
Abstract
Background/objectives
Epidemiological evidence indicates obesity in childhood and adolescence to be an independent risk factor for cancer and premature mortality in adulthood. Pathological implications from excess adiposity may begin early in life. Obesity is concurrent with a state of chronic inflammation, a well-known aetiological factor for DNA damage. In addition, obesity has been associated with micro-nutritional deficiencies. Vitamin D has attracted attention for its anti-inflammatory properties and role in genomic integrity and stability. The aim of this study was to determine a novel approach for predicting genomic instability via the combined assessment of adiposity, DNA damage, systemic inflammation, and vitamin D status.
Subjects/methods
We carried out a cross-sectional study with 132 participants, aged 10–18, recruited from schools and paediatric obesity clinics in London. Anthropometric assessments included BMI Z-score, waist and hip circumference, and body fat percentage via bioelectrical impedance. Inflammation and vitamin D levels in saliva were assessed by enzyme-linked immunosorbent assay. Oxidative DNA damage was determined via quantification of 8-hydroxy-2′-deoxyguanosine in urine. Exfoliated cells from the oral cavity were scored for genomic instability via the buccal cytome assay.
Results
As expected, comparisons between participants with obesity and normal range BMI showed significant differences in anthropometric measures (p < 0.001). Significant differences were also observed in some measures of genomic instability (p < 0.001). When examining relationships between variables for all participants, markers of adiposity positively correlated with acquired oxidative DNA damage (p < 0.01) and genomic instability (p < 0.001), and negatively correlated with vitamin D (p < 0.01). Multiple regression analyses identified obesity (p < 0.001), vitamin D (p < 0.001), and oxidative DNA damage (p < 0.05) as the three significant predictors of genomic instability.
Conclusions
Obesity, oxidative DNA damage, and vitamin D deficiency are significant predictors of genomic instability. Non-invasive biomonitoring and predictive modelling of genomic instability in young patients with obesity may contribute to the prioritisation and severity of clinical intervention measures.
Similar content being viewed by others


Introduction
There is growing epidemiological evidence that indicates obesity in childhood and adolescence to be an independent risk factor for cancer and premature mortality in adulthood. An increased BMI in childhood has been associated with an increased risk of developing lung, colorectal, kidney, cervical, ovarian and smoking-related cancers later in life [1,2,3,4]. Furthermore, a recent meta-analysis of five systematic reviews and 37 studies including 22 cohorts concluded that on average, there is a 20% increase in the odds of acquiring cancer in adulthood per standard deviation increase in childhood BMI [5]. These findings imply that prevention or early interventions with obesity in childhood may possibly reduce the rates of cancer in adulthood. However, research also suggests that childhood BMI status alone is not a useful predictor of the independent risk of morbidity in adulthood.
Obesity in childhood and adolescence is concurrent with a state of chronic, low-grade inflammation. Landgraf and colleagues report altered adipose tissue biology, including hypertrophy and hyperplasia in 6–18 years olds with obesity [6]. As adipose tissue expands to contain stores of fat, the microcirculation is disrupted, leading to adipose tissue hypoxia and cell death [7]. Adipose tissue necrosis attracts inflammatory cells and leads to the secretion of pro-inflammatory cytokines such as TNF-a [8]. Several studies have also identified an increased systemic circulation of C-reactive protein (CRP), marking low-grade inflammation as a co-feature in childhood obesity [9,10,11,12,13,14,15,16]. A recent review has described how low-grade inflammation and reduced natural killer cell functionality in obesity may promote malignancy and therefore be a possible causative mechanism for the increased risk of cancer later in life [17]. Furthermore, it is undisputed that chronic inflammation may have detrimental effects on DNA integrity and stability, and that genomic instability—a dynamic state characterised by elevated rates of genetic changes resulting from either cell-cycle dysfunctionalities or events affecting DNA integrity—is an enabling characteristic for the complex, multi-step process of tumorigenesis [18, 19].
In addition, obesity in childhood and adolescence can be associated with a state of micronutrient deficiencies. Deficiencies of several micronutrients including iron, selenium, folate, zinc, and vitamins A, D and E have been identified increasingly in children with obesity [20,21,22,23,24]. However, vitamin D deficiency has attracted the most attention, as it is being diagnosed increasingly in children within the UK [25]. Over the last few years, a large proportion of studies have identified obesity in children as a state of systemic hypovitaminosis D [26,27,28,29,30,31,32]. Furthermore, a bi-directional Mendelian randomisation analysis across 42,024 participants has demonstrated a causative association between obesity and vitamin D deficiency, such that a 10% increase in BMI may reduce levels of vitamin D by 4.2% [33]. Several studies report vitamin D to play a role in inhibiting inflammation, protecting cells from DNA damage, inducing cell-cycle arrest and promoting apoptosis [34, 35].
Links between DNA damage and genomic instability with obesity have been previously reported, with well-established, early markers of carcinogenesis found associated with excess adiposity in human participants and animal models [36]. However, so far only a few of these investigations have been conducted in children and adolescents. Higher levels of y-H2AX foci and micronuclei (MNi) were identified in peripheral blood lymphocytes (PBLs) from children with obesity compared to ‘healthy weight’ controls [37, 38]. However, findings of the same type in other tissues were inconclusive. Research in a small cohort of Mexican children did not identify an association between adiposity and nuclear anomalies in the buccal epithelium, yet a more recent study in Italian children found a significant link between childhood obesity and MNi formation in the same tissue [39, 40]. Similarly, there are discrepancies in studies of oxidative DNA damage in children with obesity; one reports positive correlations between obesity status in childhood and serum 8-OHdG, and two reports higher levels of 8-OHdG in urine samples [41,42,43], while the fourth study of urinary 8-OHdG and BMI in Italian children reports an inverse correlation [44]. Thus, comprehensive analysis of multiple markers of DNA damage and genomic instability, together with markers of systemic inflammation and micro-nutritional deficiencies, may shed light on their combined predictive value and their applicability for the early monitoring of cancer risk in relation to obesity.
To undertake this, we have adopted a non-invasive approach to testing acquired DNA damage, genomic instability, systemic inflammation and vitamin D status alongside multiple markers of adiposity in adolescents recruited from schools and paediatric obesity clinics in London.
Participants and methods
Sample size calculation
A cross-sectional study was designed to compare markers of adiposity, inflammation, vitamin D and DNA damage in adolescents aged 10–18 years. The primary endpoint was the frequency of MNi in the buccal mucosa. The required sample size was calculated by extracting data on mean values and variance in adults with normal weight (n = 21) and adults with obesity (n = 83) from a previous study [45]. These data were entered into G*Power (v3.1) software for A priori calculation of sample size based on a two-tailed, independent means test at an error rate of 1%. The required total sample size was calculated to be 80. Whilst it would have been most appropriate to source data from a study conducted in adolescents, there was a lack of literature reporting mean and variance values for buccal MNi in a cohort of adolescents with obesity. In order to account for this potential source of bias and to cover issues such as missing data, the sample size was increased by 20% with an aim to include a minimum of 96 participants.
Participant recruitment and screening
Over 200 research packs were distributed across schools in London to pursue non-selective recruitment. Paediatric clinics at St George’s London NHS Trust and King’s College Hospital London supported the recruitment of participants with obesity. In total, four schools and two NHS paediatric obesity clinics agreed to collaborate and 171 participants were screened for inclusion. The screening process required participants to complete a medical questionnaire to be assessed against the following exclusion criteria: dental treatment within last 6 weeks or local inflammation including pain, swelling and other evidence of tooth decay, consumption of medications including multivitamins, X-rays of the head and neck within the last 6 months, medical history of inflammatory conditions, cancer and other general illness (flu, cold, fever) on the day of sample collection. Overall, 132 participants met the inclusion criteria and provided all anthropometric and biological samples.
Anthropometric assessments
Obesity was defined as >98th percentile of the BMI for age and gender, in accordance with the UK-WHO classification [46]. Participant’s height was recorded using a standard, portable stadiometer (Marsden Weighing Machine Group) to the nearest mm. The TANITA BC54N body composition scales were used to determine weight and body fat percentage via bioelectrical impedance. Participants with a weight >150 kg were analysed for body fat percentage via InBody S10. Waist and hip measurements were recorded to the nearest mm using a standard measuring tape.
Biological sample collection
For the analysis of vitamin D and CRP, one saliva sample was collected from participants using the Salimetrics Oral Swab (SOS) which was placed on the floor of the oral cavity for one minute. Saliva samples were centrifuged at ×1500g and stored at −20 °C until analysis. For the analysis of 8-OHdG, participants collected 10–30 mL of a mid-stream urine sample into a polypropylene universal container. In total, 3 µL of Gentamycin was added per mL of sample and stored at −80 °C until 8-OHdG analysis. For the buccal cytome assay, cell sample collection followed the protocol of Thomas and colleagues with no modifications [47].
Analysis of inflammation and vitamin D
CRP was quantified in saliva samples using the Salimetrics CRP ELISA kit (Stratech, 1-3302). The analytical sensitivity of the kit was 0.042 pg/mL. Saliva samples were allowed to thaw before they were mixed and centrifuged at ×1500g for 15 min at room temperature, in order to remove mucins and particulate matter that can interfere with the assay. Saliva was diluted between 10 to 30-fold and assayed in duplicate. In total, four assays were run to analyse 132 saliva samples. The average R-squared value of standard curves was 0.99.
Vitamin D was quantified in saliva samples using the 25-OH Vitamin D (total) ELISA kit (DX-EIA- 5396, Oxford Biosystems). The analytical sensitivity of the kit was 2.89 ng/mL. Saliva samples were allowed to thaw before being mixed and centrifuged at ×1500g for 15 min at room temperature. Saliva was not diluted for this assay and samples were analysed in duplicate. In total, four assays were run to analyse 132 saliva samples. The average R-squared value of standard curves was 0.96. Seven saliva samples had a vitamin D level below the detection limit of the kit, which led to the exclusion of those participants from the dataset.
Analysis of DNA damage
The DNA damage EIA Kit (AD-EKS-350, Enzo Life Sciences) was used to perform the quantification of 8-OHdG in urine samples via a competitive ELISA reaction. The analytical sensitivity of the kit was 0.59 ng/mL. Prior to each assay, urine samples were allowed to thaw and centrifuged at ×2000 g for ten min at room temperature. Urine samples were diluted 20–30-fold, vortexed for 10 sec then assayed in duplicate. In total, four assays were run to analyse 132 urine samples. The average R-squared value of standard curves was 0.99. To control for intra-individual variation in urinary 8-OHdG levels, a creatinine correction was applied. Urinary creatinine was assessed by the University of Westminster Blood Testing Service (UKAS accredited). Samples were analysed in the ILab Aries based on the colorimetric methodology between the reaction of creatinine with picric acid under alkaline conditions. Urinary creatinine (mg/mL) was calculated by multiplying the concentration of creatinine (mmol/L) in urine samples by the molecular weight of creatinine then divided by one hundred. Final 8-OHdG (ng/mg creatinine) was calculated by dividing urinary 8-OHdG (ng/mL) by urinary creatinine (mg/mL).
Analysis of genomic instability by buccal cytome assay
The buccal cytome assay (BCA) was performed within one week of buccal cell sample collection according to the protocol published by Thomas et al. with some modifications [47]. Cell sample homogenisation was performed using a 25 G needle. In total, 150 µL of cell sample was added to two cytocentrifuge cups. Slides were then centrifuged at 600RPM for five min at room temperature and allowed to air dry for ten min. The slide staining process followed that of Thomas et al. [47]. Slides were examined under a Carl Zeiss Primo Star Light Microscope (37081). Unsatisfactory slides (low cell counts, debris obscuring cells or poor staining) were repeated. To score slides, the microscope was connected to a JENOPTIK ProgRes CT5 USB C Camera (D-07739 Jena) and images were captured using ProgRes Software. One thousand cells per participant were imaged at a magnification of ×1000 with immersion oil. Slides were scored blindly for their frequency of normal differentiated cells (NDCs), cells with MNi, polynucleated cells (PNCs) and cells with nuclear buds/nucleoplasmic bridges (NBUDs/NPBs). The frequency of each cell type was reported as per 1000 cells.
Statistical analysis
Data were entered into BMI SPSS version 25 and checked for outliers using boxplots. Extreme outliers (20 out of 132) were removed from subsequent analysis. Unpaired t-tests were conducted on anthropometric and biomarker data of the remaining 112 participants to test for any differences between participants with normal range BMI and those with obesity. Levene’s test for equality of variances was applied. The three genomic variables, Buccal MNi, Buccal PNCs, and Buccal NBUDs/NPBs, were combined to create a new variable: combined genomic instability score or combined GI score.
Pearson’s correlations were conducted on all variables to identify relationships. Testing for a false discovery rate was carried out to manage the risk of a Type 1 error. Two multiple linear regression models were conducted with a combined genomic instability score as the criterion variable. The first was a hierarchical linear regression analysis in which all predictor variables were included in the model: demographic variables were included in the first stage and the remaining variables in the second stage. A multiple linear regression analysis was also conducted in which only those variables that correlated with the criterion variable were included. The following assumption tests were conducted: multicollinearity, homoskedasticity, normality of errors and linearity, resulting in the removal of predictor variables: waist circumference and fat percentage.
Ethical considerations and approvals
Ethical approval was obtained from the Human Research Authority and NHS Research Ethics committee (IRAS ID: 212869) and the University of Westminster (ETH1617-1943). Data were protected in accordance with the Data Protection Act 1998 and later updated according to the General Data Protection Regulation and Data Protection Act 2018. Written consent was obtained from the parents of all participants that were screened and included in the study. Biological samples were stored in line with the Human Tissue Act 2004.
Results
Participant demographics
A contingency analysis of participants’ demographics was performed relative to the BMI category (Table 1). Fifty-nine participants were identified as with normal range BMI (<98th centile) and 53 participants were identified with obesity (>98th centile). In total, the largest proportion of participants were White British or White European (38%) and the smallest proportion was of mixed parentage (6.7%). A Chi-squared analysis revealed no significant differences in ethnic groups across BMI categories (p = 0.39). There was a fairly equal distribution of males and females across BMI categories (F: M ratio of 1: 0.84). The average age of participants across categories was 13.6 years in the normal range BMI group and 14.6 years in the group with obesity. Overall, the age range of participants—10–18 years—was the same within categories.
Obesity is associated with higher levels of genomic instability in the buccal mucosa
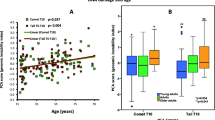
A comparison of participants’ anthropometric and biomarker measures is reported in Table 2. As expected, the average BMI Z-score (the standard deviation above or below the mean), waist to hip ratio (WHR), waist circumference and body fat percentage were all significantly higher for participants in the group with obesity (>98th centile). Unpaired t-test analysis of average levels of biomarkers between normal range BMI and participants with obesity revealed significantly higher levels of buccal cells with MNi, polynucleated buccal cells (PNCs), and nuclear buds and nucleoplasmic bridges (NBUDs/NPBs) (Fig. 1). Participants with obesity presented with higher levels of CRP and oxidative DNA damage (8-OHdG) in urine, and lower levels of vitamin D in saliva. However, those differences were not statistically significant.

Photomicrographs of exfoliated buccal mucosa cells stained with Feulgen and Light Green and viewed at 1000× magnification under transmitted light. The figure shows examples of normal differentiated cells (top row) compared to cells presenting different types of nuclear abnormalities (rows below) (buccal MNi = cells with micronuclei; PNCs = cells showing poly-nucleation or multiple nuclei; NBUDs/NPBs = cells with nuclear buds and/or nucleoplasmic bridges). Cells were scored and nuclear abnormalities classified according to the criteria defined in the ‘buccal micronucleus cytome assay’ [45].
Adiposity is correlated with levels of inflammation, vitamin D, acquired DNA damage and genomic instability
Pearson’s correlation analysis of biomarkers across all participants revealed a statistically significant correlation between all anthropometric markers (p < 0.001). BMI Z-score and body fat percentage are markers of adiposity that were positively correlated with CRP, urinary 8-OHdG and genomic instability markers (Fig. 2). Furthermore, BMI Z-score and body fat percentage were negatively correlated with salivary vitamin D. WHR and waist circumference were also negatively correlated with vitamin D and positively correlated with DNA damage and genomic instability markers, except that WHR was not correlated with the frequency of MNi. However, neither WHR nor waist circumference correlated with levels of inflammation. Interestingly, salivary CRP correlated positively with levels of cells with MNi (p = 0.029) and salivary vitamin D correlated negatively with all DNA damage and genomic instability markers.

Abbreviations used in the map: BMI cat body mass index category, WHR waist-hip-ratio, CRP C-reactive protein, 8OHdG 8-hydroxy-2′-deoxyguanosine, Buccal Mni buccal micronuclei, Buccal PNCs buccal polynucleated cells, Buccal NBUDSs buccal nuclear buds, Combined GI score combined genomic instability score.
BMI category, vitamin D and oxidative DNA damage are predictors of genomic instability (models 1 and 2)
The hierarchical multiple regression analysis in Model 1, with BMI category, age, sex and ethnicity as the predictors, explained 21.2% of the variance and was significant, F (7, 104) = 5.28, p < .001. Model 2 in which Vitamin D, CRP, 8-OHdG and waist-hip ratio were added explained significantly more variance (R2 change = 0.11, F (4, 100) = 4.21, p = 0.003). Model 2 explains 29.9% of the variance (adjusted R2 = 0.299) and was significant (F (11, 100) = 5.30, p < 0.001). The significant predictors in model 2 were BMI category, 8OHdG and vitamin D as indicated in Table 3a.
Multiple regression analysis (model 3)
Those variables that were significant predictors in model 2 were entered into a multiple linear regression using the standard method. A significant model emerged: F (3, 108) = 17.14, p < 0.001. The model explains 30.4% of the variance in the combined genomic instability score (adjusted R2 = 0.304). Table 3b gives information about regression coefficients for the predictor variables entered into the model showing that BMI category, Vitamin D and 8-OHdG were significant predictors. Therefore, participants’ predicted combined genomic instability score is equal to 0.841 + 0.487 (BMI category) −0.036 (Vitamin D) −0.001 (8-OHdG), where BMI was coded as 1 = normal range, 2 = obese.
Discussion
This research is the first to perform a combined, non-invasive investigation of inflammation, vitamin D, and multiple markers of genome damage in relation to anthropometric measurements in adolescence. Our results support the hypothesis that childhood obesity is associated with increased genomic instability and presents implications for a potential increase in the risk of cancer later in life. Most importantly, we have presented for the first time a multiple regression model for the prediction of genomic instability based on obesity, urinary 8-OHdG and salivary vitamin D.
We found that BMI Z-score and body fat percentage significantly and positively correlated with salivary CRP levels, supporting the hypothesis that low-grade inflammation may increase with adiposity. Although average salivary CRP levels were higher in participants with obesity, this was not statistically significant due to large variations across both cohorts. Several studies have found significantly higher serum CRP from children with obesity compared to controls [9,10,11,12,13,14,15,16, 48], but results in saliva are so far limited and inconclusive. Naidoo and colleagues reported a BMI above the 85th centile to be a significant predictor of elevated salivary CRP across 170 Black South African children [49]. Salivary CRP levels were also reported to be six times higher in children with a BMI above the 95th centile [50]. However, more recently Janem and colleagues reported no significant differences in salivary CRP between children with and without obesity in a study of 49 participants [51]. Nonetheless, moderate correlations between serum and salivary CRP in adolescents indicate that excess production of serum proteins may drive them to become incorporated into saliva [52]. Therefore, further studies would be useful to explore the predictive potential of inflammatory markers in saliva, in relation to cancer risk.
Furthermore, we found the frequency of cells with MNi, a type of biomarker of genomic instability, increased in correlation with salivary CRP levels. There are several indications that describe inflammation to have a causative role in the initiation of malignancy. For example, a one-unit increase in CRP levels has been linked with a 2.29-fold increase in the risk of colon cancer [53]. Chronic inflammation is a well-known aetiological factor for genetic instability and neoplastic transformations in cells and may drive the production of excess reactive oxygen and nitrogen species and cause structural modifications in DNA to promote pre-malignancy [54, 55]. A profile of circulating pro-inflammatory cytokines has been associated with pre-malignant lesions of the oral mucosa, gastric mucosa and prostate in the absence of tissue infection [56,57,58,59]. There are also reports that chronic inflammation may suppress anti-tumour defence mechanisms, lead to a loss of mitotic arrest following DNA damage and enable the accumulation of random mutations that may contribute to the genetic heterogeneity seen in cancer cells [60,61,62]. These indications coupled with the associations between excess adiposity and inflammation fuel the need for long-term monitoring of inflammation status in patients with obesity.
In addition, we found an increase in multiple markers of adiposity correlated with a significant decrease in salivary vitamin D levels, and that average vitamin D was lower in participants with obesity, although this was not statistically significant. Over the last few years, a large proportion of studies have identified obesity in children as a state of hypovitaminosis D [26,27,28,29,30,31,32]. To our knowledge, this is the first study to explore the association between adiposity in adolescents and vitamin D levels in saliva. The lack of investigations of this type so far may be due to the challenges associated with saliva as a dilute biological fluid compared to serum. Therefore, the detection of vitamin D in saliva from patients with deficient serum levels requires a high sensitivity assay. For this study, the commercially available immunoassay kit with the highest analytical sensitivity was sourced. We also obtained saliva samples using the stimulated method of saliva collection as this has been demonstrated to increase levels of salivary vitamin D [63]. However, there were seven samples that were below the detection limit of the assay, and it is important to note that six of these samples were from participants with obesity. Therefore, there is a need for the development of high-sensitive and high-throughput assays that can quantify vitamin D in saliva and be used to develop a threshold for point of care diagnosis of a deficiency, especially since a strong correlation between salivary vitamin D and serum vitamin D (R = 0.83) has been reported [64].
Most importantly, we found that salivary vitamin D may be a useful predictor of genomic instability when combined with weight status and urinary 8-OHdG levels via a multiple regression model. It is likely that deficient levels of vitamin D in obesity may exacerbate the effects of adipose tissue dysfunction and consequent DNA damage. Unlike this investigation, previous studies have not assessed vitamin D status and DNA damage simultaneously in childhood obesity. However, excess DNA damage in sperm cells has been related to vitamin D deficiency and excess adiposity in a rat model [65]. Associations between vitamin D and markers of genomic integrity and stability have been presented before and align with our findings. Sufficient serum vitamin D status has been found to modulate the effects of UV-light induced MNi formation in human lymphocytes [66]. Furthermore, vitamin D treatment has also demonstrated a reduction in MNi frequency of rat hepatocytes and in a model of murine lymphoma [67]. There are indications that vitamin D may reduce oxidative DNA damage and up-regulate DNA repair proteins to preserve genomic integrity [68,69,70, 68, 69]. Several mechanisms could explain links between vitamin D and reduced oxidative DNA damage. Vitamin D has been described as a hormone with anti-inflammatory properties by a number of studies [34, 71,72,73]. Although we did not find a correlation between salivary CRP and vitamin D, it is of interest that previous research has found vitamin D deficiency in childhood obesity to coincide with increased serum high-sensitivity CRP levels [23]. Studies in rodents imply that vitamin D deficiency can increase the secretion of pro-inflammatory cytokines [74, 75]. Overall, it can be postulated that vitamin D deficiency is a modifiable risk factor for genomic instability in children with obesity.
Another important finding is that 8-OHdG—as a measure of DNA damage—correlated significantly and positively with multiple markers of adiposity in our cohort of participants, but did not differ significantly between participants with and without obesity. To date, there are only three other investigations that we are aware of which have assessed the association between adiposity and levels of 8-OHdG in urine. An earlier report demonstrated children with obesity to have higher levels of 8-OHdG in urine [43]. However, an inverse association between BMI and urinary 8-OHdG was reported across 159 healthy Italian children [44]. Yet more recently, higher levels of urinary 8-OHdG were reported in children with obesity, corroborating the earlier findings, but these children also presented with insulin resistance [42]. Our findings support the associations between adiposity and oxidative DNA damage, similar to the findings of El Wakkad et al. [41], who reported body fat percentage assessed via bioelectrical impedance and BMI to be positively correlated with 8-OHdG in serum across 103 adolescents. These findings are important because of the role that oxidative DNA damage can play in generating genomic instability and a pre-cancerous state.
There is evidence that oxidative DNA damage can promote genomic instability. 8-OHdG can contribute to chromosomal instability by altering the maintenance of telomere length [76]. A recent review has highlighted that 8-OHdG lesions occur more frequently in telomeric DNA than bulk genomic DNA or microsatellite regions [77]. This is interesting because telomere length in children with obesity has been found to be 23% shorter compared to age-matched healthy weight controls [78]. Telomere shortening is also associated with an increase in nuclear anomalies, in particular nuclear buds and bridges [79]. These findings stress the importance of including levels of 8-OHdG to predict chromosomal instability in childhood obesity.
It is of interest that we found average levels of three different nuclear anomalies in the buccal mucosa to be significantly higher in participants with obesity compared to healthy weight controls. To the best of our knowledge, our research is the first to present significant correlations between multiple markers of adiposity and the frequency of biomarkers of genomic instability such as micronucleated cells, nuclear buds/bridges and PNCs in adolescence. In 2009, the first investigation of adiposity and nuclear anomalies in the buccal mucosa found no association but was conducted with only 20 Mexican 7–11 years olds in each weight category [39]. More recently, Idolo and colleagues investigated the impact of various lifestyle factors on nuclear anomalies in the buccal mucosa of 6–8 years old Italians and concluded obesity in children to be an independent risk factor for increased MNi frequency [40]. There is evidence that an increased prevalence of MNi in the oral mucosa may be reflective of chromosomal instability occurring in other tissues [80]. Therefore, it is unsurprising that our findings support previous research that found an increased level of MNi and chromosomal aberrations in PBLs from children with obesity [37]. Overall, these findings confer that childhood obesity is associated with an increased state of acquired DNA damage and genomic instability. Therefore, it is important to consider the potential implications of our findings in relation to cancer risk.
An increased frequency of MNi in buccal epithelial cells has been consistently identified in pre-malignancy and malignancy of the aerodigestive tract [81,82,83,84,85]. There is also evidence to suggest that an increased MNi frequency in buccal cells may be related to the risk of cancer at other sites including the breast, uterus, lung, colorectum, and bladder [86,87,88,89]. There are indications that MNi containing whole chromosomes can proceed into several cell generations and be reincorporated into the genome following further mitotic divisions [90]. MNi division cycles can lead to catastrophic genetic re-arrangements in a single or few chromosomes—a newly described mutational process called chromothripsis [91]. Such localised chromosomal re-arrangements may be transferred to daughter nuclei in subsequent mitotic cycles and play a role in generating a pre-cancerous genome. Furthermore, MNi can display a lack of nuclear envelope integrity when occurring in cancer cells [92]. Firstly, impaired nuclear envelope function has been related to an increase in DNA damage in MNi within cancer cells—a process that may also promote chromothripsis [92]. It is also likely that the nuclear envelope of a micronucleus is more likely to rupture, causing exposure of self-DNA to the cytosol. Possible immuno-stimulatory consequences of this event have recently been reported in a mouse model and human cancer cells [93]. This means that the occurrence of MNi may also drive carcinogenesis by triggering inflammation. Interestingly, we found a significant correlation in the oral cavity between levels of CRP and cells with MNi across all participants. Longitudinal studies in large cohorts may consolidate the use of the buccal cytome assay as a tool for cancer risk prediction.
Also important are the findings of excess average levels of nuclear buds and nucleoplasmic bridges in participants with obesity compared to controls. NBUDs/NPBs has been described as a consequence of unrepaired DNA damage or gene amplification and have also been linked with carcinogenesis. An increased frequency of nuclear buds and nucleoplasmic bridges was documented in PBLs from cancer patients compared to healthy controls [89]. Furthermore, nucleoplasmic bridges are associated with breakage-fusion-bridge (BFB) cycles that may drive chromosomal rearrangements seen in tumour genomes [94]. DNA amplification and chromosomal rearrangements have been noted in cancers of the lung, breast, prostate, GI tract and skin [95].
We also found a higher average frequency of PNCs in participants with obesity, which is indicative of increased cytokinesis failure. Cancer cell lines have been used to demonstrate the mutagenic consequences of binucleated cells [96]. In a recent review, tetraploidy and cytokinesis failure have been evaluated as mechanisms for aneuploidy in subsequent mitotic cycles. It has been suggested that these events can lead to genetic diversification in cancer cells that possibly provide developmental advantages [97]. An increased frequency of binucleated cells has been identified in patients with cancers of the breast, head, neck and mesothelium [98,99,100]. Furthermore, a number of different mitotic and cell cycle checkpoint proteins that regulate cytokinesis can be mutated in cancer, indicating that cytokinesis failure could possibly be an early event in tumorigenesis [101].
In summary, our results support the hypothesis that childhood obesity is associated with increased genomic instability. Importantly, we have found that obesity, vitamin D and oxidative DNA damage can together predict genomic instability. This investigation has several strengths. Firstly, we combined analysis of multiple markers of DNA damage and genomic instability which have been linked to the initiation and progression of cancer. Secondly, we investigated several parameters of adiposity in addition to traditionally reported BMI Z-scores in children and adolescents. Furthermore, we have outlined a novel, non-invasive approach for combined assessment of inflammation, micronutrition and genome health. Our findings warrant further research into the applicability of this approach as a non-invasive clinical tool for predicting early, pre-pathological genomic changes in young patients with obesity.
Conclusions
By applying a non-invasive approach for the combined assessment of parameters of adiposity, inflammation, Vitamin D status and genome damage, we have developed a multiple regression model for the prediction of genomic instability in adolescence. Further work may lead to its application in the prioritisation and provision of clinical intervention measures to prevent increased risk of malignancy in patients with obesity.
References
Jeffreys M, Davey Smith G, Martin RM, Frankel S, Gunnell D. Childhood body mass index and later cancer risk: a 50‐year follow‐up of the Boyd Orr study. Int J Cancer. 2004;112:348–51.
Bjørge T, Engeland A, Tverdal A, Smith GD. Body mass index in adolescence in relation to cause-specific mortality: a follow-up of 230,000 Norwegian adolescents. Am J Epidemiol. 2008;168:30–7.
Park MH, Falconer C, Viner RM, Kinra S. The impact of childhood obesity on morbidity and mortality in adulthood: a systematic review. Obes Rev. 2012;13:985–1000.
Simmonds M, Llewellyn A, Owen CG, Woolacott N. Predicting adult obesity from childhood obesity: a systematic review and meta-analysis. Obes Rev. 2016;17:95–107.
Simmonds M, Burch J, Llewellyn A, Griffiths C, Yang H, Owen C, et al. The use of measures of obesity in childhood for predicting obesity and the development of obesity-related diseases in adulthood: a systematic review and meta-analysis. Heal Technol Assess. 2015;19:1–336.
Landgraf K, Rockstroh D, Wagner IV, Weise S, Tauscher R, Schwartze JT, et al. Evidence of early alterations in adipose tissue biology and function and its association with obesity-related inflammation and insulin resistance in children. Diabetes. 2015;64:1249–61.
Barbarroja N, López-Pedrera R, Mayas MD, Garcia-Fuentes E, Garrido-Sánchez L, Macías-González M, et al. The obese healthy paradox: is inflammation the answer? Biochem J. 2010;430:141–9.
Bhattacharya I, Domínguez AP, Drägert K, Humar R, Haas E, Battegay EJ. Hypoxia potentiates tumor necrosis factor-α induced expression of inducible nitric oxide synthase and cyclooxygenase-2 in white and brown adipocytes. Biochem Biophys Res Commun. 2015;461:287–92.
Cook DG, Mendall MA, Whincup PH, Carey IM, Ballam L, Morris JE, et al. C-reactive protein concentration in children: relationship to adiposity and other cardiovascular risk factors. Atherosclerosis. 2000 Mar;149:139–50.
Rönnemaa T, Juonala M, Marniemi J, Raitakari OT, Taittonen L, Viikari JSA. Childhood C-reactive protein in predicting CRP and carotid intima-media thickness in adulthood. Arterioscler Thromb Vasc Biol. 2006;26:1883–8.
Schipper HS, Nuboer R, Prop S, Van Den Ham HJ, De Boer FK, Kesmir C, et al. Systemic inflammation in childhood obesity: circulating inflammatory mediators and activated CD14++ monocytes. Diabetologia. 2012;55:2800–10.
Carmona-Montesinos E, Ruiz-Fragoso Z, Ponce-Hinojosa G, Rivas-Arancibia S, CHANGES IN. C-reactive protein and biochemical profile in preschool children with obesity. Nutr Hosp. 2015;32:1548–53.
Nishide R, Ando M, Funabashi H, Yoda Y, Nakano M, Shima M. Association of serum hs-CRP and lipids with obesity in school children in a 12-month follow-up study in Japan. Environ Health Prev Med. 2015;20:116–22.
Chang C-J, Jian D-Y, Lin M-W, Zhao J-Z, Ho L-T, Juan C-C. Evidence in obese children: contribution of hyperlipidemia, obesity-inflammation, and insulin sensitivity. PLoS ONE. 2015;10:e0125935–e0125935.
Kitsios K, Papadopoulou M, Kosta K, Kadoglou N, Papagianni M, Tsiroukidou K. High-sensitivity C-reactive protein levels and metabolic disorders in obese and overweight children and adolescents. J Clin Res Pediatr Endocrinol. 2013;5:44.
Rowicka G, Dyląg H, Ambroszkiewicz J, Riahi A, Weker H, Chełchowska M. Total oxidant and antioxidant status in prepubertal children with obesity. Oxid Med Cell Longev. 2017;2017:5621989.
Weihe P, Spielmann J, Kielstein H, Henning-Klusmann J, Weihrauch-Blüher S. Childhood obesity and cancer risk in adulthood. Curr Obes Rep. 2020;9:204–12.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2016;144:646–74.
Shen Z. Genomic instability and cancer: an introduction. J Mol Cell Biol. 2011;3:1–3.
Aeberli I, Biebinger R, Lehmann R, L’allemand D, Spinas GA, Zimmermann MB. Serum retinol-binding protein 4 concentration and its ratio to serum retinol are associated with obesity and metabolic syndrome components in children. J Clin Endocrinol Metab. 2007;92:4359–65.
García OP, Ronquillo D, del Carmen Caamaño M, Martínez G, Camacho M, López V, et al. Zinc, iron and vitamins A, C and e are associated with obesity, inflammation, lipid profile and insulin resistance in Mexican school-aged children. Nutrients. 2013;5:5012–30.
Manios Y, Moschonis G, Chrousos GP, Lionis C, Mougios V, Kantilafti M, et al. The double burden of obesity and iron deficiency on children and adolescents in Greece: the Healthy Growth Study. J Hum Nutr Diet. 2013;26:470–8.
Rodriguez-Rodriguez E, Aparicio A, Andres P, Ortega RM. Moderate vitamin D deficiency and inflammation related markers in overweight/obese schoolchildren. Int J Vitam Nutr Res. 2014;84:98–107.
Ortega M, Rodríguez-Rodríguez Aparicio, Jiménez-Ortega I, Palmeros PereaM, et al. Young children with excess of weight show an impaired selenium status. Int J Vitam Nutr Res. 2012;82:121–9.
Basatemur E, Horsfall L, Marston L, Rait G, Sutcliffe A. Trends in the diagnosis of vitamin D deficiency. Pediatrics. 2017;139:e20162748.
Bellone S, Esposito S, Giglione E, Genoni G, Fiorito C, Petri A, et al. Vitamin D levels in a paediatric population of normal weight and obese subjects. J Endocrinol Investig. 2014;37:805–9.
Ekbom K, Marcus C. Vitamin D deficiency is associated with prediabetes in obese Swedish children. Acta Paediatr. 2016;105:1192–7.
Wakayo T, Whiting SJ, Belachew T. Vitamin D deficiency is associated with overweight and/or obesity among schoolchildren in central Ethiopia: a cross-sectional study. Nutrients. 2016;8:190.
Kumaratne M, Early G, Cisneros J. Vitamin D deficiency and association with body mass index and lipid levels in hispanic American adolescents. Glob Pediatr Health. 2017;4:2333794X17744141.
Alyahya KO. Vitamin D levels in schoolchildren: a cross-sectional study in Kuwait. BMC Pediatr. 2017;17:213.
Erol M, Bostan Gayret O, Hamilcikan S, Can E, Yigit OL. Vitamin D deficiency and insulin resistance as risk factors for dyslipidemia in obese children. Arch Argent Pediatr. 2017;115:133–9.
Plesner JL, Dahl M, Fonvig CE, Nielsen TRH, Kloppenborg JT, Pedersen O, et al. Obesity is associated with vitamin D deficiency in Danish children and adolescents. J Pediatr Endocrinol Metab. 2018;31:53–61.
Vimaleswaran KS, Berry DJ, Lu C, Tikkanen E, Pilz S, Hiraki LT, et al. Causal relationship between obesity and vitamin D status: bi-directional Mendelian randomization analysis of multiple cohorts. PLoS Med. 2013;10:e1001383.
Zhang Y, Leung DYM, Richers BN, Liu Y, Remigio LK, Riches DW, et al. Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol. 2012;188:2127–35.
Krishnan AV, Trump DL, Johnson CS, Feldman D. The role of vitamin D in cancer prevention and treatment. Rheum Dis Clin. 2012;38:161–78.
Usman M, Volpi EV. DNA damage in obesity: initiator, promoter and predictor of cancer. Mutat Res Rev Mutat Res. 2018;778:23–37.
Scarpato R, Verola C, Fabiani B, Bianchi V, Saggese G, Federico G. Nuclear damage in peripheral lymphocytes of obese and overweight Italian children as evaluated by the γ-H2AX focus assay and micronucleus test. FASEB J. 2011;25:685–93.
Azzarà A, Pirillo C, Giovannini C, Federico G, Scarpato R. Different repair kinetic of DSBs induced by mitomycin C in peripheral lymphocytes of obese and normal weight adolescents. Mutat Res Mol Mech Mutagen. 2016;789:9–14.
Torres-Bugarín O, Fernández-García A, Torres-Mendoza BM, Zavala-Aguirre JL, Nava-Zavala A, Zamora-Perez AL. Genetic profile of overweight and obese school-age children. Toxicol Environ Chem. 2009;91:789–95.
Idolo A, Grassi T, Bagordo F, Panico A, De Giorgi M, Serio F, et al. Micronuclei in exfoliated buccal cells of children living in a cluster area of Salento (Southern Italy) with a high incidence of lung cancer: the IMP.AIR study. Int J Environ Res Public Health. 2018;15:1659.
El Wakkad A, Giza AO, Elwakkad A, Hassan NE, Sibaii H, el Zayat S. Relationship between obesity and 8-hydroxy-2-deoxy guanosine as an oxidative marker in obese. J Med Sci. 2011;11:231–5.
Ramachandra N, Rodriguez L, Devaraj S. Urinary biomarkers of oxidative stress and insulin resistance in childhood obesity. FASEB J. 2015;1:29.
Šebeková K, Somoza V, Jarčušková M, Heidland A, Podracká L. Plasma advanced glycation end products are decreased in obese children compared with lean controls. Int J Pediatr Obes. 2009;4:112–8.
Protano C, Andreoli R, Mutti A, Petti S, Vitali M. Biomarkers of oxidative stress to nucleic acids: Background levels and effects of body mass index and life-style factors in an urban paediatric population. Sci Total Environ. 2014;500–501:44–51.
Donmez-Altuntas H, Sahin F, Bayram F, Bitgen N, Mert M, Guclu K, et al. Evaluation of chromosomal damage, cytostasis, cytotoxicity, oxidative DNA damage and their association with body-mass index in obese subjects. Mutat Res Toxicol Environ Mutagen. 2014;771:30–6.
Royal College of Paediatrics and Child Health. Body mass index UK 2–20 years. 2013;2. https://www.rcpch.ac.uk/sites/default/files/2018-03/boys_and_girls_bmi_chart.pdf
Thomas P, Holland N, Bolognesi C, Kirsch-Volders M, Bonassi S, Zeiger E, et al. Buccal micronucleus cytome assay. Nat Protoc. 2009;4:825–37.
Cohen JI, Maayan L, Convit A. Preliminary evidence for obesity-associated insulin resistance in adolescents without elevations of inflammatory cytokines. Diabetol Metab Syndr. 2012;4:26.
Naidoo T, Konkol K, Biccard B, Dudose K, McKune AJ. Elevated salivary C-reactive protein predicted by low cardio-respiratory fitness and being overweight in African children. Cardiovasc J Afr. 2012;23:501–6.
Goodson JM, Kantarci A, Hartman M-L, Denis GV, Stephens D, Hasturk H, et al. Metabolic disease risk in children by salivary biomarker analysis. PLoS ONE. 2014;9:e98799.
Janem WF, Scannapieco FA, Sabharwal A, Tsompana M, Berman HA, Haase EM, et al. Salivary inflammatory markers and microbiome in normoglycemic lean and obese children compared to obese children with type 2 diabetes. PLoS ONE. 2017;12:e0172647.
Byrne ML, O’Brien-Simpson NM, Reynolds EC, Walsh KA, Laughton K, Waloszek JM, et al. Acute phase protein and cytokine levels in serum and saliva: a comparison of detectable levels and correlations in a depressed and healthy adolescent sample. Brain Behav Immunol. 2013;34:164–75.
Izano M, Wei EK, Tai C, Swede H, Gregorich S, Harris TB, et al. Chronic inflammation and risk of colorectal and other obesity‐related cancers: the health, aging and body composition study. Int J Cancer. 2016;138:1118–28.
Tamir S, Burney S, Tannenbaum SR. DNA damage by nitric oxide. Chem Res Toxicol. 1996;9:821–7.
Hofseth LJ, Khan MA, Ambrose M, Nikolayeva O, Xu-Welliver M, Kartalou M, et al. The adaptive imbalance in base excision-repair enzymes generates microsatellite instability in chronic inflammation. J Clin Investig. 2003;112:1887–94.
Di Silverio F, Gentile V, De Matteis A, Mariotti G, Giuseppe V, Luigi PA, et al. Distribution of inflammation, pre-malignant lesions, incidental carcinoma in histologically confirmed benign prostatic hyperplasia: a retrospective analysis. Eur Urol. 2003;43:164–75.
van der Woude CJ, Kleibeuker JH, Jansen PLM, Moshage H. Chronic inflammation, apoptosis and (pre-)malignant lesions in the gastro-intestinal tract. Apoptosis. 2004;9:123–30.
Woodford D, Johnson SD, De Costa A-MA, Young MRI. An inflammatory cytokine milieu is prominent in premalignant oral lesions, but subsides when lesions progress to squamous cell carcinoma. J Clin Cell Immunol. 2014;5:230.
Tezal M, Grossi SG, Genco RJ. Is periodontitis associated with oral neoplasms? J Periodontol. 2005;76:406–10.
Cobbs CS, Whisenhunt TR, Wesemann DR, Harkins LE, Van Meir EG, Samanta M. Inactivation of wild-type p53 protein function by reactive oxygen and nitrogen species in malignant glioma cells. Cancer Res. 2003;63:8670–3.
Colotta F, Garlanda C, Allavena P, Sica A, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–81.
Grivennikov SI, Karin M. Inflammation and oncogenesis: a vicious connection. Curr Opin Genet Dev. 2010;20:65–71.
Higashi T, Hijikuro M, Yamagata K, Ogawa S. Overestimation of salivary 25-hydroxyvitamin D3 level when using stimulated saliva with gum-chewing. Steroids. 2013;78:884–7.
Higashi T, Shibayama Y, Fuji M, Shimada K. Liquid chromatography–tandem mass spectrometric method for the determination of salivary 25-hydroxyvitamin D3: a noninvasive tool for the assessment of vitamin D status. Anal Bioanal Chem. 2008;391:229–38.
Merino O, Sánchez R, Gregorio BM, Sampaio FJ, Risopatrón J. Effects of diet-induced obesity and deficient in vitamin D on spermatozoa function and DNA integrity in Sprague-Dawley rats. Biomed Res Int. 2018;2018:5479057.
Armstrong BK, Nair-Shalliker V, Fenech M, Forder PM, Clements MS. Sunlight and vitamin D affect DNA damage, cell division and cell death in human lymphocytes: a cross-sectional study in South Australia. Mutagenesis. 2012;27:609–14.
Chatterjee M. Vitamin D and genomic stability. Mutat Res Mol Mech Mutagen. 2001;475:69–87.
Wong G, Gupta R, Dixon KM, Deo SS, Choong SM, Halliday GM, et al. 1,25-Dihydroxyvitamin D and three low-calcemic analogs decrease UV-induced DNA damage via the rapid response pathway. J Steroid Biochem Mol Biol. 2004;89–90:567–70.
Fedirko V, Bostick RM, Long Q, Flanders WD, McCullough ML, Sidelnikov E, et al. Effects of supplemental vitamin D and calcium on oxidative DNA damage marker in normal colorectal mucosa: a randomized clinical trial. Cancer Epidemiol Prev Biomark. 2010;19:280–91.
Nair-Shalliker V, Armstrong BK, Fenech M. Does vitamin D protect against DNA damage? Mutat Res Mol Mech Mutagen. 2012;733:50–7.
Abbas MA. Physiological functions of Vitamin D in adipose tissue. J Steroid Biochem Mol Biol. 2017;165:369–81.
Ding C, Wilding JPH, Bing C. 1,25-dihydroxyvitamin D3 protects against macrophage-induced activation of NFkappaB and MAPK signalling and chemokine release in human adipocytes. PLoS ONE. 2013;8:e61707.
Karkeni E, Bonnet L, Marcotorchino J, Tourniaire F, Astier J, Ye J, et al. Vitamin D limits inflammation-linked microRNA expression in adipocytes in vitro and in vivo: a new mechanism for the regulation of inflammation by vitamin D. Epigenetics. 2018;13:156–62.
Chang E, Kim Y Vitamin D. Insufficiency exacerbates adipose tissue macrophage infiltration and decreases AMPK/SIRT1 activity in obese rats. Nutrients. 2017;9:338.
Karkeni E, Marcotorchino J, Tourniaire F, Astier J, Peiretti F, Darmon P, et al. Vitamin D limits chemokine expression in adipocytes and macrophage migration in vitro and in male mice. Endocrinology. 2015;156:1782–93.
Wang Z, Rhee DB, Lu J, Bohr CT, Zhou F, Vallabhaneni H, et al. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 2010;6:e1000951.
Barnes RP, Fouquerel E, Opresko PL. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech Ageing Dev. 2019;177:37–45.
Buxton JL, Walters RG, Visvikis-Siest S, Meyre D, Froguel P, Blakemore AIF. Childhood obesity is associated with shorter leukocyte telomere length. J Clin Endocrinol Metab. 2011;96:1500–5.
Pampalona J, Soler D, Genescà A, Tusell L. Telomere dysfunction and chromosome structure modulate the contribution of individual chromosomes in abnormal nuclear morphologies. Mutat Res. 2010;683:16–22.
Feki-Tounsi M, Khlifi R, Mhiri M-N, Rebai A, Hamza-Chaffai A. Cytogenetic damage in the oral mucosa cells of bladder cancer patients exposed to tobacco in Southern Tunisia. Environ Sci Pollut Res. 2014;21:12922–7.
Mandard AM, Duigou F, Marnay J, Barrellier P, Lebigot G. Analysis of the results of the micronucleus test in patients presenting upper digestive tract cancers and in non‐cancerous subjects. Int J Cancer. 1987;39:442–4.
Bloching M, Hofmann A, Lautenschläger C, Berghaus A, Grummt T. Exfoliative cytology of normal buccal mucosa to predict the relative risk of cancer in the upper aerodigestive tract using the MN-assay. Oral Oncol. 2000;36:550–5.
Ramirez A, Saldanha PH. Micronucleus investigation of alcoholic patients with oral carcinomas. Genet Mol Res. 2002;1:246–60.
Saran R, Tiwari RK, Reddy PP, Ahuja YR. Risk assessment of oral cancer in patients with pre-cancerous states of the oral cavity using micronucleus test and challenge assay. Oral Oncol. 2008;44:354–60.
Desai SS, Ghaisas SD, Jakhi SD, Bhide SV. Cytogenetic damage in exfoliated oral mucosal cells and circulating lymphocytes of patients suffering from precancerous oral lesions. Cancer Lett. 1996;109:9–14.
Flores-García A, Torres-Bugarín O, Velarde-Félix JS, Rangel-Villalobos H, Zepeda-Carrillo EA, Rodríguez-Trejo A, et al. Micronuclei and other nuclear anomalies in exfoliated buccal mucosa cells of Mexican women with breast cancer. J BUON. 2014;19:895–9.
Nersesyan AK, Adamyan RT. Micronuclei level in exfoliated buccal mucosa cells of patients with benign and malignant tumors of female reproductive organs and breasts. Cytol Genet. 2004;38:69.
Yildirim IH, Yesilada E, Yologlu S. Micronucleus frequency in peripheral blood lymphocytes and exfoliated buccal cells of untreated cancer patients. Russ J Genet. 2006;42:573–7.
Podrimaj-Bytyqi A, Borovečki A, Selimi Q, Manxhuka-Kerliu S, Gashi G, Elezaj IR. The frequencies of micronuclei, nucleoplasmic bridges and nuclear buds as biomarkers of genomic instability in patients with urothelial cell carcinoma. Sci Rep. 2018;8:17873.
Shimizu N. Molecular mechanisms of the origin of micronuclei from extrachromosomal elements. Mutagenesis. 2011;26:119–23.
Zhang C-Z, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, et al. Chromothripsis from DNA damage in micronuclei. Nature. 2015;522:179–84.
Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154:47–60.
Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548:461–5.
Zakov S, Kinsella M, Bafna V. An algorithmic approach for breakage-fusion-bridge detection in tumor genomes. Proc Natl Acad Sci USA. 2013;110:5546–51.
Fenech M. Chromosomal biomarkers of genomic instability relevant to cancer. Drug Discov Today. 2002;7:1128–37.
Nishimura K, Watanabe S, Hayashida R, Sugishima S, Iwasaka T, Kaku T. Binucleated HeLa cells are formed by cytokinesis failure in starvation and keep the potential of proliferation. Cytotechnology. 2016;68:1123–30.
Lens SMA, Medema RH. Cytokinesis defects and cancer. Nat Rev Cancer. 2019;19:32–45.
Paz MFCJ, Sobral ALP, Picada JN, Grivicich I, Júnior ALG, da Mata AMOF, et al. Persistent increased frequency of genomic instability in women diagnosed with breast cancer: before, during, and after treatments. Oxid Med Cell Longev. 2018;2018:2846819.
Khlifi R, Trabelsi-Ksibi F, Chakroun A, Rebai A, Hamza-Chaffai. A cytogenetic abnormality in exfoliated cells of buccal mucosa in head and neck cancer patients in the Tunisian population: impact of different exposure sources. Biomed Res Int. 2013;2013:905252.
Kimura N, Dota K, Araya Y, Ishidate T, Ishizaka M. Scoring system for differential diagnosis of malignant mesothelioma and reactive mesothelial cells on cytology specimens. Diagn Cytopathol. 2009;37:885–90.
Sagona AP, Stenmark H. Cytokinesis and cancer. FEBS Lett. 2010;584:2652–61.
Acknowledgements
The authors would like to acknowledge the work of Sarah March (Petchey Academy), Rosie Lloyd and Natacha Von Mitzinger (Harris Academy Battersea), Louise Thomas, Imaan Bajwa, Fatima Ahmed, Valentina Borgognoni, Dalin Murad, Winston Ekwueme, Adhe Noviello, Jenaagan Jenakendran and Mary Momoh for the recruitment of participants from schools in London. The authors would also like to acknowledge the efforts of Sophie Hawkins, Saima Haider, Anamul Hoque, Emily Shorter, William Townley, Stefan Lay and Tanya Awal in the early optimisation of laboratory protocols, and support with recruiting participants and collecting samples. The authors are indebted to Michael Fenech (Genome Health Foundation and the University of South Australia) for expert advice with the setting up of the buccal cytome assay and the scoring of nuclear abnormalities. The creatinine analysis of urine samples was performed by Chrystalla Ferrier in the School of Life Sciences, and the Authors are thankful to her and the University of Westminster, Blood Testing Service. The authors would like to thank Claire Robertson (School of Life Sciences, University of Westminster) and Roger A’Hern (Institute of Cancer Research, London) for advice on power calculations, and Matthew Madgwick (Earlham Institute) for preliminary advice on statistical modelling. This work was supported by the University of Westminster. The authors are grateful for funding received by the University through the FST Start-Up Scheme (E.V.V.) and the FST PhD Scholarship Scheme (M.U.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Usman, M., Woloshynowych, M., Britto, J.C. et al. Obesity, oxidative DNA damage and vitamin D as predictors of genomic instability in children and adolescents. Int J Obes 45, 2095–2107 (2021). https://doi.org/10.1038/s41366-021-00879-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-021-00879-2
- Springer Nature Limited
This article is cited by
-
Obesity increases genomic instability at DNA repeat-mediated endogenous mutation hotspots
Nature Communications (2024)
-
Understanding the Impact of Obesity on Ageing in the Radiance of DNA Metabolism
The Journal of nutrition, health and aging (2023)