
Abstract
In the context of the escalating global health challenge posed by Alzheimer’s disease (AD), this comprehensive review considers the potential of melatonin in both preventive and therapeutic capacities. As a naturally occurring hormone and robust antioxidant, accumulating evidence suggests melatonin is a compelling candidate to consider in the context of AD-related pathologies. The review considers several mechanisms, including potential effects on amyloid-beta and pathologic tau burden, antioxidant defense, immune modulation, and regulation of circadian rhythms. Despite its promise, several gaps need to be addressed prior to clinical translation. These include conducting additional randomized clinical trials in patients with or at risk for AD dementia, determining optimal dosage and timing, and further determining potential side effects, particularly of long-term use. This review consolidates existing knowledge, identifies gaps, and suggests directions for future research to better understand the potential of melatonin for neuroprotection and disease mitigation within the landscape of AD.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is a chronic, neurodegenerative disease and the leading cause of dementia, affecting more than 50 million people worldwide. This number is projected to be greater than 150 million by 2050 [1]. Characterized by accumulation of amyloid-beta (Aβ) plaque, neurofibrillary tangles (NFTs), and neurodegeneration, sporadic AD is understood to be the result of multiple genetic factors, as well as interactions between genes and environmental factors. See Box 1 for the definition of sporadic AD and how it differs from the familial form of AD.
Among potentially modifiable risk factors, sleep disorders, including insomnia and obstructive sleep apnea, and shortened sleep duration, have been linked to AD [2,3,4,5,6,7,8,9,10,11,12,13]. Several processes have been proposed to elucidate the mechanistic role of sleep in the context of AD. These include the clearance of potentially detrimental metabolites and protein fragments, such as soluble Aβ, from the brain [14], influencing tau protein dynamics [8], supporting the function of the blood-brain barrier (BBB) [15], maintaining synaptic integrity [16], consolidating memory [17], modulating glial activation [18], and regulating neuroinflammation [19]. Conversely, the development of AD pathology may also adversely impact sleep processes, indicating a bidirectional relationship between sleep and AD [20, 21].
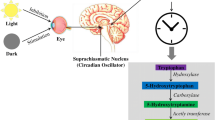
Melatonin, also known as N-acetyl-5-methoxytryptamine, is a key hormone in regulating sleep timing in humans [22] and may also play a role in the brain-health benefits associated with sleep. It is primarily produced in the pineal gland of the brain (Fig. 1) through a series of enzymatic reactions involving metabolites such as 5-hydroxytryptophan, serotonin, and N-acetylserotonin [23]. The hormone is mainly secreted into the bloodstream at night [22], affecting both peripheral organs and the central nervous system (CNS) due to its amphiphilic properties [24]. The pineal gland can also release melatonin directly into the brain ventricles, as shown by animal and human studies [25,26,27]. After being taken up by the liver from the bloodstream, melatonin undergoes enzymatic conversion to produce 6-hydroxymelatonin sulfate, which is then excreted in the urine [28].

Light, particularly in the blue wavelength range (450-490 nm), a pivotal component of sunlight, activates intrinsically photosensitive retinal ganglion cells (ipRGC) situated in the retina of the eyes. These ipRGCs transform light stimuli into action potentials transmitted to the suprachiasmatic nucleus (SCN), positioned above the optic nerve crossing [243]. Individuals with Alzheimer’s disease (AD) often exhibit a significant loss of ipRGC. The SCN comprises of approximately 20,000 neurons in each hemisphere. The interplay of core clock genes, including BMAL1, CLOCK, PER, and CRY, not only governs 24-hour processes like gene transcription in SCN neurons but also orchestrates their regulatory impact on other brain regions [244, 245]. The intricate regulation of pineal melatonin synthesis and release relies on the signaling pathway originating from the SCN in the hypothalamus, extending to the pineal gland (PG) [246]. This process involves the intermediolateral cell column in the spinal cord and sympathetic input from the superior cervical ganglion (SCG), located near the base of the skull [247]. Once released into the bloodstream by the PG, melatonin exerts its influence on target cells through specific melatonin receptors, distributed in various central and peripheral tissues [248]. Notably, melatonin is released into both the blood and directly into the brain. Several factors can disrupt the body’s natural melatonin production, including nighttime light exposure, such as the use of light-emitting devices [249]. Advanced aging [250], medications (e.g., beta blockers) [251], and various medical conditions, including dementia, pain, cancer, and type 2 diabetes mellitus [252], can also impede melatonin production. Conditions that hinder the synchronization of endogenous melatonin production with the solar day, such as blindness [253], add an additional layer of complexity.
Melatonin exerts its effects on target cells primarily through two G-protein-coupled receptors: MT1 and MT2 [29, 30]. These receptors are distributed throughout the body, including the brain [29, 30]. Activation of MT1 and MT2 receptors initiates various signaling pathways, including the inhibition of adenylate cyclase, which reduces the production of cyclic adenosine monophosphate, a crucial secondary messenger in many cellular processes [29, 30]. Beyond membrane-bound receptors, melatonin also interacts with intracellular proteins such as calmodulin and nuclear receptors, extending its biological activity [29, 30].
In addition to its role in sleep-wake regulation [22], melatonin demonstrates potential direct or indirect impacts on pathways implicated in AD pathology. As discussed in the following sections, this includes the potential to reduce the production of Aβ and the ability to counteract the effects of Aβ oligomers (Aβos), a highly pathogenic form of Aβ [31]. Additionally, melatonin may modulate the hyperphosphorylation of the microtubule-associated tau protein, a process leading to the formation of NFTs characteristic of AD [32]. Further contributing to its neuroprotective potential, melatonin reduces oxidative stress in the CNS. Its entraining effects on circadian processes in brain cells, aligning neural activity and modulating synaptic transmission with the natural day-night cycle, could also potentially mitigate the development of AD. The improved functioning of the BBB, enhanced clearance of metabolites and protein fragments implicated in AD from the brain - an astrocyte-dependent mechanism primarily occurring during sleep [14] - and enhanced insulin signaling, suggested to play a role in AD [33], present additional mechanisms by which melatonin may impact AD pathology.
Melatonin production declines with aging [34], possibly due to age-related calcification of the pineal gland [35], rather than increased clearance [34]. Notably, reduced melatonin levels and disrupted 24-hour melatonin rhythms are frequently observed in individuals with AD dementia [36,37,38,39,40]. Since there is an established link between disrupted melatonin rhythms and the risk of AD dementia [41, 42], investigating the potential therapeutic role of melatonin in addressing these issues is a promising area of research and intervention for AD. Consequently, our review delves into the diverse mechanisms through which melatonin could exert neuroprotective effects in the context of AD. Additionally, we examine current clinical trials and discuss existing gaps and opportunities that warrant further consideration.
Unlocking melatonin’s anti-amyloidogenic potential
AD is characterized by Aβ aggregation, which in turn is associated with synaptic loss, oxidative stress, and mitochondrial dysfunction in the brain [32, 43,44,45,46,47]. The neurodegenerative potential varies between different forms of Aβ peptides, for example, amyloid aggregates primarily comprise Aβ42, which has higher amyloidogenicity and lower solubility than Aβ40 [48, 49]. Amyloid aggregates form when the membrane-bound amyloid precursor protein (APP) undergoes a series of proteolytic cleavages orchestrated by β-secretase (beta-site APP-cleaving enzyme, BACE) and furthered by γ-secretase [50]. β-secretase-catalyzed cleavage produces soluble APPβ and C99, a membrane-bound fragment of APP [50]. Subsequently, γ-secretase acts on C99, releasing Aβ both outside and within the neuron [50]. In contrast, the non-amyloidogenic pathway is characterized by more active α-secretase-catalyzed cleavage (e.g., ADAM-10 and ADAM-17) of APP, releasing soluble APPα from neuronal membranes into the interstitial space of the brain. This initiates the production of C-terminal membrane-tethered α-secretase-derived fragment C83, which, upon additional enzymatic cleavage by γ-secretase, increases the concentration of p3 [50]. A third pathway involving cleavage of APP by β- and α-secretase independently of γ-secretase is also non-amyloidogenic and results in the secretion of 14-16 amino acid-long Aβ [51].
While typically not considered in conventional models of amyloid plaque formation, melatonin intersects with several pathways. For example, melatonin has been found to stimulate the α-secretase cleavage of βAPP in cultured neuronal and non-neuronal cells [52] via upregulation of the nonamyloidogenic ADAM10 and ADAM17 proteases. In turn, presence of α-secretase inhibitors in these cell lines abrogates the α-secretase-dependent activation of the non-amyloidogenic pathway by melatonin [52]. In studies using SH-SY5Y cells, recognized as a model for studying neurodegenerative processes [53], melatonin exhibits an inhibitory effect on the expression of amyloidogenic β-secretases [54]. The increased activity of α-secretases and concurrent reduction in the activity of β-secretase may partially explain why melatonin treatment results in diminished Aβ levels in the brains of both sporadic and transgenic animal models of AD [55,56,57]. It is also worth noting that Aβ peptides have been shown to significantly decrease the production of melatonin by the pineal gland [58], underscoring the bi-directional nature of these relationships.
Melatonin appears to modulate the activity of enzymes engaged in the modification of membrane-bound APP in neurons, including glycogen synthase kinase-3β (GSK3β). GSK3β, a serine-threonine kinase prominently expressed in the brain, augments the activity of β-secretase while concurrently diminishing the activity of α-secretases, thereby amplifying intra- and extracellular levels of Aβ [59, 60]. In a murine model of AD, melatonin diminished GSK3β activity [61], an effect potentially reliant on melatonin receptor MT1, as implied by findings in SH-SY5Y cells [62]. Using SH-SY5Y cells exposed to high glucose concentrations to induce hyperglycemia, disruptions in glucose sensing leads to activation of the phosphorylated protein kinase B (pAkt)/GSK-3β signaling pathway, and increased expression of BACE and Aβ42, an effect that can be reversed via pretreatment with melatonin [62]. Moreover, melatonin may impact amyloid clearance via its impact on insulin-degrading enzyme (IDE), a protease pivotal for catalyzing extracellular and intracellular Aβ degradation [63]. IDE exhibits reduced abundance concomitant with elevated CNS activity of GSK3β in diabetic murine models [64]. Significantly, a compound denoted as melatonin-trientine, covalently synthesized with melatonin and the metal ion chelator trientine, demonstrated the capacity to elevate the expression of IDE in a murine AD model, and decreased Aβ deposition and neuronal degeneration in the brains of the APP/Presenilin 1 mice [65].
Another contributing mechanism to melatonin’s anti-amyloidogenic effects may involve the enhanced clearance of Aβ from the brain. Astrocytes, positioned at the interface between the brain parenchyma and the perivascular space, which includes capillaries, larger arteries, and veins [66], play a role in the removal of Aβ from the brain via mechanisms that include enzymatic degradation of Aβ and the upregulation of efflux transporters for Aβ at the BBB [67]. Studies conducted with mouse neuroblastoma cells (Neuro-2a cells) suggest that melatonin has the potential to augment the expression of Transcription Factor EB [68]. This transcription factor acts as a master regulator of lysosome biogenesis, thereby promoting autophagosome-lysosome clearance of Aβ by astrocytes [69].
Melatonin may exert an additional impact on Aβ clearance by upregulating the expression of the low-density lipoprotein receptor-related protein 1 (LRP1) [70]. This transporter may play a crucial role in both the uptake of Aβ into astrocytes and the efflux of Aβ from the brain at the BBB [71]. Apolipoprotein E (ApoE), a protein interacting with cell surface receptors to facilitate the uptake of lipoproteins [72], is postulated to hinder the clearance of soluble Aβ from the brain by competing with soluble Aβ for LRP1–dependent cellular uptake into astrocytes [73]. Consequently, this competition heightens the likelihood of Aβ aggregation in the brain, ultimately leading to plaque formation. Notably, research involving astrocytes cultured from a transgenic AD mouse model overexpressing apoE demonstrated that melatonin reversed the Aβ aggregation-promoting activity of this protein [74]. The observation that patients with AD carrying two alleles of the APOE4 risk variant have approximately half the cerebrospinal fluid (CSF) melatonin levels compared to those with only one allele of this variant [75] could suggests that APOE4/4 carriers might particularly benefit from supplementing with exogenous melatonin.
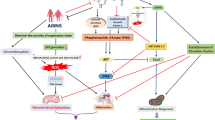
In addition to its impact on astrocytic clearance, melatonin may mitigate the burden of Aβ in the brain by promoting lymphatic clearance. In the transgenic AD mouse model Tg2576, marked by mutant APP overexpression in the brain, melatonin treatment exhibited a trend of elevated Aβ levels in cervical and axillary lymph nodes, concomitant with a decreasing trend in brain Aβ [76]. Figure 2 provides a summary of the mechanisms that may explain the anti-amyloidogenic effects of melatonin.

In the pro-amyloidogenic pathway, membrane-bound amyloid precursor protein (APP) undergoes proteolytic cleavages orchestrated by β-secretase (BACE) and γ-secretase, leading to increased intraneuronal and brain interstitial fluid amyloid-beta (Aβ) concentrations. If enzymatically cleaved first by alpha (ADAM) and then γ-secretase, no Aβ is produced. If cleaved concertedly by β- and α-secretases, short 14-16 amino acid-long Aβ fragments form but they are highly hydrophilic and not part of any amyloidogenic cascade. Through self-assembly, soluble Aβ oligomers (Aβos) are formed, known to disrupt neuronal signaling through excitotoxicity and induce oxidative stress (e.g., through microglial activation), thereby exacerbating the process of neurodegeneration. Aβos can further assemble to build fibrils, which represent a major component of Aβ plaques [254, 255]. As reviewed in Section Unlocking Melatonin’s Anti-Amyloidogenic Potential and Section Melatonin’s Neuroprotective Mechanisms against Aβ toxicity, melatonin redirects APP toward the non-amyloidogenic pathway, potentially achieved by suppressing glycogen synthase kinase-3β (GSK3β) activity. This is achieved through various mechanisms, e.g., the activation of the membrane-bound melatonin receptor 1. GSK3β is a kinase known to promote the pro-amyloidogenic pathway. Consequently, melatonin reduces Aβ burden in the brain. Melatonin may also increase the clearance of Aβ. For example, treatment with melatonin has been shown to upregulate transporters such as the low-density lipoprotein receptor-related protein 1 (LRP1), which plays a decisive role in the uptake of soluble Aβ proteins from the brain interstitial fluid into astrocytes. In animal models that overexpress apolipoprotein E (ApoE), a variant recognized for competing with Aβ for astrocytic uptake, melatonin has been shown to alleviate Aβ burden, most likely achieved through the upregulation of LRP1. Melatonin has also been demonstrated to mitigate processes involved in Aβos’ adverse effects on brain cells, such as excitotoxicity and oxidative stress.
Melatonin’s neuroprotective mechanisms against Aβ toxicity
According to the current version of the Aβ cascade hypothesis, diffusible oligomeric forms of Aβ (Aβ oligomers, Aβos) represent the most pathogenic and toxic form of Aβ [31]. Aβos, upon binding to cell-surface receptors on neurons, disrupt signal transduction, leading to a sustained interruption of intracellular Ca2+ signaling through increased N-methyl-D-aspartate (NMDA) receptor-dependent Ca2+ influx [77]. Ca2+-dependent hyperexcitability can contribute to cellular damage and cell death through various mechanisms, including mitochondrial dysfunction and apoptosis [78]. In a rat model, melatonin demonstrated inhibition of NMDA receptor activation and subsequent NMDA receptor-mediated Ca2+ entry into striatal neurons, suggesting a potential protective role [79]. However, melatonin has also been shown to upregulate NMDA receptor subunits 2A and 2B in a dose-dependent manner in the rat hippocampus [80].
In addition to excitotoxicity, Aβos can inflict injury to brain cells through oxidative stress. Specifically, Aβos possess the ability to bind to metal ions, thereby promoting the redox activity of these metals [81,82,83]. Moreover, oligomeric forms of Aβ can increase oxidative stress in neurons [84, 85]. For example, mitochondria isolated from the rat whole brain, when exposed to Aβos, produce more reactive oxygen species (ROS) [86]. Another mechanism through which Aβos contribute to oxidative stress involves the activation of toll-like receptor 4 (TLR4) on microglia [87], the brain’s resident innate immune cells. TLR4 activation triggers various pro-inflammatory processes in microglia, including an augmented production of ROS [88].
As a consequence of heightened oxidative stress induced by Aβos, the transient receptor potential melastatin 2, a Ca2+-permeable channel expressed on brain cells, opens [89]. If prolonged, this can elevate the risk of neuronal dysfunction and damage due to excessive Ca2+ influx into neurons [78]. Furthermore, escalated oxidative stress can activate GSK3β [90], contributing to elevated Aβ levels [60] (see Section Unlocking Melatonin’s Anti-Amyloidogenic Potential) and hyperphosphorylation of the microtubule-associated protein tau [91, 92] (see Melatonin’s Potential to Counteract Tau Pathology). Melatonin, known for its robust scavenging of ROS and reactive nitrogen species (RNS) [93], can effectively penetrate brain cells owing to its amphiphilic biochemical nature [94]. The reduction in oxidative stress, therefore, serves as an additional mechanism by which melatonin safeguards brain cells against the toxic effects of Aβos [95].
In addition to accumulation of amyloid and tau pathology, AD involves an immune response. Accumulation of pathology including Aβ triggers activation of microglia in the brain [96]. Activated M1 (or pro-inflammatory) microglia are part of an inflammatory cascade that includes upregulation of pro-inflammatory genes, release of pro-inflammatory cytokines, and generation of ROS, which may, under prolonged conditions, contribute to the neuroinflammation observed in AD brain [97]. Additionally, M1 microglia are often associated with reduced phagocytic activity, which may also make microglia less effective at clearing Aβos and plaques [98]. Conversely, the M2 phenotype is generally considered anti-inflammatory and has been associated with enhanced phagocytic activity, potentially reducing the risk of AD and its progression [99]. In this context, the triggering receptor expressed on myeloid cells 2 (TREM2) may play a major role in promoting the M2 microglia phenotype [99]. Consequently, a lack of function of TREM2 has previously been linked to an increased risk of AD [100]. Notably, as suggested in an animal model of neuroinflammation induced by ischemic stroke damage known to increase the prevalence of M1 microglia in the brain, pre-treatment with melatonin upregulated the expression of TREM2 while downregulating that of proinflammatory genes in microglia (e.g., inducible nitric oxide synthase) [101], suggesting that melatonin may polarize microglia towards its M2 phenotype.
The evidence outlined in the Sections Unlocking Melatonin’s Anti-Amyloidogenic Potential and Melatonin’s Neuroprotective Mechanisms against Aβ toxicity indicates that melatonin exhibits anti-amyloidogenic properties and mitigates the neurotoxic effects of Aβos, thereby potentially reducing the risk of developing AD or slowing its progression. It is imperative to emphasize that these findings predominantly originate from animal studies and experiments conducted with cell lines. As of the present moment, there are no published studies that have investigated the impact of melatonin on Aβ burden specifically in humans.
Melatonin’s potential to counteract tau pathology
The microtubule-stabilizing tau protein assumes a critical role in the assembly and stabilization of microtubules, integral components of the neuronal cytoskeleton [102]. Under normal physiological conditions, tau harbors merely 2–3 phosphate groups. However, in tauopathies such as AD, as well as in hibernation, tau undergoes hyperphosphorylation catalyzed by various protein kinases, including but not limited to glycogen GSK3β, cyclin-dependent kinase 5 (CDK5), and protein kinase A (PKA) [103,104,105], accumulating an excess of 7-10 phosphate groups [106]. As tau becomes hyperphosphorylated, its capacity to support microtubule assembly and stability diminishes, resulting in increased levels of unbound tau that may aggregate into tangles (if not secreted or degraded in the lysosome) [107]. This disruption assumes a pivotal role in the neurodegenerative processes observed in AD and other tauopathies [108, 109]. Hyperphosphorylated tau and tangle development are more closely associated with synaptic dysfunction and cognitive decline (including conversion to mild cognitive impairment and AD dementia) compared to Aβ alone [110,111,112].
As evidenced by animal studies and cell line experiments, melatonin exhibits promise in mitigating tau hyperphosphorylation. For instance, intraperitoneal pretreatment with melatonin prevented isoproterenol (a beta-receptor agonist)-induced tau hyperphosphorylation in rats [113]. Another study, involving the pharmacological blocking of endogenous melatonin production in rats, observed an increase in tau hyperphosphorylation. This effect was reversed when melatonin-deficient animals were supplemented with melatonin [114]. In a transgenic mouse model exhibiting age-associated tau pathology, melatonin treatment efficiently decreased the hyperphosphorylation of tau [115]. Additionally, melatonin significantly reduced the number of NFTs and attenuated neuronal loss in the cortex and hippocampus [115]. When SH-SY5Y cells were exposed to mercury, inducing a two-fold increase in tau hyperphosphorylation over a 9-hour period, pre-treatment with melatonin for 12 hours effectively reduced tau hyperphosphorylation due to this transition metal [116]. Lastly, among rats, the administration of wortmannin (a fungal metabolite) which induces hyperphosphorylation of tau in hippocampal pyramidal neurons, can be partly attenuated by preinjection of melatonin [117, 118], underscoring melatonin’s potential therapeutic role.
Various mechanisms may underlie the inhibitory effect of melatonin on tau hyperphosphorylation, as depicted in Fig. 3. In neuroblastoma SH-SY5Y cells exposed to hyperglycemia, a condition known to increase GSK3β activity [119], melatonin exerted an inhibitory effect on the GSK3β signaling pathway, likely through the activation of its melatonin receptor [62]. GSK3β serves as a key kinase in the hyperphosphorylation of the microtubule-associated protein tau, thereby contributing to neuronal dysfunction and degeneration [91, 92]. Stimulation of GSK3β activity can occur through exposure to oxidative stress [90]. Within neurons, oxidative stress may stem from mitochondrial dysfunction induced by the intracellular accumulation of hyperphosphorylated tau and Aβ [84, 120]. Furthermore, interactions of brain interstitial Aβ with microglia [87, 88] and exposure to transition metals [81,82,83] also contribute to oxidative stress in neurons. Thus, melatonin’s dual capacity to act both as a hormone and antioxidant may counter GSK3β-driven tau hyperphosphorylation in neurons. Notably, melatonin also diminished the activity of PKA in a rat model [121], a known trigger for tau hyperphosphorylation by GSK3β [122, 123]. Additionally, melatonin reduced the activities of other kinases involved in tau hyperphosphorylation, such as CDK5, through the upregulation of micro-RNAs [115].

Tau, a microtubule-associated protein crucial for the structural integrity of neurons, undergoes hyperphosphorylation in Alzheimer’s disease (AD), leading to the accumulation of phosphorylated tau and the formation of neurofibrillary tangles (NFTs). This hyperphosphorylation compromises tau’s ability to support microtubule assembly, contributing to the development of NFTs in neurons. Furthermore, NFTs and hyperphosphorylated tau can induce oxidative stress in neurons, primarily through mitochondrial dysfunction. Oxidative stress may also activate kinases associated with tau hyperphosphorylation, including glycogen synthase kinase-3β (GSK3β), cyclin-dependent kinase 5 (CDK5), and protein kinase A (PKA). Increased oxidative stress acting on neurons can also result from amyloid β (Aβ)-driven activation of microglia through toll-like receptor 4 (TLR4) or interactions with transition metals in the brain interstitial fluid, leading to an abundance of reactive oxygen species (ROS) and reactive nitrogen species (RNS). As elucidated in Section Melatonin’s Potential to Counteract Tau Pathology, melatonin exhibits promising potential in mitigating tau hyperphosphorylation, as demonstrated in both animal models of AD and cell line experiments. Through its robust antioxidative potential, melatonin may hinder the oxidative stress-induced activation of protein kinases involved in tau hyperphosphorylation. Additionally, the hormone, via activation of its melatonin receptor 1, reduces the activity of GSK3β, showcasing another anti-tauogenic effect. Melatonin further engages in the activation of enzymes that counteract tau hyperphosphorylation. For instance, it upregulates the activity of phosphatase 2 A (PP2A), recognized for its role in dephosphorylating tau, and elevates levels of Peptidyl-Prolyl cis-trans Isomerase NIMA-Interacting 1 (Pin1), crucial for restoring tau function.
Melatonin’s impact extends to various enzymes involved in mitigating tau hyperphosphorylation. For instance, a study in rats suggests that melatonin may counteract tau pathology by upregulating the activity of phosphatase 2A [114], an enzyme recognized for its role in dephosphorylating microtubule-associated protein tau [124]. Additionally, in SH-SY5Y cells, melatonin increased levels of Peptidyl-Prolyl cis-trans Isomerase NIMA-Interacting 1 (Pin1) [125], which has been shown to mitigate tau phosphorylation and aggregation [126]. The absence of Pin1 has been associated with age-dependent tau hyperphosphorylation and neurodegeneration [126].
Although the described findings predominantly originate from in cell line studies and animal experiments, a recent clinical study involving around 80 patients with mild cognitive impairment suggests that melatonin supplementation may also exhibit anti-tauogenic potential in humans. In particular, the study revealed that individuals who received a daily evening dose of 0.15 mg of melatonin per kg of body weight over six consecutive months exhibited CSF levels of tau proteins compared to those treated with a placebo [127].
Crossroads between melatonin and insulin
Insulin, a hormone primarily synthesized by pancreatic β-cells, has demonstrated notable efficacy in counteracting processes implicated in AD pathology upon reaching the brain [33]. Augmentation of brain insulin signaling enhanced cognitive functions in both cognitively healthy individuals and those with AD [128, 129]. Furthermore, insulin plays a crucial role in mitigating synaptic vulnerability to Aβos in hippocampal neurons [130, 131]. Supporting this, studies indicate that treatment with insulin or insulin-sensitizing drugs effectively reduced tau hyperphosphorylation in neurons [132, 133]. Conversely, a deficiency of brain insulin, as observed in transgenic mice, was associated with an increase in tau hyperphosphorylation, likely due to increased GSK3β activity [134].
Significantly, melatonin supplementation has demonstrated the capacity to alleviate oxidative stress and impede the accumulation of Aβ, as well as tau hyperphosphorylation, in the hippocampus of aged rats exhibiting brain insulin resistance induced by a high-fat diet [135]. The discovery that melatonin enhances the survival and function of insulin-producing β-cells [136] adds more support to the idea that melatonin plays a role in regulating insulin signaling. Particularly noteworthy is the discovery that melatonin signaling in β-cells led to a decrease in the activation of the stress response c-Jun N-terminal kinase [136]. This pathway is known to play a pivotal role in inducing brain insulin resistance in AD [131]. Further supporting melatonin’s potential impact on brain insulin, a rat study demonstrated that melatonin, upon binding to its membrane-bound receptors, can initiate intracellular pathways in the brain typically activated by insulin binding to the insulin receptor (e.g., AKT serine phosphorylation) [137].
As shown by animal studies, insulin may also play a crucial role in promoting melatonin synthesis in the pineal gland. Insulin can enhance norepinephrine-mediated melatonin production in the pineal gland of rats [138]. Additionally, insulin facilitates the transport of tryptophan into the brain, which is an essential precursor for melatonin synthesis [139].
Collectively, the evidence presented suggests that melatonin’s ability to mitigate AD may involve the modulation of whole-body insulin pathways, including the brain. However, it is essential to note that sustained elevation in plasma glucose concentrations due to reduced pancreatic insulin release from chronic melatonin treatment, especially when extending beyond the sleeping period [140], may compromise melatonin’s potential to ameliorate AD pathology [141, 142].
Oxidative stress - implications and mitigation with melatonin
Oxidative stress is characterized by an imbalance between the production of reactive species (e.g., ROS and RNS) and the body’s antioxidant defenses [143]. In the brain, prolonged oxidative stress initiates various pathological processes that play a pivotal role in AD pathology. Neuronal response to oxidative stress may activate GSKβ3, leading to the redirection of membrane-bound APP into the pro-amyloidogenic pathway (see Section Unlocking Melatonin’s Anti-Amyloidogenic Potential and Fig. 2). This results in an increase in intra- and extracellular Aβ. GSKβ3 also catalyzes the hyperphosphorylation of tau (see Section Melatonin’s Potential to Counteract Tau Pathology and Fig. 3), elevating the risk of NFTs. Prolonged oxidative stress can also induce apoptosis in neurons [144] and trigger neuroinflammation [145], both common characteristics in the brains of patients with AD [146, 147]. Consequently, countering prolonged oxidative stress emerges as a promising strategy to mitigate multiple processes associated with AD pathology.
In the context of CNS oxidative stress, melatonin emerges as a surprisingly prominent player. Distinguished as a potent non-enzymatic antioxidant and free radical scavenger, melatonin possesses the unique ability to neutralize free radicals independently of receptors, owing to its distinctive chemical structure [93]. This intrinsic function enables melatonin to directly detoxify ROS and RNS [93]. Leveraging its amphiphilic biochemical property [94], melatonin not only traverses the BBB but also gains access to brain cells and brain cell organelles, including mitochondria. This dual capability positions melatonin as a robust non-enzymatic antioxidant, functioning both intra- and extracellularly [93].
Furthermore, melatonin indirectly combats free radicals by chelating metal ions in the brain [148]. It also modulates the activity of pro-oxidative enzymes like nitric oxide synthase, while enhancing the activities of antioxidant enzymes, including glutathione peroxidase and superoxide dismutase in neurons [149,150,151,152]. Noteworthy findings from a mouse model of traumatic brain injury, a condition associated with increased oxidative stress in affected brain areas [153] and considered a risk factor for AD [154], underscore that melatonin treatment enhances the expression of nuclear factor erythroid 2-related factor 2 (Nrf2) [155]142. Nrf2, a redox-sensitive transcription factor, is pivotal in transcribing numerous antioxidant genes. The downregulation of Nrf2 is implicated in fostering oxidative stress in AD pathology [156].
Neuronal apoptosis also triggers oxidative stress [157]. Neuronal cell death activates microglia [158], leading to the production of more reactive species and the release of pro-inflammatory cytokines [159]. Melatonin has been shown to inhibit the expression of key signals involved in initiating apoptosis in rodent models of AD and various brain cell types. This includes Bax, a pro-apoptotic protein, and caspase-3, a key enzyme in apoptosis [160,161,162,163,164].
Notably, a study in Aβ plaque-bearing Tg2576 mice, initiated at 14 months of age, showed no significant impact on relieving oxidative damage, even with elevated plasma melatonin levels [165]. However, it was found effective in countering oxidative stress in a transgenic mouse model of amyloidosis when administered from four months of age [166]. These findings suggest that melatonin’s antioxidative effects may be more therapeutically relevant early during the disease.
Melatonin - A circadian shield
Virtually all cells within the body exhibit a robust circadian pattern, orchestrating the timing of various cellular processes throughout the day [167]. See Box 2 for a definition of circadian rhythms. Brain cells, including neurons, microglia, and astrocytes, also adhere to circadian rhythms [168,169,170]. To ensure synchronization among brain cell clocks, various mechanisms, including the release of melatonin by the pineal gland [171], come into play.
Among individuals with AD, pathways governing melatonin regulation and production may be affected. For instance, a reduction in the number of intrinsic photosensitive retinal ganglion cells has been observed in AD patients [172]. Additionally, there is evidence of structural and functional loss of neurons in the suprachiasmatic nucleus (SCN) in this disease [40, 173]. The SCN, located above the optic nerve crossing, plays a pivotal role in regulating the timing of sleep and wakefulness [174]. Neurodegeneration affecting the pineal gland could be an additional factor contributing to the observed lower melatonin levels in AD patients compared to age-matched non-demented controls [175].
Disrupted circadian rhythms in brain cells, stemming from impaired functioning of the retina-SCN-pineal axis, may contribute to both neurobehavioral symptoms and neurodegenerative processes commonly observed in AD. For instance, many AD patients display behavioral symptoms such as rest-activity rhythm fragmentation [176], sundowning (characterized by recurring confusion or agitation in the late afternoon or early evening) [177], and disturbances in sleep and wake maintenance [178]. Additionally, circadian disruption might promote neurodegenerative processes implicated in AD development, as indicated by recent animal studies. For example, the deletion of the core clock gene Bmal1 in the mouse brain led to astrocyte activation - an established marker of brain and neuronal injury - as well as synaptic degeneration [179]. Mass spectrometry analysis revealed a threefold increase in markers of neuronal membrane lipid peroxidation in Bmal1 knockout mice, reflecting heightened neuronal oxidative damage [179]. The increased oxidative stress in neurons was partially attributed to the reduced expression of redox defense genes [179]. In a separate study [180] using a mouse model of amyloidosis and disrupted circadian clock function through an inducible whole-organism deletion of the core clock gene Bmal1, these animals accumulated a higher hippocampal Aβ plaque burden. Furthermore, the deletion of Bmal1 in the brain induced apoE expression (inhibiting, e.g., astrocytic clearance of Aβ [73]), potentially elucidating how circadian disruption may increase Aβ burden in the brain.
While brain interstitial fluid tau concentrations are reported to be nearly twice as high during the active period of the rest-activity cycle in humans [8], it is noteworthy that, to the best of our knowledge, no study has investigated whether the deletion of core clock genes, simulating circadian disruption, leads to an increase in tau hyperphosphorylation. Despite this gap in knowledge, a plausible hypothesis could be formulated that either treatment with exogenous melatonin or interventions aimed at restoring endogenous melatonin secretion might offer potential in mitigating neurodegenerative processes induced by circadian disruption.
Melatonin’s role in safeguarding the BBB integrity and functionality
The BBB is a highly intricate structure composed of endothelial cells, pericytes, the capillary basement membrane, and astrocyte end-feet [181]. Its primary role is to serve as a crucial defense mechanism, preventing harmful substances from entering the brain while facilitating the transport of essential nutrients to brain tissue [181]. However, factors such as advanced aging, peripheral inflammation, and cerebral amyloid angiopathy can compromise the integrity of the BBB [182, 183]. A dysfunctional BBB can accelerate neuronal degeneration and cognitive decline, creating conditions that favor the accumulation of Aβ and the infiltration of neuroinflammatory agents and other molecules into the brain [184]. This disruption is hypothesized to contribute to the onset of various neurological disorders, including AD [185].
Melatonin demonstrates effectiveness in mitigating blood-brain barrier (BBB) damage through several mechanisms. Studies show that melatonin activates receptors to boost P-glycoprotein transporter activity, crucial for BBB function, particularly in methamphetamine-induced toxicity in rat brain endothelial cells [186]. Additionally, melatonin protects the BBB from methamphetamine damage by inhibiting nicotinamide adenine dinucleotide phosphate oxidase 2 via its receptors in these cells [187]. Melatonin’s protective effects on the BBB have also been confirmed in various experimental models. For instance, in a neonatal rat model, melatonin effectively reduced BBB damage caused by excitotoxicity [188]. Similarly, in young mice subjected to transient focal cerebral ischemia, melatonin demonstrated significant protective effects on the BBB [189, 190].
Melatonin’s potential to strengthen the BBB appears to involve the upregulation of tight junction proteins, including claudin-5, zonula occludens-1, and occluding [191], which help shield the brain from harmful substances in the bloodstream [192]. Additionally, melatonin may help maintain BBB integrity and function by interfering with angiotensin-converting enzyme 2, which acts as a receptor for viral entry. Daily injections of melatonin and melatonergic compounds have been shown to significantly reduce angiotensin-converting enzyme 2-dependent viral entry of severe acute respiratory syndrome coronavirus 2 into the brain [193]. This reduction is significant because this virus has been shown to exacerbate Aβ neurotoxicity and oxidative stress in patients with AD [194]. Melatonin also regulates matrix metalloproteinases (MMPs), enzymes known to compromise BBB integrity [195]. In a human gastric adenocarcinoma cell line, melatonin reduced MMP activity [196]. Furthermore, in aged mice treated with lipopolysaccharide to increase BBB permeability, melatonin-activated AMP-activated protein kinase in endothelial cells of the BBB [197], a kinase essential for maintaining BBB integrity [198].
In summary, these findings highlight melatonin’s role in protecting the BBB and mitigating damage across various experimental models. They underscore its potential as a therapeutic agent for maintaining BBB integrity and preventing neurological deterioration.
Melatonin and functioning of the glymphatic system
The role of sleep in brain waste clearance has been studied for over a decade, with recent studies suggesting a potential role for melatonin in glymphatic function. Originating in the subarachnoid space, CSF traverses periarterial spaces, mixes with interstitial fluid in the brain parenchyma, and ultimately exits via perivenous spaces [199]. While the first studies showing this were in mice [14], human studies have corroborated these findings [200], although its importance in relationship to traditional clearance pathways across the BBB and subarachnoidal granulations has been challenging to quantify.
Functioning as a clearance mechanism for the brain, the glymphatic system assists in removing potentially harmful waste products, including soluble Aβ, which accumulate in the interstitial fluid of the brain during the day [2, 21, 201, 202]. At the core of this process are aquaporin-4-expressing astrocytes enveloping perivascular spaces [203]. Animal studies have emphasized the efficiency of the glymphatic system in purging waste from the brain, with peak effectiveness during sleep [14], particularly at night [204], and reduced efficacy observed in animal models of hypertension [205]. However, the importance of sleep for the glymphatic system has not been confirmed by all investigations [206].
Melatonin, implicated in signaling the biological night to various cells by inducing hypothermia and promoting sleepiness [207, 208], may lower the release of the wake-promoting neuropeptide orexin, according to animal research [209]. Elevated orexin levels during sleep have been linked to increased Aβ pathology in mice brains [2, 210], possibly due to reduced glymphatic system efficacy [211]. Evidence in hypertensive patients suggests that melatonin can lower nocturnal blood pressure [212, 213]. These effects, in conjunction with potential sleep-enhancing benefits observed in patients with dementia treated with melatonin [214], provide a compelling rationale for hypothesizing that supplementing melatonin near bedtime may augment the glymphatic system’s clearance function. Supporting this hypothesis, a chronic unpredictable mild stress mouse model demonstrated that melatonin effectively restored aquaporin-4 polarization and rectified the compromised glymphatic system observed in this model [215]. Nonetheless, clinical trials involving both healthy individuals and those with mild cognitive impairment or AD are warranted to investigate whether melatonin can enhance the glymphatic system’s efficacy in reducing brain concentrations of Aβ and tau.
From animal models to clinical trials: the promise of melatonin in cognitive health
In animal models of AD, melatonin has shown promise in improving spatial memory and mitigating cognitive impairments. For example, in a sporadic AD mouse model induced by D-galactose and aluminum chloride, melatonin significantly enhanced short-term spatial memory. This improvement was linked to increased hippocampal expression of the memory-associated genes cAMP-responsive element-binding protein and brain-derived neurotrophic factor [216].
A systematic review and meta-analysis of nine studies involving 294 animals demonstrated that melatonin significantly improved learning abilities and corrected memory deficits in AD models [217]. This was evidenced by reduced escape latency, increased dwell time in the target quadrant, and more crossings over the platform location in the Morris Water Maze test. Melatonin was most effective in enhancing learning ability in senescence-related and metabolic AD models and in correcting memory deficits in toxin-induced AD models.
Studies on developing rats have also shown that melatonin can alleviate spatial learning and memory impairments by suppressing isoflurane-induced endoplasmic reticulum stress through the Sirtuin 1/Mitofusin 2/Protein kinase RNA-like endoplasmic reticulum kinase signaling pathway [218].
The positive effects of melatonin on cognition have also been documented in various human cohorts, including elderly subjects and patients with AD. A meta-analysis of 22 randomized controlled trials highlighted that patients with AD who received more than 12 weeks of melatonin treatment showed improvements in Mini-Mental State Examination scores, particularly in those with mild AD [219]. Additionally, a cross-sectional study of 1,105 community-dwelling elderly individuals found that higher physiological melatonin levels were correlated with a lower prevalence of depressed mood and cognitive impairment, independent of depressive symptoms [220].
A double-blind, placebo-controlled pilot study involving 26 healthy elderly subjects revealed that participants who received 1 mg of melatonin nightly for four weeks showed improved performance on the California Verbal Learning Test-interference subtest compared to those who received a placebo [221]. Further research demonstrated that melatonin enhances recognition memory accuracy for objects encoded under stress in healthy young men, indicating its role in central nervous system processing during stress and its potential to modulate memory consolidation [222].
A study involving individuals undergoing hemodialysis found that after six weeks of taking 3 mg of melatonin before bedtime, cognitive function significantly improved, with the Montreal Cognitive Assessment score increasing from 21.19 to 24.27 in the intervention group, compared to 22.15 in the control group [223]. Moreover, a study of 52 cognitively healthy adults, averaging 70 years of age, revealed a positive correlation between greater melatonin levels 6 hours before habitual bedtime and hippocampal volume - a region susceptible to AD pathology and integral to memory function [224, 225]. Finally, a network meta-analysis incorporating data from 50 randomized placebo-controlled trials involving approximately 20,000 AD patients demonstrated that the administration of melatonin (≤3 mg/day) over a period of 6 to 12 months was associated with improved cognitive function [226].
The evidence from both animal and human studies suggests that melatonin has significant potential as a therapeutic agent for improving cognitive function and mitigating cognitive impairments associated with AD.
Unlocking melatonin’s potential: evidence from observational studies and trials
The use of melatonin in older adults is increasing over time. In the US, the prevalence of melatonin use for sleep disorders among individuals 65 or older increased from 0.6% in 1999 to 2.1% in 2018, with a similar rise in use in UK. Melatonin appears to be a safe medication, particularly when compared to other sleep aids, but it remains understudied in older adults who show higher absorption compared to younger adults [227].
Observational studies in humans, while limited in number, provide intriguing evidence supporting a role for melatonin in the context of AD. In particular, studies showing diminished 24-hour melatonin levels or disrupted melatonin rhythm - where the primary endogenous melatonin release fails to synchronize with the dark phase of the daily light-dark cycle - have been correlated with an elevated risk of AD. Notably, a study involving approximately 276,000 participants from the UK Biobank cohort revealed that permanent night shift work, a condition acknowledged to reduce 24-hour melatonin levels [41], was associated with a 1.5-fold higher risk of developing AD during a median follow-up of 9 years [42]. Furthermore, the observation that visual loss is associated with an increased longitudinal risk of AD diagnosis, with a greater risk observed when visual loss occurs earlier in the lifespan [228], accentuates the potential significance of aligning the endogenous melatonin rhythm with the light-dark cycle for optimal brain health in humans.
Clinical investigations have shown that melatonin concentrations in the CSF of patients with AD are several-fold lower than those in age-matched control subjects without AD [75, 229]. Furthermore, lower CSF melatonin levels in AD patients are correlated with greater disease severity [229]. However, it remains unclear whether this reduction is due to decreased release from the pineal gland, increased breakdown of the hormone, or a combination of both factors.
Insights from a clinical trial involving nearly 200 nursing home residents in the Netherlands (mean age: 86; 87% with dementia) highlight the potential benefits of a daily evening regimen of melatonin (2.5 mg) combined with daytime bright light exposure over an average period of 15 months [214]. This regimen was associated with a reduction in agitated behavior - a common symptom in individuals with AD [230] - and improvements in sleep. However, some studies have failed to find an effect of melatonin on sleep in patients with AD [231, 232]. This discrepancy may be due to AD-related injury at later disease stages, emphasizing the need for clinical trials initiated prior to dementia onset. Another possibility is that patients may respond less to melatonin, particularly if they carry genetic polymorphisms associated with reduced expression of melatonin receptors [233].
As noted above, the majority of studies examining the impact of melatonin on AD-related mechanisms have been carried out in cell lines and animal models. Given the availability of biomarkers for AD that can be used to measure brain pathology in vivo (including neuroimaging with positron emission tomography, CSF, and plasma biomarker analysis), studies are needed in older adults which measure AD biomarkers in the context of melatonin use. Given its high potential for impacting AD-relevant pathologies with low risk for adverse events, additional studies of melatonin in the context of AD appear warranted.
Conclusion
AD stands as a substantial global health and economic challenge [234, 235], emphasizing the pressing need for effective therapeutic interventions and preventive strategies. Melatonin, recognized for its dual role as an antioxidant and hormone, emerges as a promising candidate for mitigating AD pathology. This review highlights its potential significance in AD prevention and potentially AD management, particularly given its high bioavailability, capacity to counteract free radicals, and its neuroprotective and chronotherapeutic properties.
However, the incorporation of melatonin into AD treatment will require additional studies. While some animal studies suggest a reduction in Aβ production with melatonin [55, 56], not all findings align with this trend. Lower melatonin doses (1.5 mg/kg/day) in a mouse model of AD (Tg2576) showed no significant modification of brain APP immunoreactivity compared to non-treated animals [165], emphasizing the need to determine efficacious doses. In this context, the consideration of dose-dependent side effects, such as exacerbating breathing difficulties in obstructive sleep apnea patients, impaired blood glucose control when consumed with carbohydrate-rich meals, and an increased risk of nighttime falls due to drowsiness, especially in the elderly, is imperative [236,237,238]. Challenges, including unsupervised medication use, discrepancies in labeled versus actual melatonin content in supplements, and variations in pharmacokinetics (immediate-release vs. extended-release formulations), underscore the need for physician supervision and strict product regulation [239, 240].
On the positive side, melatonin’s accessibility, affordability, and potential benefits position it as a promising intervention that requires further testing. Studies among both AD dementia populations as well as in preclinical asymptomatic AD coupled with biomarker testing, are needed to address remaining gaps to translation. Since peptides, proteins, and hormones can directly reach the brain when administered intranasally via transport and diffusion along the olfactory and trigeminal nerves [241], future studies should explore whether intranasal melatonin could be a viable therapeutic option for increasing brain melatonin levels in individuals at risk of developing AD. Notably, intranasal melatonin has demonstrated effectiveness in improving sleep in proof-of-concept studies [242].
References
GBD 2019 Dementia Forecasting Collaborators. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health. 2022;7:e105–e125.
Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–7.
Lim AS, Kowgier M, Yu L, Buchman AS, Bennett DA. Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep. 2013;36:1027–32.
Ju YE, McLeland JS, Toedebusch CD, Xiong C, Fagan AM, Duntley SP, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70:587–93.
Benedict C, Byberg L, Cedernaes J, Hogenkamp PS, Giedratis V, Kilander L, et al. Self-reported sleep disturbance is associated with Alzheimer’s disease risk in men. Alzheimers Dement. 2015;11:1090–7.
Ju YS, Ooms SJ, Sutphen C, Macauley SL, Zangrilli MA, Jerome G, et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain. 2017;140:2104–11.
Yeo BSY, Koh JH, Ng ACW, Loh S, See A, Seow DCC, et al. Sleep disturbances increase the risk of dementia: A systematic review and meta-analysis. Sleep Med Rev. 2018;40:4–16.
Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, et al. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019;363:880–4.
Ju YS, Zangrilli MA, Finn MB, Fagan AM, Holtzman DM. Obstructive sleep apnea treatment, slow wave activity, and amyloid-β. Ann Neurol. 2019;85:291–5.
Lucey BP, Wisch J, Boerwinkle AH, Landsness EC, Toedebusch CD, McLeland JS, et al. Sleep and longitudinal cognitive performance in preclinical and early symptomatic Alzheimer’s disease. Brain. 2021;144:2852–62.
Wang C, Nambiar A, Strickland MR, Lee C, Parhizkar S, Moore AC, et al. APOE-ε4 synergizes with sleep disruption to accelerate Aβ deposition and Aβ-associated tau seeding and spreading. J Clin Invest. 2023;133:e169131.
Parhizkar S, Gent G, Chen Y, Rensing N, Gratuze M, Strout G, et al. Sleep deprivation exacerbates microglial reactivity and Aβ deposition in a TREM2-dependent manner in mice. Sci Transl Med. 2023;15:eade6285.
Pulver RL, Kronberg E, Medenblik LM, Kheyfets VO, Ramos AR, Holtzman DM, et al. Mapping sleep’s oscillatory events as a biomarker of Alzheimer’s disease. Alzheimers Dement. 2024;20:301–15.
Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7.
Medina-Flores F, Hurtado-Alvarado G, Contis-Montes de Oca A, López-Cervantes SP, Konigsberg M, et al. Sleep loss disrupts pericyte-brain endothelial cell interactions impairing blood-brain barrier function. Brain Behav Immun. 2020;89:118–32.
de Vivo L, Bellesi M, Marshall W, Bushong EA, Ellisman MH, Tononi G, et al. Ultrastructural evidence for synaptic scaling across the wake/sleep cycle. Science. 2017;355:507–10.
Diekelmann S, Born J. The memory function of sleep. Nat Rev Neurosci. 2010;11:114–26.
Bellesi M, de Vivo L, Chini M, Gilli F, Tononi G, Cirelli C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J Neurosci. 2017;37:5263–73.
Zhu B, Dong Y, Xu Z, Gompf HS, Ward SA, Xue Z, et al. Sleep disturbance induces neuroinflammation and impairment of learning and memory. Neurobiol Dis. 2012;48:348–55.
Lew CH, Petersen C, Neylan TC, Grinberg LT. Tau-driven degeneration of sleep- and wake-regulating neurons in Alzheimer’s disease. Sleep Med Rev. 2021;60:101541.
Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci Transl Med. 2012;4:150ra122.
Arendt J, Skene DJ. Melatonin as a chronobiotic. Sleep Med Rev. 2005;9:25–39.
Hardeland R. Melatonin metabolism in the central nervous system. Curr Neuropharmacol. 2010;8:168–81.
Cipolla-Neto J, Amaral FGD. Melatonin as a hormone: new physiological and clinical insights. Endocr Rev. 2018;39:990–1028.
Skinner DC, Malpaux B. High melatonin concentrations in third ventricular cerebrospinal fluid are not due to Galen vein blood recirculating through the choroid plexus. Endocrinology. 1999;140:4399–405.
Leston J, Harthé C, Brun J, Mottolese C, Mertens P, Sindou M, et al. Melatonin is released in the third ventricle in humans. A study in movement disorders. Neurosci Lett. 2010;469:294–7.
Longatti P, Perin A, Rizzo V, Comai S, Giusti P, Costa CV. Ventricular cerebrospinal fluid melatonin concentrations investigated with an endoscopic technique. J Pineal Res. 2007;42:113–8.
Bojkowski CJ, Arendt J, Shih MC, Markey SP. Melatonin secretion in humans assessed by measuring its metabolite, 6-sulfatoxymelatonin. Clin Chem. 1987;33:1343–8.
Ahmad SB, Ali A, Bilal M, Rashid SM, Wani AB, Bhat RR, et al. Melatonin and health: insights of melatonin action, biological functions, and associated disorders. Cell Mol Neurobiol. 2023;43:2437–58.
Cecon E, Oishi A, Jockers R. Melatonin receptors: molecular pharmacology and signalling in the context of system bias. Br J Pharmacol. 2018;175:3263–80.
Cline EN, Bicca MA, Viola KL, Klein WL. The Amyloid-β Oligomer hypothesis: beginning of the third decade. J Alzheimers Dis. 2018;64:S567–S610.
Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–39.
De Felice FG, Gonçalves RA, Ferreira ST. Impaired insulin signalling and allostatic load in Alzheimer disease. Nat Rev Neurosci. 2022;23:215–30.
Sack RL, Lewy AJ, Erb DL, Vollmer WM, Singer CM. Human melatonin production decreases with age. J Pineal Res. 1986;3:379–88.
Tan DX, Xu B, Zhou X, Reiter RJ. Pineal calcification, melatonin production, aging, associated health consequences and rejuvenation of the pineal gland. Molecules. 2018;23:301.
Swaab DF, Fliers E, Partiman TS. The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 1985;342:37–44.
Skene DJ, Vivien-Roels B, Sparks DL, Hunsaker JC, Pévet P, Ravid D, et al. Daily variation in the concentration of melatonin and 5-methoxytryptophol in the human pineal gland: effect of age and Alzheimer’s disease. Brain Res. 1990;528:170–4.
Ohashi Y, Okamoto N, Uchida K, Iyo M, Mori N, Morita Y. Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer’s type. Biol Psychiatry. 1999;45:1646–52.
Wu YH, Feenstra MG, Zhou JN, Liu RY, Toranõ JS, Van Kan HJ, et al. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab. 2003;88:5898–906.
Wang JL, Lim AS, Chiang WY, Hsieh WH, Lo MT, Schneider JA, et al. Suprachiasmatic neuron numbers and rest-activity circadian rhythms in older humans. Ann Neurol. 2015;78:317–22.
Wei T, Li C, Heng Y, Gao X, Zhang G, Wang H, et al. Association between night-shift work and level of melatonin: systematic review and meta-analysis. Sleep Med. 2020;75:502–9.
Ren JJ, Zhang PD, Li ZH, Zhang XR, Zhong WF, Chen PL, et al. Association of night shifts and lifestyle risks with incident dementia. J Gerontol A Biol Sci Med Sci. 2023;78:1725–32.
Hampel H, Hardy J, Blennow K, Chen C, Perry G, Kim SH, et al. The Amyloid-β pathway in Alzheimer’s disease. Mol Psychiatry. 2021;26:5481–503.
Mucke L, Selkoe DJ. Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338.
Ashleigh T, Swerdlow RH, Beal MF. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimers Dement. 2023;19:333–42.
Bai R, Guo J, Ye XY, Xie Y, Xie T. Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease. Ageing Res Rev. 2022;77:101619.
Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–91.
Snyder SW, Ladror US, Wade WS, Wang GT, Barrett LW, Matayoshi ED, et al. Amyloid-beta aggregation: selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys J. 1994;67:1216–28.
Esbjörner EK, Chan F, Rees E, Erdelyi M, Luheshi LM, Bertoncini CW, et al. Direct observations of amyloid β self-assembly in live cells provide insights into differences in the kinetics of Aβ(1-40) and Aβ(1-42) aggregation. Chem Biol. 2014;21:732–42.
Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–66.
Portelius E, Price E, Brinkmalm G, Stiteler M, Olsson M, Persson R, et al. A novel pathway for amyloid precursor protein processing. Neurobiol Aging. 2011;32:1090–8.
Shukla M, Htoo HH, Wintachai P, Hernandez JF, Dubois C, Postina R, et al. Melatonin stimulates the nonamyloidogenic processing of βAPP through the positive transcriptional regulation of ADAM10 and ADAM17. J Pineal Res. 2015;58:151–65.
Kovalevich J, Langford D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol Biol. 2013;1078:9–21.
Panmanee J, Nopparat C, Chavanich N, Shukla M, Mukda S, Song W, et al. Melatonin regulates the transcription of βAPP-cleaving secretases mediated through melatonin receptors in human neuroblastoma SH-SY5Y cells. J Pineal Res. 2015;59:308–20.
Lahiri DK, Chen D, Ge YW, Bondy SC, Sharman EH. Dietary supplementation with melatonin reduces levels of amyloid beta-peptides in the murine cerebral cortex. J Pineal Res. 2004;36:224–31.
Rudnitskaya EA, Muraleva NA, Maksimova KY, Kiseleva E, Kolosova NG, Stefanova NA. Melatonin attenuates memory impairment, Amyloid-β accumulation, and neurodegeneration in a rat model of sporadic Alzheimer’s disease. J Alzheimers Dis. 2015;47:103–16.
Ng KM, Lau CF, Fung ML. Melatonin reduces hippocampal beta-amyloid generation in rats exposed to chronic intermittent hypoxia. Brain Res. 2010;1354:163–71.
Cecon E, Chen M, Marçola M, Fernandes PA, Jockers R, Markus RP. Amyloid β peptide directly impairs pineal gland melatonin synthesis and melatonin receptor signaling through the ERK pathway. FASEB J. 2015;29:2566–82.
Uemura K, Kuzuya A, Shimozono Y, Aoyagi N, Ando K, Shimohama S, et al. GSK3beta activity modifies the localization and function of presenilin 1. J Biol Chem. 2007;282:15823–32.
Ly PT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013;123:224–35.
Peng CX, Hu J, Liu D, Hong XP, Wu YY, Zhu LQ, et al. Disease-modified glycogen synthase kinase-3β intervention by melatonin arrests the pathology and memory deficits in an Alzheimer’s animal model. Neurobiol Aging. 2013;34:1555–63.
Nopparat C, Chaopae W, Boontem P, Sopha P, Wongchitrat P, Govitrapong P. Melatonin attenuates high glucose-induced changes in beta amyloid precursor protein processing in human neuroblastoma cells. Neurochem Res. 2022;47:2568–79.
Kurochkin IV, Guarnera E, Berezovsky IN. Insulin-degrading enzyme in the fight against Alzheimer’s disease. Trends Pharmacol Sci. 2018;39:49–58.
Jolivalt CG, Lee CA, Beiswenger KK, Smith JL, Orlov M, Torrance MA, et al. Defective insulin signaling pathway and increased glycogen synthase kinase-3 activity in the brain of diabetic mice: parallels with Alzheimer’s disease and correction by insulin. J Neurosci Res. 2008;86:3265–74.
Li LB, Fan YG, Wu WX, Bai CY, Jia MY, Hu JP, et al. Novel melatonin-trientine conjugate as potential therapeutic agents for Alzheimer’s disease. Bioorg Chem. 2022;128:106100.
Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol. 2007;28:5–11.
Thal DR. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp Neurol. 2012;236:1–5.
Li M, Pi H, Yang Z, Reiter RJ, Xu S, Chen X, et al. Melatonin antagonizes cadmium-induced neurotoxicity by activating the transcription factor EB-dependent autophagy-lysosome machinery in mouse neuroblastoma cells. J Pineal Res. 2016;61:353–69.
Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, et al. Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34:9607–20.
Promyo K, Iqbal F, Chaidee N, Chetsawang B. Aluminum chloride-induced amyloid β accumulation and endoplasmic reticulum stress in rat brain are averted by melatonin. Food Chem Toxicol. 2020;146:111829.
Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front Aging Neurosci. 2014;6:93.
Hatters DM, Peters-Libeu CA, Weisgraber KH. Apolipoprotein E structure: insights into function. Trends Biochem Sci. 2006;31:445–54.
Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci USA. 2013;110:E1807–16.
Poeggeler B, Miravalle L, Zagorski MG, Wisniewski T, Chyan YJ, Zhang Y, et al. Melatonin reverses the profibrillogenic activity of apolipoprotein E4 on the Alzheimer amyloid Abeta peptide. Biochemistry. 2001;40:14995–5001.
Liu RY, Zhou JN, van Heerikhuize J, Hofman MA, Swaab DF. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon4/4 genotype. J Clin Endocrinol Metab. 1999;84:323–7.
Pappolla MA, Matsubara E, Vidal R, Pacheco-Quinto J, Poeggeler B, Zagorski M, et al. Melatonin treatment enhances Aβ lymphatic clearance in a transgenic mouse model of amyloidosis. Curr Alzheimer Res. 2018;15:637–42.
Birnbaum JH, Bali J, Rajendran L, Nitsch RM, Tackenberg C. Calcium flux-independent NMDA receptor activity is required for Aβ oligomer-induced synaptic loss. Cell Death Dis. 2015;6:e1791.
Bano D, Ankarcrona M. Beyond the critical point: An overview of excitotoxicity, calcium overload and the downstream consequences. Neurosci Lett. 2018;663:79–85.
Escames G, León J, López LC, Acuña-Castroviejo D. Mechanisms of N-methyl-D-aspartate receptor inhibition by melatonin in the rat striatum. J Neuroendocrinol. 2004;16:929–35.
Sutcu R, Yonden Z, Yilmaz A, Delibas N. Melatonin increases NMDA receptor subunits 2A and 2B concentrations in rat hippocampus. Mol Cell Biochem. 2006;283:101–5.
Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, et al. Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274:37111–6.
Hung YH, Bush AI, Cherny RA. Copper in the brain and Alzheimer’s disease. J Biol Inorg Chem. 2010;15:61–76.
Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018;14:450–64.
Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–49.
Wang X, Su B, Perry G, Smith MA, Zhu X. Insights into amyloid-beta-induced mitochondrial dysfunction in Alzheimer disease. Free Radic Biol Med. 2007;43:1569–73.
Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem. 2002;80:91–100.
Wu L, Xian X, Xu G, Tan Z, Dong F, Zhang M, et al. Toll-like Receptor 4: A promising therapeutic target for Alzheimer’s disease. Mediators Inflamm. 2022;2022:7924199.
Simpson DSA, Oliver PL. ROS generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants. 2020;9:743.
Ostapchenko VG, Chen M, Guzman MS, Xie YF, Lavine N, Fan J, et al. The Transient Receptor Potential Melastatin 2 (TRPM2) Channel Contributes to β-Amyloid Oligomer-related neurotoxicity and memory impairment. J Neurosci. 2015;35:15157–69.
Feng Y, Xia Y, Yu G, Shu X, Ge H, Zeng K, et al. Cleavage of GSK-3β by calpain counteracts the inhibitory effect of Ser9 phosphorylation on GSK-3β activity induced by H2O2. J Neurochem. 2013;126:234–42.
Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, et al. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J Neuropathol Exp Neurol. 1999;58:1010–9.
Lauretti E, Dincer O, Praticò D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim Biophys Acta Mol Cell Res. 2020;1867:118664.
Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L. Melatonin as an antioxidant: under promises but over delivers. J Pineal Res. 2016;61:253–78.
Amaral FGD, Cipolla-Neto J. A brief review about melatonin, a pineal hormone. Arch Endocrinol Metab. 2018;62:472–9.
Ionov M, Burchell V, Klajnert B, Bryszewska M, Abramov AY. Mechanism of neuroprotection of melatonin against beta-amyloid neurotoxicity. Neuroscience. 2011;180:229–37.
Tang Y, Le W. Differential roles of M1 and M2 Microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53:1181–94.
Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3:136.
Sarlus H, Heneka MT. Microglia in Alzheimer’s disease. J Clin Invest. 2017;127:3240–9.
ElAli A, Rivest S. Microglia in Alzheimer’s disease: A multifaceted relationship. Brain Behav Immun. 2016;55:138–50.
Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–27.
Azedi F, Mehrpour M, Talebi S, Zendedel A, Kazemnejad S, Mousavizadeh K, et al. Melatonin regulates neuroinflammation ischemic stroke damage through interactions with microglia in reperfusion phase. Brain Res. 2019;1723:146401.
Maccioni RB, Cambiazo V. Role of microtubule-associated proteins in the control of microtubule assembly. Physiol Rev. 1995;75:835–64.
Horowitz JM, Horwitz BA. Extreme neuroplasticity of Hippocampal CA1 pyramidal neurons in hibernating mammalian species. Front Neuroanat. 2019;13:9.
Plattner F, Angelo M, Giese KP. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006;281:25457–65.
Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Yardin C, et al. Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res Rev. 2013;12:289–309.
Köpke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem. 1993;268:24374–84.
Holtzman DM, Carrillo MC, Hendrix JA, Bain LJ, Catafau AM, Gault LM, et al. Tau: From research to clinical development. Alzheimers Dement. 2016;12:1033–9.
Knopman DS, Amieva H, Petersen RC, Chételat G, Holtzman DM, Hyman BT, et al. Alzheimer disease. Nat Rev Dis Primers. 2021;7:33.
Yoshiyama Y, Lee VM, Trojanowski JQ. Frontotemporal dementia and tauopathy. Curr Neurol Neurosci Rep. 2001;1:413–21.
Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28.
Yin X, Qiu Y, Zhao C, Zhou Z, Bao J, Qian W. The role of amyloid-beta and tau in the early pathogenesis of Alzheimer’s disease. Med Sci Monit. 2021;27:e933084.
Ossenkoppele R, Pichet Binette A, Groot C, Smith R, Strandberg O, Palmqvist S, et al. Amyloid and tau PET-positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat Med. 2022;28:2381–7.
Wang XC, Zhang J, Yu X, Han L, Zhou ZT, Zhang Y, et al. Prevention of isoproterenol-induced tau hyperphosphorylation by melatonin in the rat. Sheng Li Xue Bao. 2005;57:7–12.
Zhu LQ, Wang SH, Ling ZQ, Wang DL, Wang JZ. Effect of inhibiting melatonin biosynthesis on spatial memory retention and tau phosphorylation in rat. J Pineal Res. 2004;37:71–7.
Chen D, Lan G, Li R, Mei Y, Shui X, Gu X, et al. Melatonin ameliorates tau-related pathology via the miR-504-3p and CDK5 axis in Alzheimer’s disease. Transl Neurodegener. 2022;11:27.
Olivieri G, Brack C, Müller-Spahn F, Stähelin HB, Herrmann M, Renard P, et al. Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SH-SY5Y neuroblastoma cells. J Neurochem. 2000;74:231–6.
Liu SJ, Wang JZ. Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol Sin. 2002;23:183–7.
Deng YQ, Xu GG, Duan P, Zhang Q, Wang JZ. Effects of melatonin on wortmannin-induced tau hyperphosphorylation. Acta Pharmacol Sin. 2005;26:519–26.
Zhang Y, Huang NQ, Yan F, Jin H, Zhou SY, Shi JS, et al. Diabetes mellitus and Alzheimer’s disease: GSK-3β as a potential link. Behav Brain Res. 2018;339:57–65.
David DC, Hauptmann S, Scherping I, Schuessel K, Keil U, Rizzu P, et al. Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J Biol Chem. 2005;280:23802–14.
Wang DL, Ling ZQ, Cao FY, Zhu LQ, Wang JZ. Melatonin attenuates isoproterenol-induced protein kinase A overactivation and tau hyperphosphorylation in rat brain. J Pineal Res. 2004;37:11–6.
Wang JZ, Wu Q, Smith A, Grundke-Iqbal I, Iqbal K. Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett. 1998;436:28–34.
Liu SJ, Zhang JY, Li HL, Fang ZY, Wang Q, Deng HM, et al. Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J Biol Chem. 2004;279:50078–88.
Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–7.
Chinchalongporn V, Shukla M, Govitrapong P. Melatonin ameliorates Aβ42 -induced alteration of βAPP-processing secretases via the melatonin receptor through the Pin1/GSK3β/NF-κB pathway in SH-SY5Y cells. J Pineal Res. 2018;64:e12470.
Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–8.
Xu L, Yu H, Sun H, Hu B, Geng Y. Dietary melatonin therapy alleviates the lamina cribrosa damages in patients with mild cognitive impairments: a double-blinded, randomized controlled study. Med Sci Monit. 2020;26:e923232.
Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J, et al. Intranasal insulin improves memory in humans. Psychoneuroendocrinology. 2004;29:1326–34.
Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A, et al. Long acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis. 2015;45:1269–70.
De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, et al. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc Natl Acad Sci USA. 2009;106:1971–6.
Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Aβ oligomers. J Clin Invest. 2012;122:1339–53.
Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem. 1997;272:19547–53.
Tokutake T, Kasuga K, Yajima R, Sekine Y, Tezuka T, Nishizawa M, et al. Hyperphosphorylation of Tau induced by naturally secreted amyloid-β at nanomolar concentrations is modulated by insulin-dependent Akt-GSK3β signaling pathway. J Biol Chem. 2012;287:35222–33.
Schechter R, Beju D, Miller KE. The effect of insulin deficiency on tau and neurofilament in the insulin knockout mouse. Biochem Biophys Res Commun. 2005;334:979–86.
Xu J, Gao H, Zhang L, Rong S, Yang W, Ma C, et al. Melatonin alleviates cognition impairment by antagonizing brain insulin resistance in aged rats fed a high-fat diet. J Pineal Res. 2019;67:e12584.
Costes S, Boss M, Thomas AP, Matveyenko AV. Activation of melatonin signaling promotes β-cell survival and function. Mol Endocrinol. 2015;29:682–92.
Anhê GF, Caperuto LC, Pereira-Da-Silva M, Souza LC, Hirata AE, Velloso LA, et al. In vivo activation of insulin receptor tyrosine kinase by melatonin in the rat hypothalamus. J Neurochem. 2004;90:559–66.
Garcia RA, Afeche SC, Scialfa JH, do Amaral FG, dos Santos SH, Lima FB, et al. Insulin modulates norepinephrine-mediated melatonin synthesis in cultured rat pineal gland. Life Sci. 2008;82:108–14.
Tagliamonte A, DeMontis MG, Olianas M, Onali PL, Gessa GL. Possible role of insulin in the transport of tyrosine and tryptophan from blood to brain. Adv Exp Med Biol. 1976;69:89–94.
Eckel RH, Depner CM, Perreault L, Markwald RR, Smith MR, McHill AW, et al. Morning Circadian Misalignment during Short Sleep Duration Impacts Insulin Sensitivity. Curr Biol. 2015;25:3004–10.
Macauley SL, Stanley M, Caesar EE, Yamada SA, Raichle ME, Perez R, et al. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J Clin Invest. 2015;125:2463–7.
Chao AC, Lee TC, Juo SH, Yang DI. Hyperglycemia increases the production of Amyloid Beta-peptide leading to decreased endothelial tight junction. CNS Neurosci Ther. 2016;22:291–7.
Storz G, Imlay JA. Oxidative stress. Curr Opin Microbiol. 1999;2:188–94.
Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–9.
Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des. 2010;16:2766–78.
Shimohama S. Apoptosis in Alzheimer’s disease–an update. Apoptosis. 2000;5:9–16.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–72.
Gulcin I, Buyukokuroglu ME, Kufrevioglu OI. Metal chelating and hydrogen peroxide scavenging effects of melatonin. J Pineal Res. 2003;34:278–81.
León J, Escames G, Rodríguez MI, López LC, Tapias V, Entrena A, et al. Inhibition of neuronal nitric oxide synthase activity by N1-acetyl-5-methoxykynuramine, a brain metabolite of melatonin. J Neurochem. 2006;98:2023–33.
Urata Y, Honma S, Goto S, Todoroki S, Iida T, Cho S, et al. Melatonin induces gamma-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic Biol Med. 1999;27:838–47.
Mayo JC, Sainz RM, Antoli I, Herrera F, Martin V, Rodriguez C. Cell Mol Melatonin regulation of antioxidant enzyme gene expression. Life Sci. 2002;59:1706–13.
Barlow-Walden LR, Reiter RJ, Abe M, Pablos M, Menendez-Pelaez A, Chen LD, et al. Melatonin stimulates brain glutathione peroxidase activity. Neurochem Int. 1995;26:497–502.
Rodríguez-Rodríguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A. Oxidative stress in traumatic brain injury. Curr Med Chem. 2014;21:1201–11.
Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396:413–46.
Wang J, Jiang C, Zhang K, Lan X, Chen X, Zang W, et al. Melatonin receptor activation provides cerebral protection after traumatic brain injury by mitigating oxidative stress and inflammation via the Nrf2 signaling pathway. Free Radic Biol Med. 2019;131:345–55.
Sharma V, Kaur A, Singh TG. Counteracting role of nuclear factor erythroid 2-related factor 2 pathway in Alzheimer’s disease. Biomed Pharmacother. 2020;129:110373.
Kannan K, Jain SK. Oxidative stress and apoptosis. Pathophysiology. 2000;7:153–63.
Minghetti L, Ajmone-Cat MA, De Berardinis MA, De Simone R. Microglial activation in chronic neurodegenerative diseases: roles of apoptotic neurons and chronic stimulation. Brain Res Brain Res Rev. 2005;48:251–6.
Radi E, Formichi P, Battisti C, Federico A. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis. 2014;42:S125–52.
Baydas G, Reiter RJ, Akbulut M, Tuzcu M, Tamer S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and anti-apoptotic protein levels. Neuroscience. 2005;135:879–86.
Feng Z, Qin C, Chang Y, Zhang JT. Early melatonin supplementation alleviates oxidative stress in a transgenic mouse model of Alzheimer’s disease. Free Radic Biol Med. 2006;40:101–9.
Das A, Belagodu A, Reiter RJ, Ray SK, Banik NL. Cytoprotective effects of melatonin on C6 astroglial cells exposed to glutamate excitotoxicity and oxidative stress. J Pineal Res. 2008;45:117–24.
Waseem M, Sahu U, Salman M, Choudhury A, Kar S, Tabassum H, et al. Melatonin pre-treatment mitigates SHSY-5Y cells against oxaliplatin induced mitochondrial stress and apoptotic cell death. PLoS One. 2017;12:e0180953.
Huang R, Xu Y, Lu X, Tang X, Lin J, Cui K, et al. Melatonin protects inner retinal neurons of newborn mice after hypoxia-ischemia. J Pineal Res. 2021;71:e12716.
Quinn J, Kulhanek D, Nowlin J, Jones R, Praticò D, Rokach J, et al. Chronic melatonin therapy fails to alter amyloid burden or oxidative damage in old Tg2576 mice: implications for clinical trials. Brain Res. 2005;1037:209–13.
Matsubara E, Bryant-Thomas T, Pacheco Quinto J, Henry TL, Poeggeler B, et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J Neurochem. 2003;85:1101–8.
Dibner C, Schibler U, Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annu Rev Physiol. 2010;72:517–49.
Herzog ED. Neurons and networks in daily rhythms. Nat Rev Neurosci. 2007;8:790–802.
Prolo LM, Takahashi JS, Herzog ED. Circadian rhythm generation and entrainment in astrocytes. J Neurosci. 2005;25:404–8.
Brécier A, Li VW, Smith CS, Halievski K, Ghasemlou N. Circadian rhythms and glial cells of the central nervous system. Biol Rev Camb Philos Soc. 2023;98:520–39.
Sack RL, Brandes RW, Kendall AR, Lewy AJ. Entrainment of free-running circadian rhythms by melatonin in blind people. N Engl J Med. 2000;343:1070–7.
La Morgia C, Ross-Cisneros FN, Koronyo Y, Hannibal J, Gallassi R, Cantalupo G, et al. Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann Neurol. 2016;79:90–109.
Zhou JN, Hofman MA, Swaab DF. VIP neurons in the human SCN in relation to sex, age, and Alzheimer’s disease. Neurobiol Aging. 1995;16:571–6.
Moore RY. Organization and function of a central nervous system circadian oscillator: the suprachiasmatic hypothalamic nucleus. Fed Proc. 1983;42:2783–9.
Wu YH, Swaab DF. The human pineal gland and melatonin in aging and Alzheimer’s disease. J Pineal Res. 2005;38:145–52.
Musiek ES, Bhimasani M, Zangrilli MA, Morris JC, Holtzman DM, Ju YS. Circadian rest-activity pattern changes in aging and preclinical Alzheimer disease. JAMA Neurol. 2018;75:582–90.
Little JT, Satlin A, Sunderland T, Volicer L. Sundown syndrome in severely demented patients with probable Alzheimer’s disease. J Geriatr Psychiatry Neurol. 1995;8:103–6.
Leng Y, Musiek ES, Hu K, Cappuccio FP, Yaffe K. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol. 2019;18:307–18.
Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest. 2013;123:5389–400.
Kress GJ, Liao F, Dimitry J, Cedeno MR, FitzGerald GA, Holtzman DM, et al. Regulation of amyloid-β dynamics and pathology by the circadian clock. J Exp Med. 2018;215:1059–68.
Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629–38.
Blanchard JW, Bula M, Davila-Velderrain J, Akay LA, Zhu L, Frank A, et al. Reconstruction of the human blood-brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat Med. 2020;26:952–63.
Sharma C, Woo H, Kim SR. Addressing blood-brain barrier impairment in Alzheimer’s disease. Biomedicines. 2022;10:742.
Kadry H, Noorani B, Cucullo L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS. 2020;17:69.
Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–50.
Jumnongprakhon P, Sivasinprasasn S, Govitrapong P, Tocharus C, Tocharus J. Toxicol Activation of melatonin receptor (MT1/2) promotes P-gp transporter in methamphetamine-induced toxicity on primary rat brain microvascular endothelial cells. In Vitro. 2017;41:42–8.
Jumnongprakho P, Govitrapong P, Tocharus C, Tocharus J. Inhibitory effect of melatonin on cerebral endothelial cells dysfunction induced by methamphetamine via NADPH oxidase-2. Brain Res. 2016;1650:84–92.
Moretti R, Zanin A, Pansiot J, Spiri D, Manganozzi L, Kratzer I, et al. Melatonin reduces excitotoxic blood-brain barrier breakdown in neonatal rats. Neuroscience. 2015;311:382–97.
Chen HY, Chen TY, Lee MY, Chen ST, Hsu YS, Kuo YL, et al. Melatonin decreases neurovascular oxidative/nitrosative damage and protects against early increases in the blood-brain barrier permeability after transient focal cerebral ischemia in mice. J Pineal Res. 2006;41:175–82.
Chen TY, Lee MY, Chen HY, Kuo YL, Lin SC, Wu TS, et al. Melatonin attenuates the postischemic increase in blood-brain barrier permeability and decreases hemorrhagic transformation of tissue-plasminogen activator therapy following ischemic stroke in mice. J Pineal Res. 2006;40:242–50.
Qin W, Li J, Zhu R, Gao S, Fan J, Xia M, et al. Melatonin protects blood-brain barrier integrity and permeability by inhibiting matrix metalloproteinase-9 via the NOTCH3/NF-κB pathway. Aging. 2019;11:11391–415.
Sugiyama S, Sasaki T, Tanaka H, Yan H, Ikegami T, Kanki H, et al. The tight junction protein occludin modulates blood-brain barrier integrity and neurological function after ischemic stroke in mice. Sci Rep. 2023;13:2892.
Cecon E, Fernandois D, Renault N, Coelho CFF, Wenzel J, Bedart C, et al. Melatonin drugs inhibit SARS-CoV-2 entry into the brain and virus-induced damage of cerebral small vessels. Cell Mol Life Sci. 2022;79:361.
Chiricosta L, Gugliandolo A, Mazzon E. SARS-CoV-2 exacerbates beta-amyloid neurotoxicity, inflammation and oxidative stress in Alzheimer’s disease patients. Int J Mol Sci. 2021;22:13603.
Rempe RG, Hartz AMS, Bauer B. Matrix metalloproteinases in the brain and blood-brain barrier: Versatile breakers and makers. J Cereb Blood Flow Metab. 2016;36:1481–507.
Rudra DS, Pal U, Maiti NC, Reiter RJ, Swarnakar S. Melatonin inhibits matrix metalloproteinase-9 activity by binding to its active site. J Pineal Res. 2013;54:398–405.
Wang X, Xue GX, Liu WC, Shu H, Wang M, Sun Y, et al. Melatonin alleviates lipopolysaccharide-compromised integrity of blood-brain barrier through activating AMP-activated protein kinase in old mice. Aging Cell. 2017;16:414–21.
Liu C, Wu J, Zou MH. Activation of AMP-activated protein kinase alleviates high-glucose-induced dysfunction of brain microvascular endothelial cell tight-junction dynamics. Free Radic Biol Med. 2012;53:1213–21.
Hablitz LM, Nedergaard M. The glymphatic system: a novel component of fundamental neurobiology. J Neurosci. 2021;41:7698–711.
Fultz NE, Bonmassar G, Setsompop K, Stickgold RA, Rosen BR, Polimeni JR, et al. Coupled electrophysiological, hemodynamic, and cerebrospinal fluid oscillations in human sleep. Science. 2019;366:628–31.
Bateman RJ, Wen G, Morris JC, Holtzman DM. Fluctuations of CSF amyloid-beta levels: implications for a diagnostic and therapeutic biomarker. Neurology. 2007;68:666–9.
Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, et al. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011;14:750–6.
Jessen NA, Munk AS, Lundgaard I, Nedergaard M. The Glymphatic system: a beginner’s guide. Neurochem Res. 2015;40:2583–99.
Hablitz LM, Plá V, Giannetto M, Vinitsky HS, Stæger FF, Metcalfe T, et al. Circadian control of brain glymphatic and lymphatic fluid flow. Nat Commun. 2020;11:4411.
Mortensen KN, Sanggaard S, Mestre H, Lee H, Kostrikov S, Xavier ALR, et al. Impaired glymphatic transport in spontaneously hypertensive rats. J Neurosci. 2019;39:6365–77.
Miao A, Luo T, Hsieh B, Edge CJ, Gridley M, Wong RTC, et al. Brain clearance is reduced during sleep and anesthesia. Nat Neurosci. 2024;27:1046–50.
Dawson D, Gibbon S, Singh P. The hypothermic effect of melatonin on core body temperature: is more better? J Pineal Res. 1996;20:192–7.
Tordjman S, Chokron S, Delorme R, Charrier A, Bellissant E, Jaafari N, et al. Melatonin: Pharmacology, Functions and Therapeutic Benefits. Curr Neuropharmacol. 2017;15:434–43.
Sharma R, Sahota P, Thakkar MM. Melatonin promotes sleep in mice by inhibiting orexin neurons in the perifornical lateral hypothalamus. J Pineal Res. 2018;65:e12498.
Roh JH, Jiang H, Finn MB, Stewart FR, Mahan TE, Cirrito JR, et al. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J Exp Med. 2014;211:2487–96.
Cedernaes J, Osorio RS, Varga AW, Kam K, Schiöth HB, Benedict C. Candidate mechanisms underlying the association between sleep-wake disruptions and Alzheimer’s disease. Sleep Med Rev. 2017;31:102–11.
Simko F, Paulis L. Melatonin as a potential antihypertensive treatment. J Pineal Res. 2007;42:319–22.
Możdżan M, Możdżan M, Chałubiński M, Wojdan K, Broncel M. The effect of melatonin on circadian blood pressure in patients with type 2 diabetes and essential hypertension. Arch Med Sci. 2014;10:669–75.
Riemersma-van der Lek RF, Swaab DF, Twisk J, Hol EM, Hoogendijk WJ, Van Someren EJ. Effect of bright light and melatonin on cognitive and noncognitive function in elderly residents of group care facilities: a randomized controlled trial. JAMA. 2008;299:2642–55.
Yao D, Li R, Hao J, Huang H, Wang X, Ran L, et al. Melatonin alleviates depression-like behaviors and cognitive dysfunction in mice by regulating the circadian rhythm of AQP4 polarization. Transl Psychiatry. 2023;13:310.
Labban S, Alshehri FS, Kurdi M, Alatawi Y, Alghamdi BS. Melatonin improves short-term spatial memory in a mouse model of Alzheimer’s disease. Degener Neurol Neuromuscul Dis. 2021;11:15–27.
Zhai Z, Xie D, Qin T, Zhong Y, Xu Y, Sun T. Effect and mechanism of exogenous melatonin on cognitive deficits in animal models of Alzheimer’s disease: a systematic review and meta-analysis. Neuroscience. 2022;505:91–110.
Fang X, Han Q, Li S, Luo A. Melatonin attenuates spatial learning and memory dysfunction in developing rats by suppressing isoflurane-induced endoplasmic reticulum stress via the SIRT1/Mfn2/PERK signaling pathway. Heliyon. 2022;8:e10326.
Sumsuzzman DM, Choi J, Jin Y, Hong Y. Neurocognitive effects of melatonin treatment in healthy adults and individuals with Alzheimer’s disease and insomnia: A systematic review and meta-analysis of randomized controlled trials. Neurosci Biobehav Rev. 2021;127:459–73.
Obayashi K, Saeki K, Iwamoto J, Tone N, Tanaka K, Kataoka H, et al. Physiological levels of melatonin relate to cognitive function and depressive symptoms: The HEIJO-KYO Cohort. J Clin Endocrinol Metab. 2015;100:3090–6.
Peck JS, LeGoff DB, Ahmed I, Goebert D. Cognitive effects of exogenous melatonin administration in elderly persons: a pilot study. Am J Geriatr Psychiatry. 2004;12:432–6.
Rimmele U, Spillmann M, Bärtschi C, Wolf OT, Weber CS, Ehlert U, et al. Melatonin improves memory acquisition under stress independent of stress hormone release. Psychopharmacology. 2009;202:663–72.
Marzieh SH, Jafari H, Shorofi SA, Setareh J, Moosazadeh M, Espahbodi F, et al. The effect of melatonin on sleep quality and cognitive function of individuals undergoing hemodialysis. Sleep Med. 2023;111:105–10.
Moon, C, Hoth, KF, Perkhounkova, Y, Zhang, M, Lee, J, Hein, M, et al. Circadian timing, melatonin and hippocampal volume in later-life adults. J Sleep Res. 2023:e14090.
Blanken AE, Hurtz S, Zarow C, Biado K, Honarpisheh H, Somme J, et al. Associations between hippocampal morphometry and neuropathologic markers of Alzheimer’s disease using 7 T MRI. Neuroimage Clin. 2017;15:56–61.
Tseng PT, Zeng BY, Chen YW, Yang CP, Su KP, Chen TY, et al. The dose and duration-dependent association between melatonin treatment and overall cognition in Alzheimer’s dementia: a network meta- analysis of randomized placebo-controlled trials. Curr Neuropharmacol. 2022;20:1816–33.
Tuft C, Matar E, Menczel Schrire Z, Grunstein RR, Yee BJ, Hoyos CM. Current insights into the risks of using melatonin as a treatment for sleep disorders in older adults. Clin Interv Aging. 2023;18:49–59.
Paik JS, Ha M, Jung YH, Kim GH, Han KD, Kim HS, et al. Low vision and the risk of dementia: a nationwide population-based cohort study. Sci Rep. 2020;10:9109.
Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF. Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res. 2003;35:125–30.
Van der Mussele S, Le Bastard N, Saerens J, Somers N, Mariën P, Goeman J, et al. Agitation-associated behavioral symptoms in mild cognitive impairment and Alzheimer’s dementia. Aging Ment Health. 2015;19:247–57.
Gehrman PR, Connor DJ, Martin JL, Shochat T, Corey-Bloom J, Ancoli-Israel S. Melatonin fails to improve sleep or agitation in double-blind randomized placebo-controlled trial of institutionalized patients with Alzheimer disease. Am J Geriatr Psychiatry. 2009;17:166–9.
Singer C, Tractenberg RE, Kaye J, Schafer K, Gamst A, Grundman M, et al. A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep. 2003;26:893–901.
Sulkava S, Muggalla P, Sulkava R, Ollila HM, Peuralinna T, Myllykangas L, et al. Melatonin receptor type 1A gene linked to Alzheimer’s disease in old age. Sleep. 2018;41:zsy103.
Dartigues JF. Alzheimer’s disease: a global challenge for the 21st century. Lancet Neurol. 2009;8:1082–3.
Rice DP, Fox PJ, Max W, Webber PA, Lindeman DA, Hauck WW, et al. The economic burden of Alzheimer’s disease care. Health Aff (Millwood). 1993;12:164–76.
Besag FMC, Vasey MJ, Lao KSJ, Wong ICK. Adverse events associated with melatonin for the treatment of primary or secondary sleep disorders: a systematic review. CNS Drugs. 2019;33:1167–86.
Garaulet M, Lopez-Minguez J, Dashti HS, Vetter C, Hernández-Martínez AM, Pérez-Ayala M, et al. Interplay of dinner timing and MTNR1B Type 2 Diabetes risk variant on glucose tolerance and insulin secretion: a randomized crossover trial. Diabetes Care. 2022;45:512–9.
Frisher M, Gibbons N, Bashford J, Chapman S, Weich S. Melatonin, hypnotics and their association with fracture: a matched cohort study. Age Ageing. 2016;45:801–6.
Cohen PA, Avula B, Wang YH, Katragunta K, Khan I. Quantity of Melatonin and CBD in Melatonin Gummies Sold in the US. JAMA. 2023;329:1401–2.
Givler D, Givler A, Luther PM, Wenger DM, Ahmadzadeh S, Shekoohi S, et al. Chronic Administration of melatonin: physiological and clinical considerations. Neurol Int. 2023;15:518–33.
Chapman CD, Frey WH 2nd, Craft S, Danielyan L, Hallschmid M, Schiöth HB, et al. Intranasal treatment of central nervous system dysfunction in humans. Pharm Res. 2013;30:2475–84.
Priprem A, Johns JR, Limsitthichaikoon S, Limphirat W, Mahakunakorn P, Johns NP. Intranasal melatonin nanoniosomes: pharmacokinetic, pharmacodynamics and toxicity studies. Ther Deliv. 2017;8:373–90.
Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295:1070–3.
Antle MC, Silver R. Orchestrating time: arrangements of the brain circadian clock. Trends Neurosci. 2005;28:145–51.
Takahashi JS. Transcriptional architecture of the mammalian circadian clock. Nat Rev Genet. 2017;18:164–79.
Borjigin J, Zhang LS, Calinescu AA. Circadian regulation of pineal gland rhythmicity. Mol Cell Endocrinol. 2012;349:13–9.
Reuss S, Concemius W, Stehle J, Seidel A, Schröder H, Vollrath L. Effects of electrical stimulation of the superior cervical ganglia on the number of “synaptic” ribbons and the activity of melatonin-forming enzymes in the rat pineal gland. Anat Embryol. 1989;179:341–5.
Pévet P. Melatonin. Dialogues Clin Neurosci. 2002;4:57–72.
Chang AM, Aeschbach D, Duffy JF, Czeisler CA. Evening use of light-emitting eReaders negatively affects sleep, circadian timing, and next-morning alertness. Proc Natl Acad Sci USA. 2015;112:1232–7.
Schmid HA, Requintina PJ, Oxenkrug GF, Sturner W. Calcium, calcification, and melatonin biosynthesis in the human pineal gland: a postmortem study into age-related factors. J Pineal Res. 1994;16:178–83.
Brismar K, Hylander B, Eliasson K, Rössner S, Wetterberg L. Melatonin secretion related to side-effects of beta-blockers from the central nervous system. Acta Med Scand. 1988;223:525–30.
Hardeland R. Neurobiology, pathophysiology, and treatment of melatonin deficiency and dysfunction. ScientificWorldJournal. 2012;2012:640389.
Arendt J, Skene DJ, Middleton B, Lockley SW, Deacon S. Efficacy of melatonin treatment in jet lag, shift work, and blindness. J Biol Rhythms. 1997;12:604–17.
Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, et al. Oligomerization of Alzheimer’s beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–9.
Masters CL, Selkoe DJ. Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006262.
Price DL, Sisodia SS. Mutant genes in familial Alzheimer’s disease and transgenic models. Annu Rev Neurosci. 1998;21:479–505.
Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain. 2015;138:2814–33.
Funding
C.B. receives financial support from the Novo Nordisk Foundation (NNF23OC0081873) and the Swedish Brain Research Foundation (FO2023-0292). X.T. research is supported by funding from ZJU 100 Young Professor Project, Rut and Arvid Wolff Memorial Foundation (2023-02467), Åke Wiberg Foundation (M19-0266), Fredrik and Ingrid Thuring Foundation (2019-00488). P.X. research is financially supported by the Åke Wiberg Foundation (M23-0040) and Tore Nilsson Foundation (2023-079). H.Z. is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2023-00356; #2022-01018 and #2019-02397), the European Union’s Horizon Europe research and innovation programme under grant agreement No 101053962, Swedish State Support for Clinical Research (#ALFGBG-71320), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), the AD Strategic Fund and the Alzheimer’s Association (#ADSF-21-831376-C, #ADSF-21-831381-C, and #ADSF-21-831377-C), the Bluefield Project, Cure Alzheimer’s Fund, the Olav Thon Foundation, the Erling-Persson Family Foundation, Stiftelsen för Gamla Tjänarinnor, Hjärnfonden, Sweden (#FO2022-0270), the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 860197 (MIRIADE), the European Union Joint Programme – Neurodegenerative Disease Research (JPND2021-00694), the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre, and the UK Dementia Research Institute at UCL (UKDRI-1003). BBB receives funding from the National Institution on Aging (R01AG070973, R01AG062285, R01AG070883, R01AG037639, R01AG059312, P30AG062715). However, the funders had no role in the study design, data collection and analysis, publication decision, or manuscript preparation. Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
Zefan Zhang and Pei Xue drafted the manuscript and figures. Xiao Tan and Christian Benedict conceived the idea for the review. All authors, including Barbara B. Bendlin, Henrik Zetterberg, and Fernanda De Felice, critically reviewed the draft prepared by Zhang and Xue and provided feedback to further improve the manuscript. All authors approve the submission of the manuscript in its present form.
Corresponding authors
Ethics declarations
Competing interests
HZ has served at scientific advisory boards and/or as a consultant for Abbvie, Acumen, Alector, Alzinova, ALZPath, Annexon, Apellis, Artery Therapeutics, AZTherapies, Cognito Therapeutics, CogRx, Denali, Eisai, Merry Life, Nervgen, Novo Nordisk, Optoceutics, Passage Bio, Pinteon Therapeutics, Prothena, Red Abbey Labs, reMYND, Roche, Samumed, Siemens Healthineers, Triplet Therapeutics, and Wave, has given lectures in symposia sponsored by Alzecure, Biogen, Cellectricon, Fujirebio, Lilly, and Roche, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB (BBS), which is a part of the GU Ventures Incubator Program (outside submitted work). BBB has served at scientific advisory boards and/or as a consultant for New Amsterdam, Cognito Therapeutics, and Merry Life and has received amyloid and tau PET tracers and precursors from AVID radiopharmaceuticals. The other authors declare there are no competing financial interests related to the content of this article.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Z., Xue, P., Bendlin, B.B. et al. Melatonin: A potential nighttime guardian against Alzheimer’s. Mol Psychiatry (2024). https://doi.org/10.1038/s41380-024-02691-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41380-024-02691-6
- Springer Nature Limited