
Abstract
Objective
This study investigated the effect of maternal obesity on aged-male offspring liver phenotype and hepatic expression of a programmed miRNA.
Methods
A mouse model (C57BL/6 J) of maternal diet-induced obesity was used to investigate fasting-serum metabolites, hepatic lipid content, steatosis, and relative mRNA levels (RT-PCR) and protein expression (Western blotting) of key components involved in hepatic and mitochondrial metabolism in 12-month-old offspring. We also measured hepatic lipid peroxidation, mitochondrial content, fibrosis stage, and apoptosis in the offspring. To investigate potential mechanisms leading to the observed phenotype, we also measured the expression of miR-582 (a miRNA previously implicated in liver cirrhosis) in 8-week-old and 12-month-old offspring.
Results
Body weight and composition was similar between 8-week-old offspring, however, 12-month-old offspring from obese mothers had increased body weight and fat mass (19.5 ± 0.8 g versus 10.4 ± 0.9 g, p < 0.001), as well as elevated serum levels of LDL and leptin and hepatic lipid content (21.4 ± 2.1 g versus 12.9 ± 1.8 g, p < 0.01). This was accompanied by steatosis, increased Bax/Bcl-2 ratio, and overexpression of p-SAPK/JNK, Tgfβ1, Map3k14, and Col1a1 in the liver. Decreased levels of Bcl-2, p-AMPKα, total AMPKα and mitochondrial complexes were also observed. Maternal obesity was associated with increased hepatic miR-582-3p (p < 0.001) and miR-582-5p (p < 0.05). Age was also associated with an increase in both miR-582-3p and miR-582-5p, however, this was more pronounced in the offspring of obese dams, such that differences were greater in 12-month-old animals (−3p: 7.34 ± 1.35 versus 1.39 ± 0.50, p < 0.0001 and −5p: 4.66 ± 1.16 versus 1.63 ± 0.65, p < 0.05).
Conclusion
Our findings demonstrate that maternal diet-induced obesity has detrimental effects on offspring body composition as well as hepatic phenotype that may be indicative of accelerated-ageing phenotype. These whole-body and cellular phenotypes were associated with age-dependent changes in expression of miRNA-582 that might contribute mechanistically to the development of metabolic disorders in the older progeny.
Similar content being viewed by others


Introduction
Global incidence of obesity has increased dramatically in all age groups, including women of reproductive age [1, 2]. A 2019 report in the United Kingdom revealed that more than half of pregnant women were overweight or obese [3]. Maternal obesity during pregnancy has been shown to have long-term “programmed” effects on the offspring [4, 5]. This involves changes in gene expression and permanent structural and functional changes in tissues that make the offspring susceptible to obesity and related diseases [6,7,8]. These effects of maternal obesity could be mediated by the maternal nutritional status and/or dietary composition [5, 9,10,11].
Epigenetic processes are thought to be major determinants of programming mechanisms. The major epigenetic determinants are DNA methylation, histone modification and noncoding RNAs, such as microRNAs (miRNAs) [5]. MiRNAs are small molecules that act to repress messenger RNA (mRNA) translation by binding to complementary sequences in the 3′-untranslated region (UTRs) of target mRNAs [12, 13]. It is predicted that at least 30% of all human genes are regulated by miRNAs and disturbances in miRNA expression have been linked to several metabolic conditions, including type-2 diabetes, cardiovascular disease (CVD), lipid-homeostasis impairment, and liver steatosis and fibrosis [14,15,16]. For example, increased hepatic miR-582 expression has been reported in response to high-fructose diet consumption [17] and cirrhotic livers [18]. Growing evidence indicates that maternal obesity and/or high-fat feeding may program the expression of different miRNAs in various tissues, directly affecting the health status of the offspring [19,20,21,22,23]. However, these studies have generally focussed on a single time point in young-adult life.
The aging process is thought to start before birth and has been associated with development of programmed phenotypes [24]. Individuals exposed to a suboptimal maternal environment (e.g., maternal obesity) during early critical periods are more susceptible to age-associated diseases in later life, such as obesity, type-2 diabetes, and CVD [24]. Rodríguez-González et al. revealed that progeny exposed to maternal obesity were predisposed to premature aging and the progression of nonalcoholic fatty liver disease (NAFLD) in a sex-specific manner [25]. However, most published animal studies on the effects of maternal obesity and/or excessive consumption of high-fat/high-sugar diets during pregnancy and lactation focused on young-adult offspring with a limited number focussing on the reproductive system and cerebrovasculature of aged offspring [26,27,28]. Therefore, this study aimed to investigate the liver phenotype of mice at 12 months of age born to obese dams and to explore if this was related to changes in the miRNA expression with advancing of age.
Materials and methods
Animals and diet
All animal experiments were conducted at the University of Cambridge according to the UK Home Office Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012, following ethical review and approval by the University of Cambridge Animal Welfare and Ethical Review Board. This study is based on an established model of maternal diet-induced obesity [29] where female C57BL/6 mice are fed either a standard laboratory chow (RM1, control diet) [~7% simple sugars, 3% fat, 50% polysaccharide, and 15% protein (wt/wt)] or an obesogenic diet [high-fat diet: ~10% simple sugars, 20% animal lard, 28% polysaccharide, and 23% protein (wt/wt) supplemented with sweetened condensed milk (~55% simple sugar, 8% fat, and 8% protein (wt/wt)) and micronutrient mineral mix] from weaning (both diets from SDS Diets, UK). Proven breeders [29] were mated when controls had a total body-fat mass less than 5 g, and the obese exceeded 10 g of total fat mass as assessed by time domain nuclear magnetic resonance (TD-NMR) (Mini-spec TD-NMR, Bruker UK Ltd). The presence of a copulatory plug indicated day 1 of pregnancy. Females remained on their respective diet throughout pregnancy and lactation under standard conditions of 12-h light and 12-h dark at 23 °C. Assignment of dietary groups was performed by a technician not involved in the molecular analysis. To control the nutritional plane, litter size was standardized on postnatal day 2 to six pups. On day 21, male offspring were weaned onto and maintained on control diet. For all outcome measurements only one animal per litter was included in the analysis, therefore, n refers both to the number of offspring included and the number of litters represented. Offspring groups are referred to as CC (offspring born to control dams) and OC (offspring born to obese dams). Body weight of male offspring was assessed from three weeks, whereas body composition by TD-NMR was measured from four weeks of age. Both were recorded weekly up to 12 weeks of age and monthly up to 52 weeks of age.
To investigate the contribution of advancing age on the expression of miR-582 (−3p and −5p), two different time points (eight weeks and 12 months of age) were included for this measurement. Mice at eight weeks of age are considered young adults (sexual maturity is attained by approximately six weeks of age), and at 12 months of age, they are deemed older adults (middle age). The average life span of C57/Bl6 laboratory mice is about 24 months of age [30]. At 12 months of age, blood glucose was measured using a glucometer (AlphaTRAK II, Abbott Logistics BV, NL). Animals were killed by rising CO2 concentration. Various tissues were collected at eight weeks of age (n = 8 for each experimental group); however, only livers and body composition were analyzed for this study. For 12-month-old offspring (n = 9 for each experimental group), blood was collected by cardiac puncture for serum-metabolite analysis. Adipose-tissue depots (intra-abdominal, retroperitoneal, and epididymal) and liver were dissected, weighed, snap-frozen, and stored at −80 °C until use.
Serum analysis
Insulin and leptin concentrations were measured using enzyme-linked immunosorbent assay (ELISA) (Crystal Chem, USA). Total cholesterol and triglycerides (TG) were measured using a Dimension RxL analyzer (Siemens Healthcare Limited, UK). HDL cholesterol was measured using a homogeneous accelerator-selective detergent assay with a Dimension RxL analyzer (Siemens Healthcare Limited, UK). LDL cholesterol was calculated using the Friedwald equation [31]. Free fatty acids were measured using the Roche Free Fatty Acid Half-Micro Kit (Roche Diagnostics Limited, UK). Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) was calculated using the formula: HOMA-IR = [fasting insulin (mU L−1) × fasting glucose (mmol L−1)]/22.5.
Glucose-tolerance test (GTT)
At 12 months of age, offspring were fasted (16 h) and tail blood glucose measured (AlphaTRAK II, Abbott Logistics BV, NL). Animals were injected intraperitoneally with a 10% (wt/vol.) glucose solution (1 g/kg body weight) and tail blood glucose measured 15, 30, 60, 120, and 180 min after injection. Area under the curve (AUC) was calculated using the trapezoids rule (GraphPad Prism 7.02 for Windows, GraphPad Software, USA).
Quantification of liver lipid content and histological analysis
Accumulation of hepatic lipid was determined using a modified Folch method [32]. For histological analysis, liver tissue was fixed in 10% neutral-buffered formalin for 48 hours before processing and paraffin embedding. For evaluation of hepatic steatosis, sections (7 µm) were stained with hematoxylin and eosin (H&E) and images were captured using a ZEISS Axio Scan.Z1 slide scanner. Steatosis was graded by a histopathologist blinded to the offspring group, and scored based on the percentage of hepatocytes showing steatosis (macro- or microvesicular) into four different categories: absent = <5% (grade 0), mild = <30% (grade 1), moderate = <60% (grade 2), and severe = >60% (grade 3) [33].
For quantification of collagen deposition, sections (5 µm) were stained with Picrosirius Red and images were captured using a ZEISS Axio Scan.Z1 slide scanner. Collagen staining was calculated by converting images to an L*a*b stack and analyzing red stain on dimension “a” using ImageJ software (version 1.52 g, National Institute of Health, USA). Five fields per section were analyzed and the average collagen staining recorded. These sections were also used to fibrosis-stage assessment (by a histopathologist blinded to the offspring group) according to the classification described by Kleiner et al. [34] as follows: absent = no increase in fibrosis, mild = centrilobular/pericellular fibrosis only, moderate = centrilobular + portal fibrosis, and severe = bridging fibrosis.
TUNEL assay
Liver apoptosis was assessed by TUNEL assay using paraffin-embedded sections. An in situ apoptosis detection kit (#MK500, Takara Bio Inc., Japan) was used to detect DNA fragments generated by apoptosis as per the manufacturer’s instructions. Images were captured using a ZEISS Axio Scan.Z1 slide scanner. TUNEL-positive cells were detected based on the average DAB (brown) staining within each cell nuclei using an embedded algorithm in QuPath v0.2.3 [35]. The percentage of positive cells was calculated by comparing the number of DAB-stained nuclei to the total nuclei number (hematoxylin-stained).
Total RNA isolation
Total RNA was isolated from frozen liver (miRNeasy Mini Kit, Qiagen, UK) and quantified using a NanoDrop spectrophotometer (Thermo Scientific, USA). RNA quality and integrity were estimated by 260/280-nm and 260/230-nm ratios, showing values between 1.8–2.0 and 1.8–2.2, respectively.
MiRNA expression
Complementary DNA (cDNA) was generated from total RNA (TaqmanTM MicroRNA Reverse Transcription Kit, Applied Biosystems, UK) and TaqmanTM MicroRNA assays (Applied Biosystems, UK). Quantitative PCR (qPCR) was performed using a TaqmanTM 2x Universal PCR Master Mix No AmpEraseTM UNG (Applied Biosystems, UK) on a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems, UK) with miR-582-3p (Assay ID: 472692_mat) and miR-582-5p (Assay ID: 471065_mat) probes (both Applied Biosystems, UK). MiRNA-expression assays were performed in duplicate, and data normalized to the geomean of miR-25-3p (hsa-miR-25 Assay ID: 000403, Applied Biosystems, UK), which displayed no differences between groups.
mRNA-level assessment
Relative mRNA levels of genes encoding proteins involved in the fibrotic process, apoptosis pathway, and mitochondrial function were determined by qPCR. cDNA was synthesized using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, UK) and qPCR performed in duplicate using Sybr Green PCR Master Mix (Applied Biosystems, UK) on a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems, UK). Target-gene expression was normalized to the geometric mean of housekeeping genes Ppia and Hprt, as their expressions were stable between groups. Primers sequences are shown in Table 1. For measurement of both mRNA and miRNA levels, the results were expressed as the relative increase according to the comparative cycle-threshold method (2−ΔΔCt) [36].
Protein extraction and Western blotting
Hepatic expression of proteins implicated in fibrosis, apoptosis, mitochondrial function, and oxidative stress was assessed by western blotting [20]. Each membrane was blotted with the respective primary antibody [p-SAPK/JNK (Thr183/Tyr185) (#9251, Cell Signaling Technology, USA), Bax (#2772, Cell Signaling Technology, USA), Bcl-2 (#2876, Cell Signaling Technology, MA, USA), Total OXPHOS (ab110413, Abcam, UK), Citrate Synthase (CS; Cat. 16131-1-AP, Proteintech, UK), Catalase (ab1877, Abcam, UK), MnSOD (06-984, Merck Millipore, UK), p-AMPKα (Thr172) (#2535, Cell Signaling Technology, USA), and AMPKα1 (ab3759, Abcam, UK)]. Proteins were detected with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody (Jackson Immuno Research, UK). Detection was performed with Super Signal West Pico Chemiluminescent substrate (Thermo Scientific, UK) using an ImageQuantTM LAS 4000 (GE Healthcare Life Sciences, UK). Band intensities were quantified by optical densitometry (Scion Image-Release Beta 3b, NIH, USA). To confirm equal protein loading, membranes were stripped (Restore™ Western Blot Stripping Buffer, Thermo Scientific, UK) and reblotted with an anti-GAPDH antibody (#2118, Cell Signaling Technology, USA). Western blotting data were calculated relative to the CC group, which were defined as 100 percent.
Relative mitochondrial DNA content
Copy number of mitochondrial DNA is one biomarker of mitochondrial dysfunction [37]. Total DNA was extracted from liver (DNeasy Blood & Tissue kit, Qiagen, UK) and total double-stranded DNA concentrations determined (Quant-iTTM PicoGreen dsDNA assay kit, Invitrogen, UK). Mitochondrial DNA content was determined by the ratio between a mitochondrial (Nd5) and nuclear (Rplp0) gene following qPCR. Primer sequences are in Table 1. The results were expressed using the following equations: ΔCT = (nuclear DNA CT - mitochondrial DNA CT) and relative mitochondrial DNA content = 2 x 2 ΔCT [38].
Determination of mitochondrial respiratory chain (MRC) enzyme activities
Hepatic MRC enzyme activities were evaluated spectrophotometrically as described previously: complex I [39], complex II/III [40], and complex IV [41]. Complex activities were expressed as a ratio to CS [42] to correct for mitochondrial enrichment of the samples [43].
Malondialdehyde (MDA) concentration
Malondialdehyde, a marker for lipid peroxidation, was quantified fluorometrically (Ex/Em=532/553 nm) (ab118970, Abcam, UK) and normalized to protein content of the sample.
Statistical analysis
Power calculations for determining adequate sample size were based on published results from our group [32], and in order to detect a 20% difference in means with a power (95% confidence and alpha = 0.05) of 0.9, the sample size required per group was determined to be n = 9 dams per group (mother is the statistical unit). Shapiro–Wilk quality test was used to assess the normality of the data. Grubbs’ test was performed and significant outlier samples were removed when the alpha value is equal to 0.05 [44]. Normally distributed data were analyzed using an unpaired two-tailed student’s t test. Data with nonnormal distribution were analyzed by the two-tailed Mann–Whitney U test or by the two-tailed Kolmogorov–Smirnov test. Two-way analysis of variance (ANOVA) was performed to estimate the effect of two independent variables (maternal diet and offspring age) followed by Bonferroni post hoc test. A chi-square one-sided test was used to analyze the binary data generated from an assessment of fibrosis by a pathologist. Pearson (normal distribution) or Spearman (nonnormal distribution) correlation coefficients were also determined. All statistical analyses were performed using GraphPad Prism 7.02 for Windows (GraphPad Software, USA). Data are presented as mean ± standard error of the mean (SEM) and differences considered significant when p < 0.05.
Results
Body weight and composition
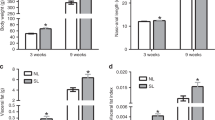
In both groups, body weight and body-fat content increased with age (effect of age: p < 0.0001), however, the effect was greater in the offspring of obese dams (interaction between maternal diet and age: p < 0.0001) (Fig. 1a). Therefore, at 12 months of age, offspring of obese dams were significantly heavier (CC 39.1 ± 1.4 g versus OC 48.8 ± 1.5 g, p < 0.0001; CC: n = 9 and OC: n = 8) and fatter (CC 10.4 ± 0.9 g versus OC 19.5 ± 0.8 g, p < 0.0001; CC: n = 9 and OC: n = 8) than control offspring. There was a small reduction in the absolute lean mass from week 7 to week 20 in the OC group; however, from 24 weeks of age until the end of experimental period, the lean mass was similar between groups (Fig. 1b). The difference in fat mass between the groups became more marked as the animals aged (Fig. 1c). There was a depot-specific difference in fat deposits, with the greatest difference observed in the intra-abdominal depot of the OC group at 12 months of age (Fig. 1d).

Average body weight (a), average absolute lean mass (b), average absolute fat mass (c), fat mass of intra-abdominal, retroperitoneal and epididymal fat depots (d), total hepatic lipid content (e), hepatic lipid content as percentage of liver weight (f), representative liver sections stained with H&E and percentage of steatosis grades in the CC group (g) and OC group (h) at 12 months of age. CC 12 m: mother received control diet and offspring-fed control diet after weaning up to 52 weeks of age; OC 12 m: mother received obesogenic diet and offspring-fed control diet after weaning up to 52 weeks of age. Values are presented as means ± SEM; CC 12 m: n = 7–13 and OC 12 m: n = 8–9 animals from independent litters for each group. Data were analyzed by unpaired student’s t test or Mann–Whitney U test. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Fasting-serum metabolites, HOMA-IR and GTT (AUC) in 12-month-old offspring
The level of circulating TG was significantly lower in the OC group compared with the CC group (Table 2). Additionally, offspring from obese dams exhibited increased serum concentrations of LDL and leptin. There were no significant differences between offspring groups for total cholesterol, HDL, FFA, insulin, fasting glucose, HOMA-IR, or glucose tolerance (Table 2).
Liver weight, hepatic lipid content, and steatosis grade in 12-month-old offspring
Absolute liver weight was increased in OC offspring compared with CC offspring at 12 months of age (CC 1.8 ± 0.2 g versus OC 2.9 ± 0.3 g, p < 0.01; CC: n = 9 and OC: n = 9). The increase remained when liver weight was presented as percentage of body weight (CC 4.36 ± 0.22% versus OC 6.36 ± 0.28%, p < 0.0001; CC: n = 9 and OC: n = 8). The offspring of obese dams showed elevated total hepatic lipid content at 12 months of age (Fig. 1e). Increased hepatic lipid content was also apparent when the results were expressed as percentage of liver weight (Fig. 1f). Blinded histopathological assessment revealed that 56% of offspring from obese dams developed severe steatosis at 12 months of age, while only 25% of control offspring had this degree of steatosis. Likewise, the percentages of animals with moderate steatosis were also greater in the OC group (22% versus 13%). Over half (62%) of control animals (CC) had absent/mild steatosis, whereas only 22% offspring of obese (OC) dams had absent/mild steatosis (Figs. 1g and 1h).
AMP-activated protein-kinase α (AMPKα) expression in the liver of 12-month-old offspring
We observed significant reductions in both phosphorylation (Thr172) and total protein levels of AMPKα in the OC group (Fig. 2a). The differences in protein expression were not accompanied by any differences in relative mRNA levels of Ampkα1 (CC 1.00 ± 0.04 versus OC 1.00 ± 0.06; CC: n = 9 and OC: n = 9) or Ampkα2 (CC 1.00 ± 0.04 versus OC 1.10 ± 0.05; CC: n = 9 and OC: n = 9). Additionally, liver lipid content strongly negatively correlated with p-AMPKα (r = −0.7375, p = 0.0005), total AMPKα (r = −0.5974, p = 0.0088), and AMPKα activity (r = −0.6697, p = 0.0033) (Supplementary Fig. 1a–c, respectively).

Protein levels of p-AMPKα (Thr172) and total AMPKα (a), representative images of protein expression (b), and relative protein expression of citrate synthase and mitochondrial OXPHOS complexes (c) in the liver of the offspring at 12 months of age. CC 12 m: mother received control diet and offspring-fed control diet after weaning up to 52 weeks of age; OC 12 m: mother received obesogenic diet and offspring-fed control diet after weaning up to 52 weeks of age. Values are presented as means ± SEM; CC 12 m: n = 9 and OC 12 m: n = 9 animals from independent litters for each group. Data were analyzed by unpaired student’s t test. *p < 0.05, **p < 0.01, and ***p < 0.001.
Mitochondrial phenotype in the liver of 12-month-old offspring
There was a significant reduction in protein expression of OXPHOS MRC complexes I (NDUFB8), III (UQCRC2), IV (MTCO1), and V (ATP5A) and an increase in hepatic protein levels of citrate synthase in the OC group (Figs. 2b and 2c). In contrast, the OC group had increased mRNA levels of Uqcrc2 (complex III: CC 1.00 ± 0.03 versus OC 1.12 ± 0.05, p < 0.05, CC: n = 9 and OC: n = 9) and Atp5a1 (complex V: CC 1.00 ± 0.04 versus OC 1.12 ± 0.04, p < 0.05; CC: n = 9 and OC: n = 9). mRNA expression of Ndufb8 (complex I: CC 1.00 ± 0.02 versus OC 1.01 ± 0.03, CC: n = 8 and OC: n = 9), Sdhb (complex II: CC 0.93 ± 0.04 versus OC 1.00 ± 0.04, CC: n = 9 and OC: n = 9), and Co1 (complex IV: CC 1.01 ± 0.08 versus OC 1.32 ± 0.20; CC: n = 9 and OC: n = 9) and Co2 (complex IV: CC 1.06 ± 0.13 versus OC 1.00 ± 0.12, CC: n = 9 and OC: n = 9) was not different between the groups. There was no change in the relative mitochondrial DNA content (CC 42.7 ± 2.7 versus OC 41.1 ± 2.0, CC: n = 9 and OC: n = 9) or in the activity of MRC complexes (complex I: CC 0.87 ± 0.05 versus OC 0.88 ± 0.05 relative to CS activity; CC: n = 9 and OC: n = 9; complexes II–III: CC 0.12 ± 0.01 versus OC 0.15 ± 0.02 relative to CS activity, CC: n = 9 and OC: n = 9; complex IV: CC 0.08 ± 0.01 versus OC 0.08 ± 0.01 k/nmol, CC: n = 9 and OC: n = 9) between groups.
We also examined hepatic lipid peroxidation and oxidative enzyme. However, there were no differences in MDA concentration (CC 0.20 ± 0.01 versus OC 0.22 ± 0.01 nmol/mg of protein, CC: n = 9 and OC: n = 9) or in protein levels of catalase (CC 100 ± 12 versus OC 92 ± 11, CC: n = 9 and OC: n = 9) or MnSOD (CC 100 ± 9 versus OC 99 ± 13, CC: n = 8 and OC: n = 9) between groups.
Hepatic fibrosis and apoptosis in 12-month-old offspring
To investigate the development of fibrosis in offspring liver at 12 months of age, quantification of collagen accumulation (Supplementary Fig. 2a and 2b) and a blinded histopathologic evaluation of fibrosis were carried out. We did not observe extensive collagen staining in either experimental group, but OC offspring exhibited a borderline significant (p = 0.0568) increase in hepatic collagen deposition (Fig. 3a). Consistent with low levels of fibrosis, histological scoring revealed no moderate or severe fibrosis in any livers of either group. A centrilobular/pericellular fibrosis (classified formally as mild) was detected in 3/9 animals from the OC group, while all control samples were deemed as fibrosis-absent (p < 0.05, CC: n = 7 and OC: n = 9). Relative mRNA levels of Col1a1 were significantly (p < 0.05) increased and Col4a1 was borderline significantly increased (p = 0.0542) in the obese offspring (Fig. 3b). Despite displaying a similar pattern, Col3a1 gene expression was not significantly changed between experimental groups (Fig. 3b). There were increased levels of phosphorylated c-Jun-N-terminal kinase (p-SAPK/JNK) (Figs. 3c and 3d), as well as relative mRNA levels of transforming growth factor-β (Tgfβ1) (CC 1.01 ± 0.07 versus OC 1.26 ± 0.09, p < 0.05; CC: n = 9 and OC: n = 9) and NF-κB-inducing kinase (Map3k14) (CC 1.02 ± 0.08 versus OC 1.38 ± 0.09, p < 0.01; CC: n = 9 and OC: n = 9) in the OC group. We also investigated some key mediators of the apoptotic pathway. Offspring from obese dams showed decreased protein expression of Bcl-2 (Figs. 3c and 3d) and an increased Bax/Bcl-2 ratio (CC 1.02 ± 0.07 versus OC 1.64 ± 0.24, p < 0.05; CC: n = 9 and OC: n = 8). Concomitantly, there was a negative correlation between Bcl-2 protein levels and hepatic collagen deposition (r = −0.6436, p = 0.0053) (Supplementary Fig. 1d). Protein levels of Bax (Figs. 3c and 3d) and transcript levels of both Bax (CC 1.00 ± 0.03 versus OC 1.08 ± 0.03; CC: n = 9 and OC: n = 9) and Bcl-2 (CC 1.03 ± 0.09 versus OC 1.18 ± 0.12; CC: n = 9 and OC: n = 9) were not significantly different between CC and OC groups. The percentage of TUNEL-positive cells was low in both groups and not significantly different (p = 0.0807) (Fig. 3e).

Quantification of Picrosirius red staining (a), relative mRNA levels of Col1a1, Col3a1 and Col4a1 (b), representative images of protein expression (c), relative protein expression of p-SAPK/JNK, Bax and Bcl-2 (d), and percentage of TUNEL-positive cells (e) in the liver of the offspring at 12 months of age. CC 12 m: mother received control diet and offspring-fed control diet after weaning up to 52 weeks of age; OC 12 m: mother received obesogenic diet and offspring fed control diet after weaning up to 52 weeks of age. Values are presented as means ± SEM; CC 12 m: n = 7–9 and OC 12 m: n = 8–9 animals from independent litters for each group. Data were analyzed by unpaired student’s t test or Mann–Whitney U test or Kolmogorov–Smirnov test. *p < 0.05 and **p < 0.01.
MiR-582 expression in the liver of 12-month-old offspring
Maternal diet-induced obesity during pregnancy and lactation resulted in increased expression of both miR-582-3p and miR-582-5p in the older-programmed offspring (Fig. 4a). There was also a significant effect of age to increase both miR-582-3p and miR-582-5p. However, in both cases, the impact of age was greater in the offspring of obese dams compared with controls, meaning that the greatest effect of maternal diet was at 12 months of age when hepatic expression of miR-582-3p and miR-582-5p was substantially higher in the offspring of the obese dams (Fig. 4a). MiR-582 is intragenic, located within the gene phosphodiesterase 4D (Pde4d). Similarly, to the miRNA expression, expression of Pde4d was increased in the 12-month-old offspring from obese dams (Fig. 4b). However, the magnitude of increase in the host mRNA was much smaller than that of the miRNAs. Both strands of mmu-miR-582 (-3p and -5p) were positively correlated with hepatic lipid accumulation (r = 0.7549, p = 0.0007 and r = 0.7420, p = 0.0004, respectively) (Supplementary Fig. 1e and 1f).

Relative expressions of miR-582-3p and miR-582-5p in the liver of the offspring at eight weeks and 12 months of age (a) and relative mRNA expression of phosphodiesterase 4D in the liver of the offspring at 12 months of age (b). CC 8w: mother received control diet and offspring-fed control diet after weaning up to eight weeks of age; OC 8w: mother received obesogenic diet and offspring-fed control diet after weaning up to eight weeks of age. CC 12 m: mother received control diet and offspring-fed control diet after weaning up to 52 weeks of age; OC 12 m: mother received obesogenic diet and offspring-fed control diet after weaning up to 52 weeks of age. Values are presented as means ± SEM; CC 8w: n = 8 and OC 8w: n = 8; CC 12 m: n = 8–9 and OC 12 m: n = 9 animals from independent litters for each group. Data were analyzed by Mann–Whitney U test or two-way ANOVA. *p < 0.05 and ****p < 0.0001.
Discussion
Our study revealed that maternal diet-induced obesity caused by consumption of a high-fat/high-sugar diet adversely programs growth trajectory, adiposity, hepatic lipid accumulation, and moderate/severe steatosis in 12-month-old offspring fed a control diet from weaning. Our experimental design does not allow for the differentiation of effects resulting from maternal obesity per se and those caused by maternal consumption of a high-fat/high-sugar diet. However, in the human situation, these often occur in parallel. Our findings highlight the importance of advancing age in the development of programmed phenotype, with longitudinal data demonstrating that the effects on body weight and body composition become more pronounced with age. Consistent with this observation, there is an interaction between maternal diet and age on expression of miR-582 with the effect of age being exaggerated in the male offspring exposed to maternal obesity. These observations highlight the importance of studying the impact of programming across the life course.
Previous studies from our research group using the same model demonstrated that chow-fed offspring have similar body weights up to 12 weeks of age [22, 45,46,47]. In the current study, we observed a significant impact of age, with the offspring of obese dams displaying increased body weight and fat mass from around 20 weeks of age and that the magnitude of effect increased up to 12 months of age. Consistent with these findings, Rodríguez-González et al. [25] demonstrated that postnatal day 450 was the first age at which body weight and adiposity index were greater in male-rat offspring of a similar maternal obesity model. In addition to the effects on adiposity, the current study also showed that programmed changes in metabolites were associated with advancing age. Previous studies on 8-week-old mice showed no effects of maternal obesogenic diets on serum LDL cholesterol and leptin levels in offspring of obese dams [47]. However, both were elevated as a consequence of maternal obesity at 12 months of age. Persistent elevation in circulating LDL increases the risk of CVD development and LDL-particle concentration has been positively associated with NAFLD [48, 49]. Therefore, high LDL concentration observed in the older offspring may contribute to programming of cardiovascular and hepatic diseases.
NAFLD refers to excessive intrahepatocellular fat deposition and is considered to be the liver manifestation of metabolic syndrome [50]. NAFLD is one of the most prevalent liver diseases in the world and evidence suggests that an adverse early life environment can program offspring susceptibility to the development and progression of this condition [9]. In the current study, increased hepatic lipid content, liver weight, and percentage of animals with severe-to-moderate steatosis (78% in total) were accompanied by increased body weight and adiposity in the older progeny exposed to a maternal obesogenic environment, indicating the presence of NAFLD independently of changes in the fasting concentrations of FFA, total cholesterol, HDL, insulin, or glucose tolerance. Despite increased hepatic lipid accumulation, offspring of obese dams had reduced serum levels of circulating triglycerides. The underlying mechanisms are unknown but may indicate an inability to export triglycerides from the liver into the circulation or an enhanced uptake of triglycerides by adipose tissue. Similar observations have been made in other mouse models of nonalcoholic steatohepatitis, such as those using a choline-deficient L-amino acid-defined high-fat diet [51].
Given that strong negative correlations were found between hepatic p-AMPKα protein levels and liver lipid content, as well as between the p-AMPKα/AMPKα ratio and liver lipid content, we speculate that the hepatic fat accumulation observed in these offspring can be partially explained by defects in AMPKα activation and activity. It has been shown previously that AMPK activation declines with obesity [52] and aging [53], which is consistent with the offspring of obese dams displaying an obese, prematurely metabolic aging phenotype. Furthermore, it has been reported that exogenous AMPK activation can be a viable strategy for the treatment of obesity and NAFLD [54]. In the current study, the absence of differences in the relative mRNA levels of Ampkα1 or Ampkα2 may suggest a post-transcriptional mechanism mediating the expression of AMPK.
Mitochondria dysfunction has been implicated in developmental programming and age-related diseases [55,56,57]. Previous studies showed decreased protein levels of mitochondrial respiratory chain complex II, complex III, and complex V in the liver [57] and skeletal muscle [55] of young offspring (at weaning [57] and eight weeks of age [55]) born to obese dams. In the current study, we demonstrated reductions in a number of proteins within these complexes; however, there were no differences in mitochondrial copy number or enzyme activity. Protein subunits NDUFB8 (complex I) [58] and UQCRC2 (complex III) [59] are structural subunits without involvement in the catalytic activity. Therefore, although there was a significant decrease in protein levels of NDUFB8 and UQCRC2, it may not impact on the MRC enzyme activity but impacts on other aspects of mitochondrial biology. Our findings also revealed that increased mRNA levels of Uqcrc2 and Atp5a1 may indicate a compensatory response to the reduction in the protein expression of these MRC complexes in the obese offspring. In parallel, the absence of significant changes in the mRNA levels of Ndufb8, Co1, and Co2 again suggests that differences in protein expression may arise from post-transcriptional mechanisms.
The spectrum of NAFLD in humans ranges from nonalcoholic fatty liver (NAFL, simple hepatic steatosis) to NASH (severe form of the disease). NAFL can progress to NASH, which is characterized by hepatocellular injury and inflammation with or without fibrosis [50]. Liver fibrosis results from an excessive accumulation of collagen and other extracellular-matrix proteins in the affected tissue [60]. Mice models generally do not develop NASH and moderate or severe fibrosis is rarely reported [61], consistent with the absence of moderate/severe fibrosis in the current study. However, we demonstrated increased collagen deposition, gene overexpression of Col1a1, and mild centrilobular/pericellular fibrosis in some animals, indicating that maternal diet-induced obesity leads to increased risk of liver fibrosis in the older offspring at 12 months of age, which could progress if the animals were aged further. Leptin, obesity, and TGFβ play a crucial role in the profibrogenic responses within the liver [60, 62, 63]. Leptin is thought to upregulate the expression of TGFβ, which in turn activates JNK, leading to fibrosis and apoptosis [62, 63]. Consistent with this, we revealed increased circulating leptin and increased hepatic TGFβ as well as increased phosphorylated JNK.
Apoptosis is considered a key mediator of NAFLD progression and the degree of apoptosis is inversely associated with the level of Bcl-2 [64, 65]. The Bcl-2 family includes three groups: anti-apoptotic proteins (Bcl-2), pro-apoptotic pore formers (Bax) and pro-apoptotic BH3-only proteins [66]. The balance between anti- and pro-apoptotic members is crucial [66] and a high Bax/Bcl-2 ratio, as observed in the livers from the offspring of obese dams, suggests a pro-apoptotic phenotype [67]. Although we detected decreased protein level of Bcl-2 (an important apoptosis-regulatory protein) and high Bax/Bcl-2 ratio in the animals from OC group, the percentage of TUNEL-positive cells was low and did not differ between groups at 12 months of age. These findings may suggest that hepatic cells from obese offspring are more susceptible to apoptotic stimuli; however, the time point investigated was too early to detect differences in the number of apoptotic cells by TUNEL assay. Furthermore, the strong negative correlation between Bcl-2 protein expression and collagen deposition in the liver supports an interaction between liver fibrosis and apoptosis.
We demonstrated that maternal diet-induced obesity leads to differences in hepatic expression of miR-582 (-3p and -5p) in the 12-month-old offspring of obese dams. This miRNA has not previously been implicated in the programming of liver, but has been shown to be increased in response to high-fructose-diet consumption [17] and in cirrhotic livers [18]. It has also been shown to be increased in the cardiac left ventricle of gestational protein-restricted offspring at 12 days of age [68] and in maternal plasma from women with preeclampsia [69]. In the current study, we demonstrated an impact of advancing age on hepatic miR-582-3p and miR-582-5p with levels increasing between offspring at eight weeks (young adult just after sexual maturity) and 12 months (older adults at middle age). However, the magnitude of the increase was much more marked in the offspring of obese dams compared with control offspring. This indicates that disturbances in the expression of these miRNAs are also consistent with an accelerated metabolic aging process triggered by a maternal obesogenic environment prior to and during pregnancy and lactation.
There are some limitations to acknowledge in the current study. First, there is growing evidence that male and female fetuses respond differently to a suboptimal in utero environment, leading to sexually dimorphic programmed responses [70]. In the current study, only males were included and, therefore, it remains unknown if the changes observed would also occur in female offspring. Second, although studying mice at an older age (12 months) than most other programming studies, this still represents approximately half of the mouse lifespan. Therefore, further studies are required to establish if age-associated changes become even great as the animals near their expected lifespan.
In conclusion, we demonstrate that maternal diet-induced obesity unfavorably modulates body composition and fatty-liver phenotype in exposed offspring that is exaggerated with age. This is accompanied by an accelerated age-associated increase in hepatic levels of miR-582, which may contribute mechanistically to the development of hepatic fatty liver. Our findings support the hypothesis that accelerated metabolic aging may contribute to the development of programmed phenotypes and highlights the possibility that there may be a critical time for intervention after the suboptimal exposure and prior to the development of programmed metabolic dysfunction.
References
Apovian CM. Obesity: definition, comorbidities, causes, and burden. Am J Manag Care. 2016;22:s176–s185.
NHS Digital. Statistics on Obesity, Physical Activity and Diet, England, 2019. Part 3: Adult overweight and obesity. https://digital.nhs.uk/data-and-information/publications/statistical/statistics-on-obesity-physical-activity-and-diet/statistics-on-obesity-physical-activity-and-diet-england-2019/part-3-adult-obesity (2019). Accessed 27 Apr 2020.
NMPA Project Team. National Maternity and Perinatal Audit: Clinical Report 2019. Based on births in NHS maternity services between 1 April 2016 and 31 March 2017. London: RCOG; 2019.
Nicholas LM, Morrison JL, Rattanatray L, Zhang S, Ozanne SE, McMillen IC. The early origins of obesity and insulin resistance: timing, programming and mechanisms. Int J Obes. 2016;40:229–38.
Elshenawy S, Simmons R. Maternal obesity and prenatal programming. Mol Cell Endocrinol. 2016;435:2–6.
Barker DJ. In utero programming of chronic disease. Clin Sci. 1998;95:115–28.
Godfrey KM. Maternal regulation of fetal development and health in adult life. Eur J Obstet Gynecol Reprod Biol. 1998;78:141–50.
Langley-Evans SC. Nutrition in early life and the programming of adult disease: A review. J Hum Nutr Diet. 2015;28:1–14.
Lynch C, Chan CS, Drake AJ. Early life programming and the risk of non-alcoholic fatty liver disease. J Dev Orig Health Dis. 2017;8:263–72.
Fernandez-Twinn DS, Hjort L, Novakovic B, Ozanne SE, Saffery R. Intrauterine programming of obesity and type 2 diabetes. Diabetologia. 2019;62:1789–801.
Lomas-Soria C, Reyes-Castro LA, Rodríguez-González GL, Ibáñez CA, Bautista CJ, Cox LA, et al. Maternal obesity has sex-dependent effects on insulin, glucose and lipid metabolism and the liver transcriptome in young adult rat offspring. J Physiol. 2018;596:4611–28.
Wilczynska A, Bushell M. The complexity of miRNA-mediated repression. Cell Death Differ. 2015;22:22–33.
Landrier JF, Derghal A, Mounien L. MicroRNAs in obesity and related metabolic disorders. Cells. 2019;8:E859.
Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol. 2013;10:542–52.
Gonz lez-S nchez LE, Ortega-Camarillo C, Contreras-Ramos A, Barajas-Nava LA.miRNAs as biomarkers for diagnosis of type 2 diabetes: A systematic review.J Diabetes.2021;13:792–816.
Li M, Marin-Muller C, Bharadwaj U, Chow K, Yao Q, Chen C. MicroRNAs: control and loss of control in human physiology and disease. World J Surg. 2009;33:667–84.
Sud N, Zhang H, Pan K, Cheng X, Cui J, Su Q. Aberrant expression of microRNA induced by high-fructose diet: implications in the pathogenesis of hyperlipidemia and hepatic insulin resistance. J Nutr Biochem. 2017;43:125–31.
Vuppalanchi R, Liang T, Goswami CP, Nalamasu R, Li L, Jones D, et al. Relationship between differential hepatic microRNA expression and decreased hepatic cytochrome P450 3A activity in cirrhosis. PLoS ONE. 2013;8:e74471.
Benatti RO, Melo AM, Borges FO, Ignacio-Souza LM, Simino LAP, Milanski M, et al. Maternal high-fat diet consumption modulates hepatic lipid metabolism and microRNA-122 (miR-122) and microRNA-370 (miR-370) expression in offspring. Br J Nutr. 2014;111:2112–22.
Fernandez-Twinn DS, Alfaradhi MZ, Martin-Gronert MS, Duque-Guimaraes DE, Piekarz A, Ferland-McCollough D, et al. Downregulation of IRS-1 in adipose tissue of offspring of obese mice is programmed cell-autonomously through post-transcriptional mechanisms. Mol Metab. 2014;3:325–33.
Alfaradhi MZ, Kusinski LC, Fernandez-Twinn DS, Pantaleão LC, Carr SK, Ferland-McCollough D, et al. Maternal obesity in pregnancy developmentally programs adipose tissue inflammation in young, lean male mice offspring. Endocrinology. 2016;157:4246–56.
De Almeida Faria J, Duque-guimarães D, Carpenter AAM, Loche E, Ozanne SE. A post-weaning obesogenic diet exacerbates the detrimental effects of maternal obesity on offspring insulin signaling in adipose tissue. Sci Rep. 2017;7:44949.
Sun Y, Wang Q, Zhang Y, Geng M, Wei Y, Liu Y, et al. Multigenerational maternal obesity increases the incidence of HCC in offspring via miR-27a-3p. J Hepatol. 2020;73:603–15.
Tarry-Adkins JL, Ozanne SE. Poor early growth and age-associated disease. Subcell Biochem. 2019;91:1–19.
Rodríguez-González GL, Reyes-Castro LA, Bautista CJ, Beltrán AA, Ibáñez CA, Vega CC, et al. Maternal obesity accelerates rat offspring metabolic ageing in a sex-dependent manner. J Physiol. 2019;597:5549–63.
Bautista CJ, Rodríguez-González GL, Morales A, Lomas-Soria C, Cruz-Pérez F, Reyes-Castro LA, et al. Maternal obesity in the rat impairs male offspring aging of the testicular antioxidant defence system. Reprod Fertil Dev. 2017;29:1950–7.
Rodríguez-González GL, Vega CC, Boeck L, Vázquez M, Bautista CJ, Reyes-Castro LA, et al. Maternal obesity and overnutrition increase oxidative stress in male rat offspring reproductive system and decrease fertility. Int J Obes. 2015;39:549–56.
Contu L, Nizari S, Heath CJ, Hawkes CA. Pre- and post-natal high fat feeding differentially affects the structure and integrity of the neurovascular unit of 16-month old male and female mice. Front Neurosci. 2019;13:1045.
Fernandez-twinn DS, Gascoin G, Musial B, Carr S, Duque-Guimaraes D, Blackmore HL, et al. Exercise rescues obese mothers’ insulin sensitivity, placental hypoxia and male offspring insulin sensitivity. Sci Rep. 2017;7:44650.
Ozanne S, Hales N. Lifespan: catch-up growth and obesity in male mice. Nature. 2004;427:411–2.
Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502.
Alfaradhi MZ, Fernandez-Twinn DS, Martin-Gronert MS, Musial B, Fowden A, Ozanne SE. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am J Physiol Regul Integr Comp Physiol. 2014;307:26–34.
Bedossa P, Poitou C, Veyrie N, Bouillot J, Basdevant A, Paradis V, et al. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology. 2012;56:1751–9.
Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21.
Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7:16878.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT Method. Methods. 2001;25:402–8.
Castellani CA, Longchamps RJ, Sun J, Guallar E, Arking DE. Thinking outside the nucleus: mitochondrial DNA copy number in health and disease. Mitochondrion. 2020;53:214–23.
Rooney J, Ryde I, Sanders L, Howlett E, Colton M, Germ K, et al. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol Biol. 2015;1241:23–38.
Ragan C, Wilson M, Darley-Usmar V, Lowe P. Mitochondria, a Practical Approach. Oxford, UK: IRL Press at Oxford University Press; 1987.
King T. Methods in Enzymology. Volume 10. Cambridge, MA, USA: Academic Press; 1967. pp. 217–35.
Wharton DC, Tzagoloff A Cytochrome oxidase from beef heart mitochondria. In: Methods in Enzymology. Volume 10. Amsterdam, The Netherlands: Elsevier BV; 1967. pp. 245–50.
Shepherd J, Garland P Methods in Enzymology. Volume 13. Cambridge, MA, USA: Academic Press; 1969. pp. 11–9.
Selak MA, De Chadarevian JP, Melvin JJ, Grover WD, Salganicoff L, Kaye EM. Mitochondrial activity in Pompe’s disease. Pediatr Neurol. 2000;23:54–57.
Grubbs F. Procedures for detecting outlying observations in samples. Technometrics. 1969;11:1–21.
Blackmore HL, Niu Y, Fernandez-Twinn DS, Tarry-Adkins JL, Giussani DA, Ozanne SE. Maternal diet-induced obesity programs cardiovascular dysfunction in adult male mouse offspring independent of current body weight. Endocrinology. 2014;155:3970–80.
Beeson JH, Blackmore HL, Carr SK, Dearden L, Duque-guimarães DE, Kusinski LC, et al. Maternal exercise intervention in obese pregnancy improves the cardiovascular health of the adult male offspring. Mol Metab. 2018;16:35–44.
Loche E, Blackmore HL, Carpenter AA, Beeson JH, Pinnock A, Ashmore TJ, et al. Maternal diet-induced obesity programmes cardiac dysfunction in male mice independently of post-weaning diet. Cardiovasc Res. 2018;114:1372–84.
Wadhera RK, Steen DL, Khan I, Giugliano RP, Foody JM. A review of low-density lipoprotein cholesterol, treatment strategies, and its impact on cardiovascular disease morbidity and mortality. J Clin Lipidol. 2016;10:472–89.
DeFilippis AP, Blaha MJ, Martin SS, Reed RM, Jones SR, Nasir K, et al. Nonalcoholic fatty liver disease and serum lipoproteins: the multi-ethnic study of atherosclerosis. Atherosclerosis. 2013;227:429–36.
Bugianesi E, Moscatiello S, Ciaravella MF, Marchesini G. Insulin resistance in nonalcoholic fatty liver disease. Curr Pharm Des. 2010;16:1941–51.
Yasuda D, Torii H, Shimizu R, Hiraoka Y, Kume N. Reduced serum cholesterol and triglyceride levels in a choline-deficient L-Amino acid-defined high-fat diet (CDAHFD)-induced mouse model of non-alcoholic steatohepatitis (NASH). Biol Pharm Bull. 2020;43:616–8.
Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med. 2016;48:e245.
Salminen A, Kaarniranta K, Kauppinen A. Age-related changes in AMPK activation: Role for AMPK phosphatases and inhibitory phosphorylation by upstream signaling pathways. Ageing Res Rev. 2016;28:15–26.
Smith BK, Marcinko K, Desjardins EM, Lally JS, Ford RJ, Steinberg GR. Treatment of nonalcoholic fatty liver disease: role of AMPK. Am J Physiol Endocrinol Metab. 2016;311:E730–E740.
Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, et al. Maternal metabolic syndrome programs mitochondrial dysfunction via germline changes across three generations. Cell Rep. 2016;16:1–8.
Jaeger K, Saben JL, Moley KH. Transmission of metabolic dysfunction across generations. Physiology. 2017;32:51–59.
Borengasser SJ, Lau F, Kang P, Blackburn ML, Ronis MJJ, Badger TM, et al. Maternal obesity during gestation impairs fatty acid oxidation and mitochondrial SIRT3 expression in rat offspring at weaning. PLoS ONE. 2011;6:2–10.
Vogel RO, Smeitink JAM, Nijtmans LGJ. Human mitochondrial complex I assembly: a dynamic and versatile process. Biochim Biophys Acta. 2007;1767:1215–27.
Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, et al. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71.
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–18.
Im YR, Hunter H, de Gracia Hahn D, Duret A, Cheah Q, Dong J, et al. A systematic review of animal models of NAFLD finds high-fat, high-fructose diets most closely resemble human NAFLD. Hepatology. 2021;0:1–18.
Seki E, Brenner DA, Karin M. A liver full of JNK: Signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology. 2012;143:307–20.
Marra F. Leptin and liver fibrosis: a matter of fat. Gastroenterology. 2002;122:1529–32.
El Bassat H, Ziada DH, Hasby EA, Nagy H, Abo Ryia MH. Apoptotic and anti-apoptotic seromarkers for assessment of disease severity of non-alcoholic steatohepatitis. Arab J Gastroenterol. 2014;15:6–11.
Afonso MB, Castro RE, Rodrigues CMP. Processes exacerbating apoptosis in non-alcoholic steatohepatitis. Clin Sci. 2019;133:2245–64.
Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80.
Perlman H, Zhang X, Chen MW, Walsh K, Buttyan R. An elevated bax/bcl-2 ratio corresponds with the onset of prostate epithelial cell apoptosis. Cell Death Differ. 1999;6:48–54.
Assalin HB, Gontijo JAR, Boer PA. miRNAs, target genes expression and morphological analysis on the heart in gestational protein-restricted offspring. PLoS ONE. 2019;14:e0210454.
Tsai P, Li S, Chen W, Tsai H, Su M. Differential miR-346 and miR-582-3p expression in association with selected maternal and fetal complications. Int J Mol Sci. 2017;18:E1570.
Dearden L, Bouret SG, Ozanne SE. Sex and gender differences in developmental programming of metabolism. Mol Metab. 2018;15:8–19.
Acknowledgements
The authors gratefully acknowledge São Paulo Research Foundation (FAPESP grant #2017/16637-9, #2014/10683-0, and #2019/22635-4), the Medical Research Council (MC_UU_12012/4, MC_UU_00014/4, and MC_UU_00014/5), the British Heart Foundation (RG/17/12/33167 and FS/16/53/32729), and the National Institute for Health Research (clinical lectureship: CL-2019-14-504). Also, the authors would like to thank Claire Custance for expert technical assistance and James Warner from the MRC Metabolic Diseases Unit for his histological expertise (MRC Metabolic Diseases Unit [MC_UU_00014/5]). Serum-lipid analyses were carried out by the Core Biochemical Assay Laboratory, Cambridge University Hospitals NHS Foundation Trust, Cambridge, UK.
Author information
Authors and Affiliations
Contributions
LVM performed experiments, analyzed data, and wrote and edited the paper. AAMC, LCP, DSF, and LPP performed experiments, analyzed data, and edited and reviewed the paper. EL, JMS, HLB, TJA, JAT, and IH performed experiments and edited and reviewed the paper. SEO designed the study, supervised the project, interpreted data, and wrote, reviewed, and edited the paper. All authors have read, reviewed, and agreed to the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mennitti, L.V., Carpenter, A.A.M., Loche, E. et al. Effects of maternal diet-induced obesity on metabolic disorders and age-associated miRNA expression in the liver of male mouse offspring. Int J Obes 46, 269–278 (2022). https://doi.org/10.1038/s41366-021-00985-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-021-00985-1
- Springer Nature Limited
This article is cited by
-
MicroRNA-582-3p knockdown alleviates non-alcoholic steatohepatitis by altering the gut microbiota composition and moderating TMBIM1
Irish Journal of Medical Science (1971 -) (2024)
-
Maternal obesity and programming of metabolic syndrome in the offspring: searching for mechanisms in the adipocyte progenitor pool
BMC Medicine (2023)
-
Childhood obesity, metabolic syndrome, and oxidative stress: microRNAs go on stage
Reviews in Endocrine and Metabolic Disorders (2023)
-
Maternal obesity induces liver lipid accumulation of offspring through the lncRNA Lockd/mTOR autophagy pathway
Molecular Genetics and Genomics (2022)
-
Sex-specific effects of maternal metformin intervention during glucose-intolerant obese pregnancy on body composition and metabolic health in aged mouse offspring
Diabetologia (2022)