliv. Rev. 176, 113904 (2021).
Morey, M., O’Gaora, P., Pandit, A. & Hélary, C. Hyperglycemia acts in synergy with hypoxia to maintain the pro-inflammatory phenotype of macrophages. PLoS One 14, e0220577 (2019).
Yang, N. et al. Dectin-1 deficiency alleviates diabetic cardiomyopathy by attenuating macrophage-mediated inflammatory response. Biochim. Biophys. Acta Mol. Basis Dis. 1869, 166710 (2023).
Zhu, N., Zhu, L., Huang, B., Xiang, W. & Zhao, X. Galectin-3 inhibition ameliorates streptozotocin-induced diabetic cardiomyopathy in mice. Front. Cardiovasc. Med. 9, 868372 (2022).
Wu, W., Chai, Q. & Zhang, Z. Inhibition of SGLT1 alleviates the glycemic variability-induced cardiac fibrosis via inhibition of activation of macrophage and cardiac fibroblasts. Mol. Cell. Biol. 42, e0028221 (2022).
Widiapradja, A. et al. Replacement of lost substance P reduces fibrosis in the diabetic heart by preventing adverse fibroblast and macrophage phenotype changes. Cells 10, 2659 (2021).
Govindappa, P. K. et al. Targeting exosome-associated human antigen R attenuates fibrosis and inflammation in diabetic heart. FASEB J. 34, 2238–2251 (2020).
Privratsky, J. R., Wold, L. E., Sowers, J. R., Quinn, M. T. & Ren, J. AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension 42, 206–212 (2003).
Caglayan, E. et al. Differential roles of cardiomyocyte and macrophage peroxisome proliferator-activated receptor gamma in cardiac fibrosis. Diabetes 57, 2470–2479 (2008).
Qi, G. M., Jia, L. X., Li, Y. L., Li, H. H. & Du, J. Adiponectin suppresses angiotensin II-induced inflammation and cardiac fibrosis through activation of macrophage autophagy. Endocrinology 155, 2254–2265 (2014).
Hotta, K. et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler. Thromb. Vasc. Biol. 20, 1595–1599 (2000).
Chen, M. S., Lee, R. T. & Garbern, J. C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 118, 1173–1187 (2022).
Xie, S., Xu, S. C., Deng, W. & Tang, Q. Metabolic landscape in cardiac aging: insights into molecular biology and therapeutic implications. Signal Transduct. Target. Ther. 8, 114 (2023).
Pinto, A. R. et al. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging 6, 399–413 (2014).
Molawi, K. et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 211, 2151–2158 (2014).
Shirakabe, A., Ikeda, Y., Sciarretta, S., Zablocki, D. K. & Sadoshima, J. Aging and autophagy in the heart. Circ. Res. 118, 1563–1576 (2016).
Trial, J., Heredia, C. P., Taffet, G. E., Entman, M. L. & Cieslik, K. A. Dissecting the role of myeloid and mesenchymal fibroblasts in age-dependent cardiac fibrosis. Basic Res. Cardiol. 112, 34 (2017).
Cieslik, K. A., Trial, J. & Entman, M. L. Aicar treatment reduces interstitial fibrosis in aging mice: suppression of the inflammatory fibroblast. J. Mol. Cell. Cardiol. 111, 81–85 (2017).
Sundaresan, N. R. et al. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Investig. 119, 2758–2771 (2009).
Orlandi, A., Francesconi, A., Marcellini, M., Ferlosio, A. & Spagnoli, L. G. Role of ageing and coronary atherosclerosis in the development of cardiac fibrosis in the rabbit. Cardiovasc. Res. 64, 544–552 (2004).
Chiao, Y. A. et al. Matrix metalloproteinase-9 deletion attenuates myocardial fibrosis and diastolic dysfunction in ageing mice. Cardiovasc. Res. 96, 444–455 (2012).
Toba, H. et al. Transgenic overexpression of macrophage matrix metalloproteinase-9 exacerbates age-related cardiac hypertrophy, vessel rarefaction, inflammation, and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 312, H375–H383 (2017).
Westermann, D. et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ. Heart Fail. 4, 44–52 (2011).
Wynn, T. A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 (2008).
Ma, Y. et al. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc. Res. 106, 421–431 (2015).
Toba, H. et al. Secreted protein acidic and rich in cysteine facilitates age-related cardiac inflammation and macrophage M1 polarization. Am. J. Physiol. Cell Physiol. 308, C972–C982 (2015).
Mehdizadeh, M., Aguilar, M., Thorin, E., Ferbeyre, G. & Nattel, S. The role of cellular senescence in cardiac disease: basic biology and clinical relevance. Nat. Rev. Cardiol. 19, 250–264 (2022).
Gianopoulos, I. & Daskalopoulou, S. S. Macrophage profiling in atherosclerosis: understanding the unstable plaque. Basic Res. Cardiol. 119, 35–56 (2024).
Bäck, M., Yurdagul, A., Tabas, I., Öörni, K. & Kovanen, P. T. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat. Rev. Cardiol. 16, 389–406 (2019).
Weber, B. N., Giles, J. T. & Liao, K. P. Shared inflammatory pathways of rheumatoid arthritis and atherosclerotic cardiovascular disease. Nat. Rev. Rheumatol. 19, 417–428 (2023).
Kim, K.-W., Ivanov, S. & Williams, J. W. Monocyte recruitment, specification, and function in atherosclerosis. Cells 10, 15 (2020).
Murphy, A. J. et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 121, 4138–4149 (2011).
Tolani, S. et al. Hypercholesterolemia and reduced HDL-C promote hematopoietic stem cell proliferation and monocytosis: studies in mice and FH children. Atherosclerosis 229, 79–85 (2013).
Winter, C. et al. Chrono-pharmacological targeting of the CCL2-CCR2 axis ameliorates atherosclerosis. Cell Metab. 28, 175–182 (2018).
Gilbert, J. et al. Effect of CC chemokine receptor 2 CCR2 blockade on serum C-reactive protein in individuals at atherosclerotic risk and with a single nucleotide polymorphism of the monocyte chemoattractant protein-1 promoter region. Am. J. Cardiol. 107, 906–911 (2011).
van Gils, J. M. et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat. Immunol. 13, 136–143 (2012).
Robbins, C. S. et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 19, 1166–1172 (2013).
Farahi, L., Sinha, S. K. & Lusis, A. J. Roles of macrophages in atherogenesis. Front. Pharmacol. 12, 785220 (2021).
Lin, P., Ji, H., Li, Y.-J. & Guo, S.-D. Macrophage plasticity and atherosclerosis therapy. Front. Mol. Biosci. 8, 679797 (2021).
Doran, A. C. Inflammation resolution: implications for atherosclerosis. Circ. Res. 130, 130–148 (2022).
Poznyak, A. V. et al. Anti-inflammatory therapy for atherosclerosis: focusing on cytokines. Int. J. Mol. Sci. 22, 7061 (2021).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 466, 652–652 (2010).
Hettwer, J. et al. Interleukin-1β suppression dampens inflammatory leucocyte production and uptake in atherosclerosis. Cardiovasc. Res. 118, 2778–2791 (2021).
Abbate, A. et al. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular disease. Circ. Res. 126, 1260–1280 (2020).
Carmi, Y. et al. The role of macrophage-derived IL-1 in induction and maintenance of angiogenesis1. J. Immunol. 183, 4705–4714 (2009).
Kamari, Y. et al. Reduced atherosclerosis and inflammatory cytokines in apolipoprotein-E-deficient mice lacking bone marrow-derived interleukin-1α. Biochem. Biophys. Res. Commun. 405, 197–203 (2011).
Wainstein, M. V. et al. Elevated serum interleukin-6 is predictive of coronary artery disease in intermediate risk overweight patients referred for coronary angiography. Diabetol. Metab. Syndr. 9, 67 (2017).
Tyrrell, D. J. & Goldstein, D. R. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat. Rev. Cardiol. 18, 58–68 (2020).
Goodwin, B. L., Pendleton, L. C., Levy, M. M., Solomonson, L. P. & Eichler, D. C. Tumor necrosis factor-α reduces argininosuccinate synthase expression and nitric oxide production in aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 293, H1115–H1121 (2007).
Boesten, L. S. M. et al. Tumor necrosis factor-alpha promotes atherosclerotic lesion progression in APOE*3-Leiden transgenic mice. Cardiovasc. Res. 66, 179–185 (2005).
Kampschulte, M. et al. Thalidomide influences atherogenesis in aortas of ApoE−/−/LDLR−/− double knockout mice: a nano-CT study. Int. J. Cardiovasc. Imaging 30, 795–802 (2014).
Ohta, H. et al. Disruption of tumor necrosis factor-α gene diminishes the development of atherosclerosis in ApoE-deficient mice. Atherosclerosis 180, 11–17 (2005).
Oberoi, R. et al. Anti-tumor necrosis factor-α therapy increases plaque burden in a mouse model of experimental atherosclerosis. Atherosclerosis 277, 80–89 (2018).
Gerszten, R. E. et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature 398, 718–723 (1999).
Moreno Velásquez, I. et al. Serum IL8 is not associated with cardiovascular events but with all-cause mortality. BMC Cardiovasc. Disord. 19, 34 (2019).
Davenport, P. & Tipping, P. G. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am. J. Pathol. 163, 1117–1125 (2003).
Zhang, X. et al. Interleukin 12 induces T-cell recruitment into the atherosclerotic plaque. Circ. Res. 98, 524–531 (2006).
Jefferis, B. J. et al. Interleukin 18 and coronary heart disease: prospective study and systematic review. Atherosclerosis 217, 227–233 (2011).
Bhat, O. M. et al. Interleukin-18-induced atherosclerosis involves CD36 and NF-κB crosstalk in Apo E−/− mice. J. Cardiol. 66, 28–35 (2015).
Dinarello, C. A., Novick, D., Kim, S. & Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 4, 289 (2013).
Fourman, T. et al. Anti-Inflammatory interleukin 10 inversely relates to coronary atherosclerosis in persons with human immunodeficiency virus. J. Infect. Dis. 221, 510–515 (2020).
Jiang, Y. et al. Deficiency of programmed cell death 4 results in increased IL-10 expression by macrophages and thereby attenuates atherosclerosis in hyperlipidemic mice. Cell. Mol. Immunol. 13, 524–534 (2016).
Han, X. & Boisvert, W. A. Interleukin-10 protects against atherosclerosis by modulating multiple atherogenic macrophage function. Thromb. Haemost. 113, 505–512 (2014).
Han, X., Kitamoto, S., Lian, Q. & Boisvert, W. A. Interleukin-10 facilitates both cholesterol uptake and efflux in macrophages. J. Biol. Chem. 284, 32950–32958 (2009).
Rubic, T. & Lorenz, R. L. Downregulated CD36 and oxLDL uptake and stimulated ABCA1/G1 and cholesterol efflux as anti-atherosclerotic mechanisms of interleukin-10. Cardiovasc. Res. 69, 527–535 (2006).
Panousis, C., Evans, G. F. & Zuckerman, S. H. TGF-β increases cholesterol efflux and ABC-1 expression in macrophage-derived foam cells: opposing the effects of IFN-γ. J. Lipid Res. 42, 856–863 (2001).
Mallat, Z. et al. Inhibition of transforming growth factor-beta signaling accelerates atherosclerosis and induces an unstable plaque phenotype in mice. Circ. Res. 89, 930–934 (2001).
Low, E. L., Baker, A. H. & Bradshaw, A. C. TGFβ, smooth muscle cells and coronary artery disease: a review. Cell. Signal. 53, 90–101 (2019).
de Jager, S. C. A. et al. Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis. J. Exp. Med 208, 217–225 (2011).
Chen, P. Y. et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 1, 912–926 (2019).
Guo, L. et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investig. 128, 1106–1124 (2018).
Pourcet, B. & Staels, B. Alternative macrophages in atherosclerosis: not always protective! J. Clin. Investig. 128, 910–912 (2018).
van Tits, L. J. H. et al. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: a crucial role for Krüppel-like factor 2. Atherosclerosis 214, 345–349 (2011).
Maguire, E. M., Pearce, S. W. A. & Xiao, Q. Foam cell formation: a new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 112, 54–71 (2019).
Kim, K. et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ. Res. 123, 1127–1142 (2018).
Manning-Tobin, J. J. et al. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler. Thromb. Vasc. Biol. 29, 19–26 (2009).
Mäkinen, P. I. et al. Silencing of either SR-A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc. Res. 88, 530–538 (2010).
Febbraio, M., Guy, E. C. & Silverstein, R. L. Stem cell transplantation reveals that absence of macrophage CD36 is protective against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 2333–2338 (2004).
Chen, Y., Zhang, J., Cui, W. & Silverstein, R. L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 219, e20211314 (2022).
Inoue, K., Arai, Y., Kurihara, H., Kita, T. & Sawamura, T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E–null mice. Circ. Res. 97, 176–184 (2005).
Mehta, J. L. et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ. Res. 100, 1634–1642 (2007).
Akhmedov, A. et al. Lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1): a crucial driver of atherosclerotic cardiovascular disease. Eur. Heart J. 42, 1797–1807 (2021).
Hoebe, K. et al. CD36 is a sensor of diacylglycerides. Nature 433, 523–527 (2005).
Gao, J.-H. et al. CXCL12 promotes atherosclerosis by downregulating ABCA1 expression via the CXCR4/GSK3β/β-cateninT120/TCF21 pathway. J. Lipid Res. 60, 2020–2033 (2019).
Xia, X.-D. et al. Myocardin suppression increases lipid retention and atherosclerosis via downregulation of ABCA1 in vascular smooth muscle cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866, 158824 (2021).
Zhao, Y.-Y. et al. Hypocholesterolemia, foam cell accumulation, but no atherosclerosis in mice lacking ABC-transporter A1 and scavenger receptor BI. Atherosclerosis 218, 314–322 (2011).
Demetz, E. et al. Inhibition of hepatic scavenger receptor-class B type I by RNA interference decreases atherosclerosis in rabbits. Atherosclerosis 222, 360–366 (2012).
Zhao, Y. et al. Enhanced foam cell formation, atherosclerotic lesion development, and inflammation by combined deletion of ABCA1 and SR-BI in Bone marrow-derived cells in LDL receptor knockout mice on western-type diet. Circ. Res. 107, e20–e31 (2010).
Meurs, I. M. et al. The effect of ABCG1 deficiency on atherosclerotic lesion development in LDL receptor knockout mice depends on the stage of atherogenesis. Atherosclerosis 221, 41–47 (2012).
van Eck, M. et al. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am. J. Pathol. 165, 785–794 (2004).
Boyle, J. J. et al. Activating transcription factor 1 directs mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ. Res. 110, 20–33 (2012).
Saeed, O. S. et al. Pharmacologic suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 299–307 (2012).
Li, J. J. et al. Hepcidin destabilizes atherosclerotic plaque via overactivating macrophages after erythrophagocytosis. Arterioscler. Thromb. Vasc. Biol. 32, 1158–1166 (2012).
Yurdagul, A. Metabolic consequences of efferocytosis and its impact on atherosclerosis. Immunometabolism 3, e210017 (2021).
Kojima, Y., Weissman, I. L. & Leeper, N. J. The role of efferocytosis in atherosclerosis. Circulation 135, 476–489 (2017).
Singh, B. et al. Defective efferocytosis of vascular cells in heart disease. Front. Cardiovasc. Med. 9, 1031293 (2022).
Tajbakhsh, A. et al. Autoantigen-specific immune tolerance in pathological and physiological cell death: Nanotechnology comes into view. Int. Immunopharmacol. 90, 107177 (2020).
Tao, H. et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J. Lipid Res. 56, 1449–1460 (2015).
Chinetti-Gbaguidi, G. et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPAR&ggr; and LXR&agr; pathways. Circ. Res. 108, 985–995 (2011).
Zizzo, G., Hilliard, B., Monestier, M. & Cohen, P. L. Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J. Immunol. 189, 3508–3520 (2012).
Cai, B. et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J. Clin. Investig. 127, 564–568 (2017).
Schrijvers, D. M., De Meyer, G. R. Y., Kockx, M. M., Herman, A. G. & Martinet, W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 25, 1256–1261 (2005).
González, L. & Trigatti, B. L. Macrophage apoptosis and necrotic core development in atherosclerosis: a rapidly advancing field with clinical relevance to imaging and therapy. Can. J. Cardiol. 33, 303–312 (2017).
Jinnouchi, H. et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell. Mol. Life Sci. 77, 1919–1932 (2019).
Yurdagul, A., Doran, A. C., Cai, B., Fredman, G. & Tabas, I. A. Mechanisms and consequences of defective efferocytosis in atherosclerosis. Front. Cardiovasc. Med. 4, 86 (2018).
Dhawan, U. K., Singhal, A. & Subramanian, M. Dead cell and debris clearance in the atherosclerotic plaque: mechanisms and therapeutic opportunities to promote inflammation resolution. Pharmacol. Res. 170, 105699 (2021).
Yin, C. et al. Efferocytic defects in early atherosclerosis are driven by GATA2 overexpression in macrophages. Front. Immunol. 11, 594136 (2020).
Wang, Y. C. et al. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell 171, 331–345.e22 (2017).
Chen, L. et al. Platelet membrane-coated nanocarriers targeting plaques to deliver anti-CD47 antibody for atherosclerotic therapy. Research 2022, 9845459 (2022).
Kojima, Y. et al. CD47 blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 536, 86–90 (2016).
Singla, B. et al. Loss of myeloid cell-specific SIRPα, but not CD47, attenuates inflammation and suppresses atherosclerosis. Cardiovasc. Res. 118, 3097–3111 (2021).
Otsuka, F., Sakakura, K., Yahagi, K., Joner, M. D. & Virmani, R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler. Thromb. Vasc. Biol. 34, 724–736 (2014).
Reith, S., Milzi, A., Dettori, R., Marx, N. & Burgmaier, M. Predictors for target lesion microcalcifications in patients with stable coronary artery disease: an optical coherence tomography study. Clin. Res. Cardiol. 107, 763–771 (2018).
Burgmaier, M. et al. Co-localization of plaque macrophages with calcification is associated with a more vulnerable plaque phenotype and a greater calcification burden in coronary target segments as determined by OCT. PLoS One 13, e0205984 (2018).
Shioi, A. & Ikari, Y. Plaque calcification during atherosclerosis progression and regression. J. Atheroscler. Thromb. 25, 294–303 (2017).
Nadra, I. J. et al. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ. Res. 96, 1248–1256 (2005).
Shioi, A. et al. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: roles of tumor necrosis factor-&agr; and oncostatin M derived from macrophages. Circ. Res. 91, 9–16 (2002).
Ceneri, N. M. et al. Rac2 modulates atherosclerotic calcification by regulating macrophage interleukin-1&bgr; production. Arterioscler. Thromb. Vasc. Biol. 37, 328–340 (2017).
Kakutani, Y. et al. Oncostatin M promotes osteoblastic differentiation of human vascular smooth muscle cells through JAK3‐STAT3 pathway. J. Cell. Biochem. 116, 1325–1333 (2015).
Williams, J. W. et al. Limited proliferation capacity of aorta intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 21, 1194–1204 (2020).
Ivan, E. et al. Expansive arterial remodeling is associated with increased neointimal macrophage foam cell content: the murine model of macrophage-rich carotid artery lesions. Circulation 105, 2686–2691 (2002).
Peeters, W. et al. Collagenase matrix metalloproteinase-8 expressed in atherosclerotic carotid plaques is associated with systemic cardiovascular outcome. Eur. Heart J. 32, 2314–2325 (2010).
Scholtes, V. P. W. et al. Carotid atherosclerotic plaque matrix metalloproteinase-12–positive macrophage subpopulation predicts adverse outcome after endarterectomy. J. Am. Heart Assoc. 1, e001040 (2012).
Newby, A. C. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol. Rev. 85, 1–31 (2005).
Newby, A. C. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler. Thromb. Vasc. Biol. 28, 2108–2114 (2008).
Huang, W.-C., Sala-Newby, G. B., Susana, A., Johnson, J. L. & Newby, A. C. Classical macrophage activation up-regulates several matrix metalloproteinases through mitogen activated protein kinases and nuclear factor-κB. PLoS One 7, e42507 (2012).
Erbel, C. et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. 21, 255–265 (2015).
Liu, S.-l et al. Cardiovascular protection in females linked to estrogen-dependent inhibition of arterial stiffening and macrophage MMP12. JCI insight 4, e122742 (2019).
Komukai, K. et al. Effect of atorvastatin therapy on fibrous cap thickness in coronary atherosclerotic plaque as assessed by optical coherence tomography: the EASY-FIT study. J. Am. Coll. Cardiol. 64, 2207–2217 (2014).
Cardilo-Reis, L. et al. Interleukin-13 protects from atherosclerosis and modulates plaque composition by skewing the macrophage phenotype. EMBO Mol. Med. 4, 1072–1086 (2012).
Khallou-Laschet, J. et al. Macrophage plasticity in experimental atherosclerosis. PLoS One 5, e8852 (2010).
Bi, Y. et al. M2 macrophages as a potential target for antiatherosclerosis treatment. Neural Plast. 2019, 6724903 (2019).
Josefs, T. et al. Atherosclerosis regression and cholesterol efflux in hypertriglyceridemic mice. Circ. Res. 128, 690–705 (2021).
Barrett, T. J. Macrophages in atherosclerosis regression. Arterioscler. Thromb. Vasc. Biol. 40, 20–33 (2019).
Visseren, F. L. J. et al. 2021 ESC guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 42, 3227–3337 (2021).
Härdtner, C. et al. Inhibition of macrophage proliferation dominates plaque regression in response to cholesterol lowering. Basic Res. Cardiol. 115, 78 (2020).
Tang, J. et al. Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci. Adv. 1, e1400223 (2015).
Llodrá, J. et al. Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl Acad. Sci. USA 101, 11779–11784 (2004).
Feig, J. E. et al. Statins promote the regression of atherosclerosis via activation of the CCR7-dependent emigration pathway in macrophages. PLoS One 6, e28534 (2011).
Trogan, E. et al. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl Acad. Sci. USA 103, 3781–3786 (2006).
Mueller, P. A. et al. Deletion of macrophage low-density lipoprotein receptor-related protein 1 (LRP1) accelerates atherosclerosis regression and increases C-C chemokine receptor type 7 (CCR7) expression in plaque macrophages. Circulation 138, 1850–1863 (2018).
Ramsey, S. A. et al. Epigenome-guided analysis of the transcriptome of plaque macrophages during atherosclerosis regression reveals activation of the wnt signaling pathway. PLoS Genet 10, e1004828 (2014).
Wang, F. et al. Myeloid β-catenin deficiency exacerbates atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 38, 1468–1478 (2018).
Wanschel, A. C. B. A. et al. Neuroimmune guidance cue semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler. Thromb. Vasc. Biol. 33, 886–893 (2013).
Potteaux, S. et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J. Clin. Investig. 121, 2025–2036 (2011).
Feig, J. E. et al. Regression of atherosclerosis is characterized by broad changes in the plaque macrophage transcriptome. PLoS One 7, e39790 (2012).
Sharma, M. et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ. Res. 127, 335–353 (2020).
Rayner, K. J. et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 121, 2921–2931 (2011).
Ouimet, M. et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Investig. 125, 4334–4348 (2015).
Feig, J. E. et al. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc. Natl Acad. Sci. USA 108, 7166–7171 (2011).
Barrett, T. J. et al. Apolipoprotein AI) promotes atherosclerosis regression in diabetic mice by suppressing myelopoiesis and plaque inflammation. Circulation 140, 1170–1184 (2019).
Sanson, M., Distel, E. & Fisher, E. A. HDL induces the expression of the M2 macrophage markers arginase 1 and Fizz-1 in a STAT6-dependent process. PLoS One 8, e74676 (2013).
Sha, H., Zhang, D., Zhang, Y., Wen, Y. & Wang, Y. ATF3 promotes migration and M1/M2 polarization of macrophages by activating tenascin‑C via Wnt/β‑catenin pathway. Mol. Med. Rep. 16, 3641–3647 (2017).
Cho, M. J., Lee, M.-R. & Park, J.-G. Aortic aneurysms: current pathogenesis and therapeutic targets. Exp. Mol. Med. 55, 2519–2530 (2023).
Yuan, Z. et al. Abdominal aortic aneurysm: roles of inflammatory cells. Front. Immunol. 11, 609161 (2021).
Boytard, L. et al. Role of proinflammatory CD68(+) mannose receptor(−) macrophages in peroxiredoxin-1 expression and in abdominal aortic aneurysms in humans. Arterioscler. Thromb. Vasc. Biol. 33, 431–438 (2013).
Mellak, S. et al. Angiotensin II mobilizes spleen monocytes to promote the development of abdominal aortic aneurysm in Apoe-/- mice. Arterioscler. Thromb. Vasc. Biol. 35, 378–388 (2015).
Ishibashi, M. et al. Bone marrow-derived monocyte chemoattractant protein-1 receptor CCR2 is critical in angiotensin II-induced acceleration of atherosclerosis and aneurysm formation in hypercholesterolemic mice. Arterioscler. Thromb. Vasc. Biol. 24, e174–e178 (2004).
Zhang, J. et al. Chemokine (C-C motif) receptor 2 mediates mast cell migration to abdominal aortic aneurysm lesions in mice. Cardiovasc. Res. 96, 543–551 (2012).
Boytard, L. et al. Lung-derived HMGB1 is detrimental for vascular remodeling of metabolically imbalanced arterial macrophages. Nat. Commun. 11, 4311 (2020).
McCormick, M. L., Gavrila, D. & Weintraub, N. L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 27, 461–469 (2007).
Raffort, J. et al. Monocytes and macrophages in abdominal aortic aneurysm. Nat. Rev. Cardiol. 14, 457–471 (2017).
Thomas, M. et al. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation 114, 404–413 (2006).
Wang, K. C. et al. Membrane-bound thrombomodulin regulates macrophage inflammation in abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 35, 2412–2422 (2015).
Sun, W. et al. Macrophage inflammasome mediates hyperhomocysteinemia-aggravated abdominal aortic aneurysm. J. Mol. Cell. Cardiol. 81, 96–106 (2015).
Ju, X. et al. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler. Thromb. Vasc. Biol. 33, 1612–1621 (2013).
Batra, R. et al. IL-1β (interleukin-1β) and TNF-α (Tumor Necrosis Factor-α) impact abdominal aortic aneurysm formation by differential effects on macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 38, 457–463 (2018).
Zhang, Y. et al. S-Nitrosylation of Septin2 exacerbates aortic aneurysm and dissection by coupling the TIAM1-RAC1 axis in macrophages. Circulation https://doi.org/10.1161/CIRCULATIONAHA.123.066404 (2024).
Cai, D., Sun, C., Murashita, T., Que, X. & Chen, S. Y. ADAR1 non-editing function in macrophage activation and abdominal aortic aneurysm. Circ. Res. 132, e78–e93 (2023).
Wenjing, F. et al. The role of IL-1β in aortic aneurysm. Clin. Chim. Acta 504, 7–14 (2020).
Wu, D. et al. NLRP3 (nucleotide oligomerization domain-like receptor family, pyrin domain containing 3)-caspase-1 inflammasome degrades contractile proteins: implications for aortic biomechanical dysfunction and aneurysm and dissection formation. Arterioscler. Thromb. Vasc. Biol. 37, 694–706 (2017).
Tazume, H. et al. Macrophage-derived angiopoietin-like protein 2 accelerates development of abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 32, 1400–1409 (2012).
Fu, M. & Song, J. Single-cell transcriptomics reveals the cellular heterogeneity of cardiovascular diseases. Front. Cardiovasc. Med. 8, 643519 (2021).
Li, B. et al. Single-cell transcriptome profiles reveal fibrocytes as potential targets of cell therapies for abdominal aortic aneurysm. Front. Cardiovasc. Med. 8, 753711 (2021).
Li, Y. et al. Single-cell transcriptome analysis reveals dynamic cell populations and differential gene expression patterns in control and aneurysmal human aortic tissue. Circulation 142, 1374–1388 (2020).
Anzai, A. et al. Adventitial CXCL1/G-CSF expression in response to acute aortic dissection triggers local neutrophil recruitment and activation leading to aortic rupture. Circ. Res. 116, 612–623 (2015).
Tieu, B. C. et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Invest. 119, 3637–3651 (2009).
Gao, J.-P. & Guo, W. Mechanisms of abdominal aortic aneurysm progression: a review. Vasc. Med. 27, 88–96 (2021).
Rateri, D. L. et al. Prolonged infusion of angiotensin II in apoE−/− mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am. J. Pathol. 179, 1542–1548 (2011).
Hwang, J. S. et al. PPARδ reduces abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice by regulating extracellular matrix homeostasis and inflammatory responses. Int. J. Cardiol. 174, 43–50 (2014).
Son, B. K. et al. Granulocyte macrophage colony-stimulating factor is required for aortic dissection/intramural haematoma. Nat. Commun. 6, 6994 (2015).
Sharma, N. et al. Deficiency of IL12p40 (Interleukin 12 p40) promotes Ang II (Angiotensin II)-induced abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 39, 212–223 (2019).
Pyo, R. et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J. Clin. Investig. 105, 1641–1649 (2000).
Longo, G. M. et al. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Investig. 110, 625–632 (2002).
Luo, W. et al. Critical role of cytosolic DNA and its sensing adaptor STING in aortic degeneration, dissection, and rupture. Circulation 141, 42–66 (2020).
Saraff, K., Babamusta, F., Cassis, L. A. & Daugherty, A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 23, 1621–1626 (2003).
Rizas, K. D., Ippagunta, N. & Tilson, M. D. 3rd Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol. Rev. 17, 201–210 (2009).
Findeisen, H. M. et al. Telomerase deficiency in bone marrow-derived cells attenuates angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 31, 253–260 (2011).
Wang, Y. et al. Involvement of macrophage-derived exosomes in abdominal aortic aneurysms development. Atherosclerosis 289, 64–72 (2019).
Sho, E. et al. Hemodynamic forces regulate mural macrophage infiltration in experimental aortic aneurysms. Exp. Mol. Pathol. 76, 108–116 (2004).
Nakahashi, T. K. et al. Flow loading induces macrophage antioxidative gene expression in experimental aneurysms. Arterioscler. Thromb. Vasc. Biol. 22, 2017–2022 (2002).
Kiema, M. et al. Wall shear stress predicts media degeneration and biomechanical changes in thoracic aorta. Front. Physiol. 13, 934941 (2022).
Márquez-Sánchez, A. C. & Koltsova, E. K. Immune and inflammatory mechanisms of abdominal aortic aneurysm. Front. Immunol. 13, 989933 (2022).
Barp, C. G., Bonaventura, D. & Assreuy, J. NO, ROS, RAS, and PVAT: more than a soup of letters. Front. Physiol. 12, 640021 (2021).
Police, S. B., Thatcher, S. E., Charnigo, R., Daugherty, A. & Cassis, L. A. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 29, 1458–1464 (2009).
Thanassoulis, G. et al. Periaortic adipose tissue and aortic dimensions in the Framingham Heart Study. J. Am. Heart Assoc. 1, e000885 (2012).
Michel, J. B. et al. Novel aspects of the pathogenesis of aneurysms of the abdominal aorta in humans. Cardiovasc. Res. 90, 18–27 (2010).
Miranda, A. M. A. et al. Single-cell transcriptomics for the assessment of cardiac disease. Nat. Rev. Cardiol. 20, 289–308 (2023).
Li, L. et al. Single-cell transcriptome sequencing of macrophages in common cardiovascular diseases. J. Leukoc. Biol. 113, 139–148 (2023).
Longo, S. K., Guo, M. G., Ji, A. L. & Khavari, P. A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 22, 627–644 (2021).
Jung, S.-H. et al. Spatiotemporal dynamics of macrophage heterogeneity and a potential function of Trem2hi macrophages in infarcted hearts. Nat. Commun. 13, 4580 (2022).
Rizzo, G. et al. Dynamics of monocyte-derived macrophage diversity in experimental myocardial infarction. Cardiovasc. Res. 119, 772–785 (2023).
Jin, K. et al. Single-cell RNA sequencing reveals the temporal diversity and dynamics of cardiac immunity after myocardial infarction. Small Methods 6, e2100752 (2022).
Kim, S. H., Lee, K. Y. & Chang, K. The protective role of TREM2 in the heterogenous population of macrophages during post-myocardial infarction inflammation. Int. J. Mol. Sci. 24, 5556 (2023).
King, K. R. et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23, 1481–1487 (2017).
Jung, S. H. et al. Spatiotemporal dynamics of macrophage heterogeneity and a potential function of Trem2(hi) macrophages in infarcted hearts. Nat. Commun. 13, 4580 (2022).
Zhang, K. et al. TREM2(hi) resident macrophages protect the septic heart by maintaining cardiomyocyte homeostasis. Nat. Metab. 5, 129–146 (2023).
Rao, M. et al. Resolving the intertwining of inflammation and fibrosis in human heart failure at single-cell level. Basic Res. Cardiol. 116, 55 (2021).
Smart, C. D. et al. Immune profiling of murine cardiac leukocytes identifies Trem2 as a novel mediator of hypertensive heart failure. Cardiovasc. Res. 119, 2312–2328 (2023).
Hulsmans, M. et al. Recruited macrophages elicit atrial fibrillation. Science 381, 231–239 (2023).
Fu, M. et al. Single-cell RNA sequencing in donor and end-stage heart failure patients identifies NLRP3 as a therapeutic target for arrhythmogenic right ventricular cardiomyopathy. BMC Med. 22, 11 (2024).
Amrute, J. M. et al. Defining cardiac functional recovery in end-stage heart failure at single-cell resolution. Nat. Cardiovasc. Res. 2, 399–416 (2023).
Xu, Y. et al. A transient wave of Bhlhe41+ resident macrophages enables remodeling of the developing infarcted myocardium. Cell Rep. 42, 113174 (2023).
Zernecke, A. et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ. Res. 127, 402–426 (2020).
Depuydt, M. A. C. et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ. Res. 127, 1437–1455 (2020).
Cochain, C. et al. Single-cell RNA-seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ. Res. 122, 1661–1674 (2018).
Winkels, H. et al. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ. Res. 122, 1675–1688 (2018).
Fernandez, D. M. et al. Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 25, 1576–1588 (2019).
Fernandez, D. M. & Giannarelli, C. Immune cell profiling in atherosclerosis: role in research and precision medicine. Nat. Rev. Cardiol. 19, 43–58 (2021).
Willemsen, L. & de Winther, M. P. J. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol. 250, 705–714 (2020).
Zhao, G. et al. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc. Res. 117, 1402–1416 (2021).
Boytard, L. et al. Role of proinflammatory CD68(+) mannose receptor(-) macrophages in peroxiredoxin-1 expression and in abdominal aortic aneurysms in humans. Arterioscler. Thromb. Vasc. Biol. 33, 431–438 (2013).
Lin, J.-D. et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. Jci. Insight 4, e124574 (2019).
Hu, Z. et al. Single-cell transcriptomic atlas of different human cardiac arteries identifies cell types associated with vascular physiology. Arterioscler. Thromb. Vasc. Biol. 41, 1408–1427 (2021).
Lim, H. Y. et al. Hyaluronan receptor LYVE-1-expressing macrophages maintain arterial tone through hyaluronan-mediated regulation of smooth muscle cell collagen. Immunity 49, 326–341.e7 (2018).
Wu, H. et al. Comparative analysis of thoracic and abdominal aortic aneurysms across the segment and species at the single-cell level. Front. Pharmacol. 13, 1095757 (2023).
Reardon, C. A. et al. Obesity and insulin resistance promote atherosclerosis through an IFNγ-regulated macrophage protein network. Cell Rep. 23, 3021–3030 (2018).
Leipner, J. et al. Myeloid cell-specific Irf5 deficiency stabilizes atherosclerotic plaques in Apoe–/– mice. Mol. Metab. 53, 101250 (2021).
Edsfeldt, A. et al. Interferon regulatory factor-5-dependent CD11c+ macrophages contribute to the formation of rupture-prone atherosclerotic plaques. Eur. Heart J. 43, 1864–1877 (2022).
Wang, Y., Wang, Q. & Xu, D. New insights into macrophage subsets in atherosclerosis. J. Mol. Med. 100, 1239–1251 (2022).
Lin, C.-S. et al. The CCL5/CCR5 axis promotes vascular smooth muscle cell proliferation and atherogenic phenotype switching. Cell. Physiol. Biochem. 47, 707–720 (2018).
Schlegel, M. et al. Silencing myeloid netrin-1 induces inflammation resolution and plaque regression. Circ. Res. 129, 530–546 (2021).
Hadi, T. et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat. Commun. 9, 5022 (2018).
Afonso, M. S. et al. miR-33 silencing reprograms the immune cell landscape in atherosclerotic plaques. Circ. Res. 128, 1122–1138 (2021).
Zhang, X. et al. Targeted suppression of miRNA-33 using pHLIP improves atherosclerosis regression. Circ. Res. 131, 77–90 (2022).
Flores, A. M. et al. Pro-efferocytic nanoparticles are specifically taken up by lesional macrophages and prevent atherosclerosis. Nat. Nanotechnol. 15, 154–161 (2020).
Wu, X. & Zhang, H. Omics approaches unveiling the biology of human atherosclerotic plaques. Am. J. Pathol. 194, 482–498 (2024).
Schneider, M. et al. Combined near infrared photoacoustic imaging and ultrasound detects vulnerable atherosclerotic plaque. Biomaterials 302, 122314 (2023).
Sun, J. et al. Spatial transcriptional mapping reveals site-specific pathways underlying human atherosclerotic plaque rupture. J. Am. Coll. Cardiol. 81, 2213–2227 (2023).
Mentkowski, K. I., Euscher, L. M., Patel, A., Alevriadou, B. R. & Lang, J. K. Monocyte recruitment and fate specification after myocardial infarction. Am. J. Physiol. Cell Physiol. 319, C797–c806 (2020).
Muller, W. A. Mechanisms of transendothelial migration of leukocytes. Circ. Res. 105, 223–230 (2009).
Chen, W. et al. Macrophage-targeted nanomedicine for the diagnosis and treatment of atherosclerosis. Nat. Rev. Cardiol. 19, 228–249 (2022).
Georgakis, M. K., Bernhagen, J., Heitman, L. H., Weber, C. & Dichgans, M. Targeting the CCL2-CCR2 axis for atheroprotection. Eur. Heart J. 43, 1799–1808 (2022).
Boring, L., Gosling, J., Cleary, M. & Charo, I. F. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 394, 894–897 (1998).
Gu, L. et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 2, 275–281 (1998).
Gosling, J. et al. MCP-1 deficiency reduces susceptibility to atherosclerosis in mice that overexpress human apolipoprotein B. J. Clin. Invest. 103, 773–778 (1999).
Aiello, R. J. et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 19, 1518–1525 (1999).
Combadière, C. et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657 (2008).
Saederup, N., Chan, L., Lira, S. A. & Charo, I. F. Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2-/- mice: evidence for independent chemokine functions in atherogenesis. Circulation 117, 1642–1648 (2008).
Pickett, J. R., Wu, Y., Zacchi, L. F. & Ta, H. T. Targeting endothelial vascular cell adhesion molecule-1 in atherosclerosis: drug discovery and development of vascular cell adhesion molecule-1-directed novel therapeutics. Cardiovasc. Res. 119, 2278–2293 (2023).
Lee, S. K. et al. PAR4 inhibition reduces coronary artery atherosclerosis and myocardial fibrosis in SR-B1/LDLR double knockout mice. Arterioscler. Thromb. Vasc. Biol. 43, 2165–2178 (2023).
Imanparast, F. et al. Potential of mZD7349-conjugated PLGA nanoparticles for selective targeting of vascular cell-adhesion molecule-1 in inflamed endothelium. Microvasc. Res. 106, 110–116 (2016).
Ostermann, G. et al. Involvement of JAM-A in mononuclear cell recruitment on inflamed or atherosclerotic endothelium: inhibition by soluble JAM-A. Arterioscler. Thromb. Vasc. Biol. 25, 729–735 (2005).
Zernecke, A., Bernhagen, J. & Weber, C. Macrophage migration inhibitory factor in cardiovascular disease. Circulation 117, 1594–1602 (2008).
Sinitski, D. et al. Macrophage migration inhibitory factor (MIF)-based therapeutic concepts in atherosclerosis and inflammation. Thromb. Haemost. 119, 553–566 (2019).
Burger-Kentischer, A. et al. Reduction of the aortic inflammatory response in spontaneous atherosclerosis by blockade of macrophage migration inhibitory factor (MIF). Atherosclerosis 184, 28–38 (2006).
Xu, Y. J., Zheng, L., Hu, Y. W. & Wang, Q. Pyroptosis and its relationship to atherosclerosis. Clin. Chim. Acta 476, 28–37 (2018).
Yu, X. H., Fu, Y. C., Zhang, D. W., Yin, K. & Tang, C. K. Foam cells in atherosclerosis. Clin. Chim. Acta 424, 245–252 (2013).
Kunjathoor, V. V. et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 277, 49982–49988 (2002).
McLaren, J. E., Michael, D. R., Ashlin, T. G. & Ramji, D. P. Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog. Lipid Res. 50, 331–347 (2011).
Chistiakov, D. A., Bobryshev, Y. V. & Orekhov, A. N. Macrophage-mediated cholesterol handling in atherosclerosis. J. Cell. Mol. Med. 20, 17–28 (2016).
Perrey, S. et al. Preferential pharmacological inhibition of macrophage ACAT increases plaque formation in mouse and rabbit models of atherogenesis. Atherosclerosis 155, 359–370 (2001).
Yang, L. et al. Enhancement of human ACAT1 gene expression to promote the macrophage-derived foam cell formation by dexamethasone. Cell Res. 14, 315–323 (2004).
Li, A. C. et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARalpha, beta/delta, and gamma. J. Clin. Investig. 114, 1564–1576 (2004).
Sun, K., Li, Y. Y. & Jin, J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct. Target. Ther. 6, 79 (2021).
De Meyer, G. R. Y., Zurek, M., Puylaert, P. & Martinet, W. Programmed death of macrophages in atherosclerosis: mechanisms and therapeutic targets. Nat. Rev. Cardiol. https://doi.org/10.1038/s41569-023-00957-0 (2024).
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T. & Kroemer, G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 (2010).
Shi, J., Gao, W. & Shao, F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254 (2017).
Liu, Y. & Levine, B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 22, 367–376 (2015).
Stockwell, B. R. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Robinson, N. et al. Programmed necrotic cell death of macrophages: focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 26, 101239 (2019).
Majno, G. & Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 146, 3–15 (1995).
Sansonetti, M., Al Soodi, B., Thum, T. & Jung, M. Macrophage-based therapeutic approaches for cardiovascular diseases. Basic Res. Cardiol. 119, 1–33 (2024).
De Meyer, I., Martinet, W. & De Meyer, G. R. Therapeutic strategies to deplete macrophages in atherosclerotic plaques. Br. J. Clin. Pharmacol. 74, 246–263 (2012).
Shoulders, H., Garner, K. H. & Singla, D. K. Macrophage depletion by clodronate attenuates bone morphogenetic protein-7 induced M2 macrophage differentiation and improved systolic blood velocity in atherosclerosis. Transl. Res. 203, 1–14 (2019).
Secchiero, P. et al. Systemic tumor necrosis factor-related apoptosis-inducing ligand delivery shows antiatherosclerotic activity in apolipoprotein E-null diabetic mice. Circulation 114, 1522–1530 (2006).
Karunakaran, D. et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2, e1600224 (2016).
Luo, X. et al. MCL attenuates atherosclerosis by suppressing macrophage ferroptosis via targeting KEAP1/NRF2 interaction. Redox Biol. 69, 102987 (2024).
Xu, J., Han, X., Xia, N., Zhao, Q. & Cheng, Z. IL‑37 suppresses macrophage ferroptosis to attenuate diabetic atherosclerosis via the NRF2 pathway. Exp. Ther. Med. 25, 289 (2023).
Verheye, S. et al. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J. Am. Coll. Cardiol. 49, 706–715 (2007).
Wang, X. et al. Knockdown of mTOR by lentivirus‑mediated RNA interference suppresses atherosclerosis and stabilizes plaques via a decrease of macrophages by autophagy in apolipoprotein E‑deficient mice. Int. J. Mol. Med. 32, 1215–1221 (2013).
Zhai, C. et al. Selective inhibition of PI3K/Akt/mTOR signaling pathway regulates autophagy of macrophage and vulnerability of atherosclerotic plaque. PLoS One 9, e90563 (2014).
van Amerongen, M. J., Harmsen, M. C., van Rooijen, N., Petersen, A. H. & van Luyn, M. J. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am. J. Pathol. 170, 818–829 (2007).
Frantz, S. et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 27, 871–881 (2013).
Ishikawa, S. et al. Apoptosis inhibitor of macrophage depletion decreased M1 macrophage accumulation and the incidence of cardiac rupture after myocardial infarction in mice. PLoS One 12, e0187894 (2017).
Sager, H. B. et al. Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation 132, 1880–1890 (2015).
Abbate, A. et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117, 2670–2683 (2008).
Han, J. et al. Dual roles of graphene oxide to attenuate inflammation and elicit timely polarization of macrophage phenotypes for cardiac repair. ACS Nano. 12, 1959–1977 (2018).
Wang, D. et al. Dual delivery of an NF-κB inhibitor and IL-10 through supramolecular hydrogels polarizes macrophages and promotes cardiac repair after myocardial infarction. Acta Biomater. 164, 111–123 (2023).
Chen, W. et al. A matrix-metalloproteinase-responsive hydrogel system for modulating the immune microenvironment in myocardial infarction. Adv. Mater. 35, e2209041 (2023).
Forsberg, M. H., Kink, J. A., Hematti, P. & Capitini, C. M. Mesenchymal stromal cells and exosomes: progress and challenges. Front. Cell Dev. Biol. 8, 665 (2020).
Shao, L. et al. Inflammation in myocardial infarction: roles of mesenchymal stem cells and their secretome. Cell Death Discov. 8, 452 (2022).
Liao, Y. et al. Cardiac nestin(+) mesenchymal stromal cells enhance healing of ischemic heart through periostin-mediated M2 macrophage polarization. Mol. Ther. 28, 855–873 (2020).
Podaru, M. N. et al. Reparative macrophage transplantation for myocardial repair: a refinement of bone marrow mononuclear cell-based therapy. Basic Res. Cardiol. 114, 34 (2019).
Zhu, Y. et al. Hypoxia-primed monocytes/macrophages enhance postinfarction myocardial repair. Theranostics 12, 307–323 (2022).
Rizzacasa, B., Amati, F., Romeo, F., Novelli, G. & Mehta, J. L. Epigenetic modification in coronary atherosclerosis: JACC review topic of the week. J. Am. Coll. Cardiol. 74, 1352–1365 (2019).
Caescu, C. I. et al. Colony stimulating factor-1 receptor signaling networks inhibit mouse macrophage inflammatory responses by induction of microRNA-21. Blood 125, e1–e13 (2015).
Jin, H. et al. Local delivery of miR-21 stabilizes fibrous caps in vulnerable atherosclerotic lesions. Mol. Ther. 26, 1040–1055 (2018).
Wei, Y. et al. Regulation of Csf1r and Bcl6 in macrophages mediates the stage-specific effects of microRNA-155 on atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 35, 796–803 (2015).
Price, N. L. et al. Genetic dissection of the impact of miR-33a and miR-33b during the progression of atherosclerosis. Cell Rep. 21, 1317–1330 (2017).
Hu, Y. W. et al. An agomir of miR-144-3p accelerates plaque formation through impairing reverse cholesterol transport and promoting pro-inflammatory cytokine production. PLoS One 9, e94997 (2014).
Wang, Y. et al. Macrophage-targeting gene silencing orchestrates myocardial microenvironment remodeling toward the anti-inflammatory treatment of ischemia-reperfusion (IR) injury. Bioact. Mater. 17, 320–333 (2022).
Li, Y. et al. Injectable hydrogel with MSNs/microRNA-21-5p delivery enables both immunomodification and enhanced angiogenesis for myocardial infarction therapy in pigs. Sci. Adv. 7, eabd6740 (2021).
Gabunia, K. et al. IL-19 halts progression of atherosclerotic plaque, polarizes, and increases cholesterol uptake and efflux in macrophages. Am. J. Pathol. 186, 1361–1374 (2016).
Dragoljevic, D. et al. Inhibition of interleukin-1β signalling promotes atherosclerotic lesion remodelling in mice with inflammatory arthritis. Clin. Transl. Immunol. 9, e1206 (2020).
Bhaskar, V. et al. Monoclonal antibodies targeting IL-1 beta reduce biomarkers of atherosclerosis in vitro and inhibit atherosclerotic plaque formation in apolipoprotein E-deficient mice. Atherosclerosis 216, 313–320 (2011).
Lee, T. M., Chang, N. C. & Lin, S. Z. Dapagliflozin, a selective SGLT2 Inhibitor, attenuated cardiac fibrosis by regulating the macrophage polarization via STAT3 signaling in infarcted rat hearts. Free Radic. Biol. Med. 104, 298–310 (2017).
Wu, Q. et al. Dapagliflozin protects against chronic heart failure in mice by inhibiting macrophage-mediated inflammation, independent of SGLT2. Cell Rep. Med. 4, 101334 (2023).
Abdollahi, E. et al. Dapagliflozin exerts anti-inflammatory effects via inhibition of LPS-induced TLR-4 overexpression and NF-κB activation in human endothelial cells and differentiated macrophages. Eur. J. Pharmacol. 918, 174715 (2022).
Liu, Z. et al. Impact of sodium glucose cotransporter 2 (SGLT2) inhibitors on atherosclerosis: from pharmacology to pre-clinical and clinical therapeutics. Theranostics 11, 4502–4515 (2021).
Tokutome, M. et al. Peroxisome proliferator-activated receptor-gamma targeting nanomedicine promotes cardiac healing after acute myocardial infarction by skewing monocyte/macrophage polarization in preclinical animal models. Cardiovasc. Res. 115, 419–431 (2019).
Brenner, C. et al. DPP-4 inhibition ameliorates atherosclerosis by priming monocytes into M2 macrophages. Int. J. Cardiol. 199, 163–169 (2015).
Zhang, X. et al. Rosuvastatin exerts anti-atherosclerotic effects by improving macrophage-related foam cell formation and polarization conversion via mediating autophagic activities. J. Transl. Med. 19, 62 (2021).
Ridker, P. M. et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N. Engl. J. Med. 377, 1119–1131 (2017).
Toldo, S. et al. Targeting the NLRP3 inflammasome in cardiovascular diseases. Pharmacol. Ther. 236, 108053 (2022).
Angelidis, C. et al. Colchicine pharmacokinetics and mechanism of action. Pharmacol. Ther. 24, 659–663 (2018).
Tardif, J. C. et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N. Engl. J. Med. 381, 2497–2505 (2019).
Nidorf, S. M. et al. Colchicine in patients with chronic coronary disease. N. Engl. J. Med. 383, 1838–1847 (2020).
Ridker, P. M. & Rane, M. Interleukin-6 signaling and anti-interleukin-6 therapeutics in cardiovascular disease. Circ. Res. 128, 1728–1746 (2021).
Broch, K. et al. Randomized trial of interleukin-6 receptor inhibition in patients with acute ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 77, 1845–1855 (2021).
Kharbanda, R. K. et al. Systemic Acyl-CoA:cholesterol acyltransferase inhibition reduces inflammation and improves vascular function in hypercholesterolemia. Circulation 111, 804–807 (2005).
Meuwese, M. C. et al. ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: the CAPTIVATE randomized trial. JAMA 301, 1131–1139 (2009).
Nissen, S. E. et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N. Engl. J. Med. 354, 1253–1263 (2006).
Tardif, J. C. et al. Effects of the acyl coenzyme A: cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation 110, 3372–3377 (2004).
Colombo, A. et al. A double-blind randomised study to evaluate the efficacy and safety of bindarit in preventing coronary stent restenosis. EuroIntervention 12, e1385–e1394 (2016).
Sharma, T. S. et al. Hydroxychloroquine use is associated with decreased incident cardiovascular events in rheumatoid arthritis patients. J. Am. Heart Assoc. 5, e002867 (2016).
Johnson, T. M. et al. Investigating changes in disease activity as a mediator of cardiovascular risk reduction with methotrexate use in rheumatoid arthritis. Ann. Rheum. Dis. 80, 1385–1392 (2021).
Shi, H. T., Huang, Z. H., Xu, T. Z., Sun, A. J. & Ge, J. B. New diagnostic and therapeutic strategies for myocardial infarction via nanomaterials. EBioMedicine 78, 103968 (2022).
Ma, Y., Gu, T., He, S., He, S. & Jiang, Z. Development of stem cell therapy for atherosclerosis. Mol. Cell. Biochem. https://doi.org/10.1007/s11010-023-04762-8 (2023).
Wang, L. L. et al. Cell therapies in the clinic. Bioeng. Transl. Med. 6, e10214 (2021).
Machtakova, M., Thérien-Aubin, H. & Landfester, K. Polymer nano-systems for the encapsulation and delivery of active biomacromolecular therapeutic agents. Chem. Soc. Rev. 51, 128–152 (2022).
Zhao, L., Ren, T. H. & Wang, D. D. Clinical pharmacology considerations in biologics development. Acta Pharmacol. Sin. 33, 1339–1347 (2012).
Song, S. et al. Functional nanoprobes for ultrasensitive detection of biomolecules. Chem. Soc. Rev. 39, 4234–4243 (2010).
Wang, W., Lu, K. J., Yu, C. H., Huang, Q. L. & Du, Y. Z. Nano-drug delivery systems in wound treatment and skin regeneration. J. Nanobiotechnol. 17, 82 (2019).
Spada, A., Emami, J., Tuszynski, J. A. & Lavasanifar, A. The uniqueness of albumin as a carrier in nanodrug delivery. Mol. Pharm. 18, 1862–1894 (2021).
Li, W., Gonzalez, K. M., Chung, J., Kim, M. & Lu, J. Surface-modified nanotherapeutics targeting atherosclerosis. Biomater. Sci. 10, 5459–5471 (2022).
Fredman, G. et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci. Transl. Med. 7, 275ra220 (2015).
Schoenmaker, L. et al. mRNA-lipid nanoparticle COVID-19 vaccines: structure and stability. Int. J. Pharm. 601, 120586 (2021).
Ackermann, M. et al. Continuous human iPSC-macrophage mass production by suspension culture in stirred tank bioreactors. Nat. Protoc. 17, 513–539 (2022).
Zangi, L. et al. Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells 27, 2865–2874 (2009).
Cui, L. L. et al. The cerebral embolism evoked by intra-arterial delivery of allogeneic bone marrow mesenchymal stem cells in rats is related to cell dose and infusion velocity. Stem Cell Res. Ther. 6, 11 (2015).
Nauta, A. J. et al. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood 108, 2114–2120 (2006).
Røsland, G. V. et al. Long-term cultures of bone marrow-derived human mesenchymal stem cells frequently undergo spontaneous malignant transformation. Cancer Res. 69, 5331–5339 (2009).
Pan, Y., Wu, W., Jiang, X. & Liu, Y. Mesenchymal stem cell-derived exosomes in cardiovascular and cerebrovascular diseases: from mechanisms to therapy. Biomed. Pharmacother. 163, 114817 (2023).
Hwang, M. W. et al. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J. Am. Coll. Cardiol. 38, 1546–1553 (2001).
Liberale, L., Montecucco, F., Schwarz, L., Lüscher, T. F. & Camici, G. G. Inflammation and cardiovascular diseases: lessons from seminal clinical trials. Cardiovasc. Res. 117, 411–422 (2021).
Lam, C. S. P. et al. Recent successes in heart failure treatment. Nat. Med. 29, 2424–2437 (2023).
Heidenreich, P. A. et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: executive summary: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 145, e876–e894 (2022).
Doster, R. S., Rogers, L. M., Gaddy, J. A. & Aronoff, D. M. Macrophage extracellular traps: a scoping review. J. Innate Immun. 10, 3–13 (2018).
Rasmussen, K. H. & Hawkins, C. L. Role of macrophage extracellular traps in innate immunity and inflammatory disease. Biochem. Soc. Trans. 50, 21–32 (2022).
Deniset, J. F. et al. Gata6+ pericardial cavity macrophages relocate to the injured heart and prevent cardiac fibrosis. Immunity 51, 131–140.e5 (2019).
Jin, H. et al. Genetic lineage tracing of pericardial cavity macrophages in the injured heart. Circ. Res. 130, 1682–1697 (2022).
Mourad, O., Yee, R., Li, M. & Nunes, S. S. Modeling heart diseases on a chip: advantages and future opportunities. Circ. Res. 132, 483–497 (2023).
Ravassa, S. et al. Cardiac Fibrosis in heart failure: Focus on non-invasive diagnosis and emerging therapeutic strategies. Mol. Asp. Med. 93, 101194 (2023).
Wang, C. et al. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol. Ther. 25, 192–204 (2017).
Razin, T. et al. Interleukin-1α dependent survival of cardiac fibroblasts is associated with StAR/STARD1 expression and improved cardiac remodeling and function after myocardial infarction. J. Mol. Cell. Cardiol. 155, 125–137 (2021).
Humeres, C. et al. Smad7 effects on TGF-β and ErbB2 restrain myofibroblast activation and protect from postinfarction heart failure. J. Clin. Investig. 132, e146926 (2022).
Verma, S. K. et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-κB. Circulation 126, 418–429 (2012).
Shimojo, N. et al. Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin αVβ3/nuclear factor-κB/interleukin-6 axis. Hypertension 66, 757–766 (2015).
Khalil, H. et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Invest. 127, 3770–3783 (2017).
Suetomi, T. et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca(2+)/calmodulin-dependent protein kinase II δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 138, 2530–2544 (2018).
Souissi, I. J., Billiet, L., Cuaz-Pérolin, C., Slimane, M. N. & Rouis, M. Matrix metalloproteinase-12 gene regulation by a PPAR alpha agonist in human monocyte-derived macrophages. Exp. Cell Res. 314, 3405–3414 (2008).
Yan, H. et al. Interleukin-12 and -23 blockade mitigates elastase-induced abdominal aortic aneurysm. Sci. Rep. 9, 10447 (2019).
Yang, H., Zhou, T., Sorenson, C. M., Sheibani, N. & Liu, B. Myeloid-derived TSP1 (Thrombospondin-1) contributes to abdominal aortic aneurysm through suppressing tissue inhibitor of metalloproteinases-1. Arterioscler. Thromb. Vasc. Biol. 40, e350–e366 (2020).
Salarian, M. et al. Homeostatic, non-canonical role of macrophage elastase in vascular integrity. Circ. Res. 132, 432–448 (2023).
Davis, F. M. et al. The histone methyltransferase SETDB2 modulates tissue inhibitors of metalloproteinase–matrix metalloproteinase activity during abdominal aortic aneurysm development. Ann. Surg. 278, 426–440 (2023).
Ye, B. et al. Macrophage-derived GSDMD promotes abdominal aortic aneurysm and aortic smooth muscle cells pyroptosis. Int. Immunopharmacol. 128, 111554 (2024).
Wang, J. et al. Effect of CCR2 inhibitor-loaded lipid micelles on inflammatory cell migration and cardiac function after myocardial infarction. Int. J. Nanomed. 13, 6441–6451 (2018).
Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Das Pradhan, A. et al. Triglyceride lowering with pemafibrate to reduce cardiovascular risk. N. Engl. J. Med. 387, 1923–1934 (2022).
Puato, M. et al. Atorvastatin reduces macrophage accumulation in atherosclerotic plaques: a comparison of a nonstatin-based regimen in patients undergoing carotid endarterectomy. Stroke 41, 1163–1168 (2010).
Elkhawad, M. et al. Effects of p38 mitogen-activated protein kinase inhibition on vascular and systemic inflammation in patients with atherosclerosis. Jacc. Cardiovasc. Imaging 5, 911–922 (2012).
O’Donoghue, M. L. et al. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: a randomized clinical trial. JAMA 315, 1591–1599 (2016).
Newby, L. K. et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet 384, 1187–1195 (2014).
Fox, K. et al. Ivabradine in stable coronary artery disease without clinical heart failure. N. Engl. J. Med. 371, 1091–1099 (2014).
Rodriguez, A. E. et al. Randomized comparison of cost-saving and effectiveness of oral rapamycin plus bare-metal stents with drug-eluting stents: three-year outcome from the randomized oral rapamycin in Argentina (ORAR) III trial. Catheter. Cardiovasc. Interv. 80, 385–394 (2012).
Stähli, B. E. et al. Mammalian target of rapamycin inhibition in patients with ST-segment elevation myocardial infarction. J. Am. Coll. Cardiol. 80, 1802–1814 (2022).
Jamialahmadi, T. et al. The effect of trehalose administration on vascular inflammation in patients with coronary artery disease. Biomed. Pharmacother. 147, 112632 (2022).
Ridker, P. M. et al. Low-dose methotrexate for the prevention of atherosclerotic events. N. Engl. J. Med. 380, 752–762 (2019).
Razavi, M. K., Donohoe, D., D’Agostino, R. B. Jr., Jaff, M. R. & Adams, G. Adventitial drug delivery of dexamethasone to improve primary patency in the treatment of superficial femoral and popliteal artery disease: 12-month results from the DANCE clinical trial. Jacc. Cardiovasc. Interv. 11, 921–931 (2018).
Abbate, A. et al. Interleukin-1 blockade inhibits the acute inflammatory response in patients with ST-segment-elevation myocardial infarction. J. Am. Heart Assoc. 9, e014941 (2020).
Kron, J. et al. Interleukin-1 blockade in cardiac sarcoidosis: study design of the multimodality assessment of granulomas in cardiac sarcoidosis: Anakinra randomized trial (MAGiC-ART). J. Transl. Med. 19, 460 (2021).
El Sayed, H., Kerensky, R., Stecher, M., Mohanty, P. & Davies, M. A randomized phase II study of Xilonix, a targeted therapy against interleukin 1α, for the prevention of superficial femoral artery restenosis after percutaneous revascularization. J. Vasc. Surg. 63, 133–141.e131 (2016).
Ridker, P. M. et al. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397, 2060–2069 (2021).
Kleveland, O. et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur. Heart J. 37, 2406–2413 (2016).
Meyer, M. A. S. et al. Treatment effects of interleukin-6 receptor antibodies for modulating the systemic inflammatory response after out-of-hospital cardiac arrest (the IMICA trial): a double-blinded, placebo-controlled, single-center, randomized, clinical trial. Circulation 143, 1841–1851 (2021).
Acknowledgements
XX, SH, RC, HZ, BT, YL, YY, XZ, SC are the Small Small bird (SSb) group members and acknowledge the strong support from SSb. HZ, BT and RC were supported by grants from the National Students’ Platform for Innovation and Entrepreneurship Training Program of China (No. 202212121004 and No. 202212121023) and the Guangdong Students’ Platform for Innovation and Entrepreneurship Training Program (No. S202212121074). CL was supported by the National Natural Science Foundation of China (82170231, 82370410) and Guangdong Basic and Applied Basic Research Foundation (2023B1515020103).
Author information
Authors and Affiliations
Contributions
S.H., X.X. and C.L. contributed to conception and manuscript design. R.C., H.Z., B.T., Y.L. and S.H. drafted the manuscript. R.C., H.Z., B.T., Y.L., Y.Y. and S.H. prepared the tables and figures. S.H., R.C., H.Z., B.T. and Y.L. collected the related references. S.H., X.X., R.C., H.Z., B.T., Y.L., X.Z., S.C. and C.L. participated in the revision of the manuscript. C.L. was involved in funding acquisition. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, R., Zhang, H., Tang, B. et al. Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets. Sig Transduct Target Ther 9, 130 (2024). https://doi.org/10.1038/s41392-024-01840-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-024-01840-1
- Springer Nature Limited