
Abstract
Endothelial cell (EC) barrier disruption and inflammation are the pathological hallmarks of vascular disorders and acute infectious diseases and related conditions, including the coronavirus disease 2019 (COVID-19) and sepsis. Ubiquitination plays a critical role in regulating the stability, intracellular trafficking, and enzymatic activity of proteins and is reversed by deubiquitinating enzymes (DUBs). The role of DUBs in endothelial biology is largely unknown. In this study, we report that USP40, a poorly characterized DUB, prevents EC barrier disruption through reductions in the activation of RhoA and phosphorylation of myosin light chain (MLC) and cofilin. Furthermore, USP40 reduces EC inflammation through the attenuation of NF-ĸB activation, ICAM1 expression, and leukocyte-EC adhesion. We further show that USP40 activity and expression are reduced in response to endotoxin challenge. Global depletion of USP40 and EC-targeted USP40 depletion in mice exacerbated experimental lung injury, whereas lentiviral gene transfer of USP40 protected against endotoxin-induced lung injury. Using an unbiased approach, we discovered that the protective effect of USP40 occurs through the targeting of heat shock protein 90β (HSP90β) for its deubiquitination and inactivation. Together, these data reveal a critical protective role of USP40 in vascular injury, identifying a unique mechanistic pathway that profoundly impacts endothelial function via DUBs.
Similar content being viewed by others

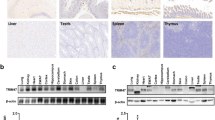

Introduction
Vascular ECs maintain vessel integrity. EC hyperpermeability and inflammation are important pathogenic features of inflammatory diseases such as acute lung injury and sepsis1,2. Loss of endothelial barrier integrity leads to an influx of protein-rich edema fluid into the interstitial tissue2,3. Stress fibers are formed by contractile actin-myosin bundles in ECs under inflammatory conditions such as bacterial or viral infection3,4,5. Contractile forces disrupt cell‒cell junctions and increase EC permeability6,7. RhoA, a small GTPase, plays a central role in stress fiber assembly and EC hyperpermeability by regulating two pathways6,8. Activation of RhoA triggers the phosphorylation of myosin light chain (MLC), leading to stress fiber formation. Additionally, RhoA-dependent kinase (ROCK) activates LIMK1 (LIM domain kinase 1), resulting in the phosphorylation and inactivation of cofilin, an actin depolymerizing factor, thereby stabilizing stress fibers and promoting EC barrier disruption9,10. EC inflammation entails EC dysfunction and tissue damage by promoting neutrophil infiltration to the site of inflammation11. Increased expression of adhesion molecules, such as ICAM1, on the EC surface is required for neutrophil adhesion to ECs and transmigration across the endothelial barrier12,13. Proinflammatory stimuli induce ICAM1 expression in ECs through the NF-κB pathway, which is activated by IKKβ-mediated phosphorylation of I-κB14,15.
HSP90, a chaperone protein, activates RhoA. Inhibition of Hsp90 prevents LPS-induced endothelial barrier disruption by attenuating RhoA signaling16. Moreover, inhibition of HSP90 decreases NF-ĸB activation in response to LPS or tumor necrosis factor α (TNFα) stimulation17,18. Thus, HSP90 plays a central role in the regulation of EC barrier disruption and inflammation16. Understanding the molecular regulation of HSP90 activation is important for the development of a therapeutic strategy to maintain endothelial function. Acetylation of HSP90 results in a reduction in its activity and its dissociation from its binding partners, thus suppressing HSP90-mediated biological functions19,20. Ubiquitination is a major posttranslational modification involved in protein localization, protein‒protein interactions, and enzymatic activity21,22. Ubiquitin E3 ligases mediate ubiquitination, which can be reversed by DUBs. The ubiquitin E3 ligases CHIP and Hectd1 mediate HSP90 ubiquitination23,24; however, a DUB for HSP90 has not been identified.
USP40, a newly recognized DUB, has been shown to regulate glomerular permeability in zebrafish. USP40 gene knockdown disrupts glomerular barrier integrity by reducing the expression of nestin, a filament protein25. However, a study from the same group demonstrated that nestin expression was upregulated in the glomeruli of USP40KO mice. However, unlike in zebrafish, no apparent phenotypic changes were observed in USP40KO mice25,26. These contrasting findings indicate that the molecular mechanism of USP40-mediated EC biological functions differs across species. Through an unbiased screening approach, we found that USP40 attenuates LPS- or thrombin-induced human lung microvascular endothelial cell (HLMVEC) barrier disruption and LPS-induced EC inflammatory responses, such as ICAM1 expression and neutrophil-EC interactions. USP40 exhibits protective effects on ECs, but our findings indicate that the underlying molecular mechanisms differ from those found in a previous study25. No reduction in nestin expression was found in USP40-deficient HLMVECs or in the lungs of USP40KO mice. Furthermore, our data reveal that USP40 deubiquitinates HSP90, thereby increasing its acetylation and reducing its activation. Our observations are the first to reveal that USP40 reduces the severity of acute lung injury by reducing HSP90 ubiquitination, thereby attenuating the activation of RhoA and NF-κB, EC permeability, and inflammation.
Materials and methods
Cell culture and reagents
Human lung microvascular endothelial cells (HLMVECs), THP-1 cells, MLE12 cells, and Raw 264.7 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). HLMVECs were cultured with EGM-2MV microvascular endothelial growth medium containing 5% fetal bovine serum (FBS) at 37 °C in 5% CO2. LPS (E. coli O55:B5), thrombin, and the anti-β-actin antibody were purchased from Sigma–Aldrich (St. Louis, MO, USA). Antibodies against VE-cadherin, USP40, ICAM1, VCAM1, Lamin A/C, and GAPDH and immobilized protein A/G beads were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against pMLC, MLC, the Flag tag, the V5 tag, pLimk1, Limk1, p-cofilin 1, cofilin 1, pIκBα, p65, p-p65, HSP90β, K63-linked ubiquitin, ubiquitin, and acetylated lysine (AcK) were purchased from Cell Signaling (Beverly, MA, USA). ELISA kits for quantifying TNFα, IL-6, and IL-8 were obtained from eBioscience (San Diego, CA), and mouse IL-1β, KC/CXCL1 ELISA kits, and human recombinant TNFα protein were purchased from R&D Systems (Minneapolis, MN, USA). The anti-CD31-FITC antibody was obtained from Biolegend (San Diego, CA, USA). GeneJet™ reagent and GeneMute siRNA transfection reagent were purchased from SignaGen (Frederick, MD, USA). All materials used in the experiments were of the highest grade commercially available.
Animals and LPS administration
Male and female C57BL/6 J mice were housed and cared for in the specific pathogen-free animal care facility at The Ohio State University in accordance with institutional guidelines and the guidelines of the US National Institutes of Health. All animal experiments were approved by the Animal Care and Use Committee at The Ohio State University and were performed in accordance with the guidelines outlined by the committee. The CRISPR/Cas9 system was used by the Innovative Technologies Development Core Facility at the University of Pittsburgh to generate USP40 global knockout (USP40−/−) and USP40-Loxp mice27,28. USP40 EC knockout mice were generated by crossing USP40floxp/floxp mice with Tek-Cre or Cdh5-Cre transgenic mice (B6.Cg-TgTek-Cre1Ywa/J or B6.Cg-TgCdh5-cre7Mlia/J, respectively; The Jackson Laboratory). Lung injury models were established by intratracheal administration of LPS (2 mg/kg body weight) for 24 h. For construction of the lentiviral vector delivery system, human USP40 cDNA was inserted into the pLVX-IRES-tdTomato vector (Clontech, Palo Alto, CA, USA). C57BL/6 J mice were intravenously administered lentiviral vectors or lenti-USP40 (5 × 107 plaque-forming units per mouse) for 7 days before intratracheal injection of LPS. BALF and lung tissues were collected for further evaluation by ELISA, western blotting, H&E staining, wet/dry ratio measurement, immunohistochemical staining, and immunofluorescence staining.
Cecal ligation and puncture (CLP)-induced polymicrobial sepsis
Mice were anesthetized by intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg). A 3-cm longitudinal incision was made in the lower abdomen; the cecum with the adjoining intestine was then externalized and ligated 0.5 cm from its end with a 3.0 silk suture. Then, the ligated cecum was punctured with an 18-gauge needle, allowing entrapped fecal material to leak into the normally sterile peritoneal cavity. The cecum was then repositioned in the peritoneal cavity, and the abdomen was closed. Sham-operated animals received laparotomy only.
Flow cytometric analysis of mouse lung cells
Isolated single lung cells were incubated with a rat anti-mouse CD16/32 antibody (Biolegend) for 30 min to block Fc receptors. Fluorophore-conjugated antibodies were added at the recommended dilutions, and the cells were then incubated with the specific antibodies in the dark for an additional 30 min. The following anti-mouse antibodies were used for cell surface marker staining: anti-CD31-PE/Cy7 (endothelial cell marker), anti-CD54-PE (a surrogate marker of antigen-presenting cell activation), anti-CD11c-FITC, and anti-Ly6G-APC/Cy7 (expressed on neutrophils) antibodies purchased from Biolegend (San Diego, CA); an anti-CD326-APC (epithelial cell surface marker) antibody purchased from eBioscience (San Diego, CA); and an anti-CD45-Percp (leukocyte common antigen) antibody purchased from BD Bioscience (San Jose, CA). Data acquisition and data analysis were performed with an Agilent NovoCyte flow cytometer and NovoExpress Software, respectively. We defined cell compartments as follows: endothelial, CD326− CD45−/CD31+; and neutrophil, CD45+/Ly6G+.
Measurement of TEER (transendothelial electrical resistance) by an electrical cell-substrate impedance sensing system (ECIS)
HLMVECs were grown on gold electrodes. Resistance changes were monitored in real time using the ECIS (Applied Biophysics) at 4000 Hz. TEER values for each microelectrode were pooled at discrete time points and plotted against time as the mean ± S.E.M. values.
Immunoblot analysis
HLMVECs and lung tissues were lysed in cell lysis buffer containing 20 mM Tris HCl (pH 7.4), 150 mM NaCl, 2 mM EGTA, 5 mM β-glycerophosphate, 1 mM MgCl2, 1% Triton X-100, 1 mM sodium orthovanadate, 10 μg/ml protease inhibitors, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. Equal amounts of protein were separated by SDS‒PAGE. Immunoblotting with primary and secondary antibodies was performed as described above29. Signals on the membrane were detected by an Azure c600 imaging system. The intensities of the bands were determined by ImageJ software.
Ubiquitination assay
Cells were harvested in cold PBS. Cell pellets were suspended in 50–80 μl of 2% SDS lysis buffer with 1 μl of ubiquitin aldehyde and 1 μl of NEM. After sonication, the cell lysates were boiled at 100 °C for 10 min. The samples were diluted with 500–800 μl of 1× Tris-buffered saline and subjected to immunoprecipitation (IP) with an anti-HSP90β antibody followed by an anti-ubiquitin or anti-K63-linked ubiquitin antibody.
DUB activity assay
Cells transfected with empty vector or the Flag-USP40 plasmid were collected, and Flag-USP40 was immunoprecipitated with FLAG-M2 affinity gel (Sigma). The beads were resuspended in DUB assay buffer (40 mM Tris (pH 7.1), 100 mM NaCl, and 5 mM DTT) and divided into 10 µl aliquots after washing with TBS buffer containing 0.1% Triton X-100. Ten microliters of Flag-USP40-conjugated beads and 2 µg of purified K63-linked or K48-linked di-Ub in 40 µl of reaction buffer were incubated at 37 °C for 2 h with gentle mixing. The DUB cleavage products of K63-linked or K48-linked di-Ub were visualized by immunoblotting with an anti-ubiquitin antibody after the reactions were terminated with SDS sample buffer. DUB activity was continuously monitored by incubation of Flag-USP40 beads with Ubiquitin-AMC (UBPBio, Inc., Aurora, CO, USA), a fluorogenic substrate. DUB activity was determined using fluorescence spectrophotometry with an excitation wavelength of 360 and an emission wavelength of 460 nm.
ATPase activity assay
Cells cotransfected with HSP90-HA and Flag-USP40 or empty vector were collected, and HA-HSP90 was immunoprecipitated with HA affinity gel (Sigma). Hsp90 ATPase activity was determined by a Transcreener ADP² Assay (BellBrook Labs, Madison WI, USA), a direct method for measuring the amount of ADP generated during a kinase reaction. Fluorescence polarization (FP) measurement with Alexa Fluor® 633 was performed on a CLARIOstar Plus plate reader.
Proteomic analysis of USP40-interacting proteins by immunoprecipitation and mass spectrometry
USP40-interacting proteins were immunoprecipitated from Flag-USP40-transfected HLMVECs using FLAG-M2 affinity gel (Sigma). For proteomic analysis, the USP40-interacting proteins were eluted from the beads under reducing conditions, separated by SDS‒PAGE, and stained with Coomassie blue. The protein bands were excised, and in-gel trypsin digestion was performed as described previously30. LC‒MS analysis was performed at the core facility of the University of Pittsburgh.
Statistical analysis
All data were subjected to statistical analysis using one-way or two-way ANOVA followed by Tukey’s post hoc test or unpaired Student’s t test to compare continuous variables or the Mantel‒Cox (log-rank) test to compare survival curves. Data are expressed as the mean ± SEM of triplicate samples from at least three independent experiments. Values of p < 0.05 were considered statistically significant.
Results
USP40 plays a protective role in EC barrier function
DUBs negatively regulate ubiquitination, thus modulating protein stability or enzyme activity. The effect of DUBs on endothelial barrier integrity has not been well studied. To identify which DUB affects endothelial barrier function, more than 15 plasmids encoding different DUBs were transfected into HLMVECs, and the cells were then treated with thrombin. Phosphorylation of MLC triggers actin stress fiber formation, resulting in EC contraction and hyperpermeability. Among the overexpressed DUBs, only USP40 attenuated thrombin-induced phosphorylation of MLC (Supplementary Fig. 1a). Transendothelial resistance (TEER) was measured using the ECIS system. Overexpression of USP40 attenuated the decreases in TEER induced by LPS (Fig. 1a) and thrombin (Supplementary Fig. 1b). Downregulation of USP40 exacerbated LPS-induced permeability (Fig. 1b) and intercellular gap formation (Fig. 1c), as well as LPS- and TNFα-induced phosphorylation of MLC (Fig. 1d, Supplementary Fig. 1c) in HLMVECs. The specificity of the USP40 siRNA was confirmed as shown in Supplementary Fig. 1f. Overexpression of USP40 attenuated stress fiber formation induced by thrombin (Fig. 1e), LPS and TNFα (Supplementary Fig. 1d). LPS treatment of HLMVECs increased the phosphorylation of MLC, while it was reduced by USP40-V5 overexpression (Fig. 1f), suggesting that USP40 negatively regulates the phosphorylation of MLC. LIMK1-mediated phosphorylation of cofilin reduces actin filament depolymerization, thus promoting stress fiber stabilization31. LPS treatment of HLMVECs induced the phosphorylation of LIMK1 and cofilin 1, while these effects were attenuated by overexpression of USP40 (Fig. 1g, h). Overexpression of USP40 also reduced thrombin-induced phosphorylation of cofilin 1 (Supplementary Fig. 1e), suggesting an inhibitory role of USP40 in stress fiber stabilization. RhoA plays a central role in the regulation of MLC and LIMK1 phosphorylation. To understand the mechanisms by which USP40 inhibits stress fiber formation, RhoA activity was measured. As shown in Fig. 1i, overexpression of USP40 attenuated LPS- and thrombin-induced activation of RhoA, indicating that USP40 protects against proinflammatory agonist-induced EC barrier disruption by preventing RhoA-mediated stress fiber formation and stabilization.

a HLMVECs were transfected with empty vector or the USP40-V5 plasmid for 48 h and were then treated with LPS (0.2 μg/ml). TEER was measured by the ECIS system. Normalized resistance was recorded at 4000 Hz. Each line indicates the mean ± SEM at the specified time points. The area under the curve (AUC) bar plots show the quantification of the AUCs for changes in barrier resistance. *: Significant difference (p < 0.001) versus the values in untreated Vector and USP40-V5 cells as well as LPS-treated USP40-V5 cells. b HLMVECs grown in transwell inserts (0.4 μm) were transfected with control siRNA (siCont) or USP40 siRNA (siUSP40) for 72 h and then treated with LPS for 6 h. Leakage of FITC-labeled dextran was measured and quantified. *: Significant difference (p < 0.001) versus the values in untreated siCont and siUSP40 cells and (p < 0.05) between LPS-treated siCont and siUSP40 cells, as determined by one-way ANOVA. c HLMVECs grown on glass-bottom dishes were transfected with control siRNA or USP40 siRNA for 72 h and then treated with LPS for 6 h. Cells were subjected to immunofluorescence staining with an anti-VE-cadherin antibody (green). Nuclei were stained with DAPI (blue). The red arrows indicate gap formation. Scale bars = 100 µm. The effect of USP40 siRNA was confirmed by immunoblotting. d HLMVECs were transfected with control siRNA or USP40 siRNA for 72 h and then treated with LPS (0.2 μg/ml) for 3 h. Immunoblot analysis was performed with the indicated antibodies. Quantification of the pMLC protein level relative to the MLC level was performed by densitometry. *: Significant difference (p < 0.001) versus the values in untreated siCont and siUSP40 cells as well as LPS-treated siUSP40 cells, as determined by one-way ANOVA with Tukey’s multiple comparison test. e HLMVECs grown on glass-bottom dishes were transfected with empty vector or USP40-V5 for 48 h and then treated with thrombin (1 U/ml) for 0.5 h. F-actin and stress fibers were fluorescently stained with phalloidin. Scale bars = 50 µm. f HLMVECs were transfected with empty vector or Flag-USP40 for 48 h and then treated with LPS (0.2 μg/ml) for 1 and 3 h. Immunoblot analysis was performed with the indicated antibodies. Quantification of the pMLC protein level relative to the MLC level was performed by densitometry (n = 3). Significant differences (p < 0.001) between the two groups were determined by two-way ANOVA. g HLMVECs were transfected with empty vector or USP40-V5 for 48 h and then treated with LPS (0.2 μg/ml) for 3 h. Immunoblot analysis was performed with the indicated antibodies. Quantification of the pLIMK1 protein level relative to the LIMK1 level was performed by densitometry. *: Significant difference (p < 0.001) versus the values in untreated Vector and USP40-V5 cells and (p < 0.01) between LPS-treated Vector and USP40-V5 cells, as determined by one-way ANOVA with Tukey’s multiple comparison test. h HLMVECs were transfected with empty vector or Flag-USP40 for 48 h and then treated with LPS (0.2 μg/ml) for 0.5–3 h. Immunoblot analysis was performed with the indicated antibodies. Quantification of the pCofilin1 protein level relative to the cofilin 1 level was performed by densitometry. Fold changes in the pCofilin 1 level were analyzed. Significant differences (p < 0.001) between the two groups were determined by two-way ANOVA. i HLMVECs were transfected with empty vector or USP40-V5 for 48 h and then treated with thrombin (1 U/ml, 0.5 h) or LPS (0.2 μg/ml, 1 h). RhoA activity was measured and quantified according to the manufacturer’s instructions.
USP40 attenuates TNFα- and LPS-induced EC inflammation
NF-ĸB-driven ICAM1 and VCAM1 expression contributes to neutrophil adhesion to ECs in response to proinflammatory agonists32. Downregulation of USP40 increased TNFα-induced ICAM1 expression (Fig. 2a), while overexpression of wild-type USP40 but not an enzyme active site mutant of USP40 (USP40C625SH317A) attenuated TNFα-induced ICAM1 expression in HLMVECs (Fig. 2b). LPS increased ICAM1 and VCAM1 expression, and these effects were attenuated in USP40-overexpressing HLMVECs (Fig. 2c, d). In addition to downregulating USP40 by siRNA transfection, we generated USP40-deficient mice (USP40−/−) (Fig. 2e). Depletion of USP40 in the lungs was confirmed by immunoblotting (Fig. 2f). Wild-type (USP40+/+) and USP40−/− mice were used to establish a murine model of intratracheal LPS-induced acute lung injury. Depletion of USP40 further elevated LPS-induced ICAM1 expression in the lungs without altering nestin expression (Fig. 2g and Supplementary Fig. 2a). ICAM1 and VCAM1 play vital roles in immune cell adherence to the endothelium33. Furthermore, we found that downregulation of USP40 in HLMVECs promoted THP-1 cell adherence to HLMVECs (Fig. 2h), while overexpression of USP40 resulted in the opposite effect (Supplementary Fig. 2b). In addition to ICAM1 and VCAM1 expression, USP40 overexpression attenuated LPS-induced IL-8, IL-6, and KC expression in different cell types: HLMVECs, mouse macrophages (RAW264.7), and mouse lung epithelial (MLE12) cells (Fig. 2i, Supplementary Fig. 2c, d). Downregulation of USP40 increased LPS-induced IL-6 secretion by RAW264.7 cells (Supplementary Fig. 2e), indicating that USP40 exhibits anti-inflammatory properties.

a HLMVECs were transfected with control siRNA or USP40 siRNA for 72 h and then treated with TNFα (10 ng/ml) for 3 h. Immunoblot analysis was performed with the indicated antibodies. Fold changes in the ICAM1 protein level relative to the β-actin level were assessed by densitometry (n = 3). b HLMVECs were transfected with empty vector, Flag-USP40, or Flag-USP40C62SH317A for 48 h and then treated with TNFα (10 ng/ml) for 1–3 h. Immunoblot analysis was performed with the indicated antibodies. Fold changes in the ICAM1 protein level relative to the β-actin level were assessed by densitometry (n = 3). *p < 0.01 for Flag-USP40 cells versus TNFα-treated Vector and Flag-USP40C62SH317A cells and #p < 0.001 for Flag-USP40C62SH317A cells versus TNFα-treated Vector and Flag-USP40 cells, as determined by two-way ANOVA with Tukey’s multiple comparison test. c HLMVECs grown on glass-bottom dishes were transfected with empty vector or the USP40-V5 plasmid and then treated with LPS (0.2 μg/ml) for 6 h. Cells were subjected to immunofluorescence staining with an anti-ICAM1 antibody (red). Scale bars = 50 µm. d HLMVECs were transfected with empty vector or the Flag-USP40 plasmid for 48 h and then treated with LPS (0.2 µg/ml) for 6 h. Immunoblot analysis was performed with the indicated antibodies. Fold changes in the VCAM1 protein level relative to the β-actin level were assessed by densitometry (n = 3). e Mouse Usp40 exon 3 (chr1: 88003332-88006310) was deleted with the CRISPR/Cas9 system. f Immunoblot analysis of lung tissues from wild-type (USP40+/+), USP40+/−, and USP40−/− mice. g USP40+/+ and USP40−/− mice were challenged by intratracheal (i.t.) instillation of LPS (4 mg/kg) for 24 h. ICAM1 and nestin levels in lung tissues were analyzed by immunoblotting. Fold changes in the ICAM1/β-actin ratio were analyzed (n = 4–8). h HLMVECs were transfected with control siRNA or USP40 siRNA for 72 h. Cells were treated with LPS (0.2 μg/ml) for 6 h, and the adhesion of fluorescently labeled THP-1 cells to HLMVECs was then measured by fluorescence microscopy and quantified. i HLMVECs were transfected with empty vector or the Flag-USP40 plasmid for 48 h and then treated with LPS (0.2 µg/ml) for 6 h. The IL-8 concentration in the medium was measured by ELISA (n = 3–6).
USP40 negatively regulates the NF-κB pathway in HLMVECs
To investigate the effect of USP40 on NF-κB activity, we examined the phosphorylation of I-κBα and the nuclear translocation and phosphorylation of NF-κB65. USP40-V5 overexpression attenuated NF-κBp65 nuclear translocation, p65 phosphorylation, and I-ĸBα phosphorylation in response to TNFα or LPS stimulation (Supplementary Fig. 3a–d). Together, these findings indicate that USP40 negatively regulates the NF-κB pathway.
USP40 activity and expression are decreased in response to LPS exposure
To investigate whether USP40 plays a role in the pathogenesis of acute inflammatory diseases, we examined the activity and expression of USP40 in response to LPS. To determine whether USP40 catalyzes the cleavage of K48- and K63-linked polyubiquitin chains, USP40 was immunoprecipitated and then incubated with K48- or K63-linked diubiquitin. As shown in Fig. 3a, USP40 catalyzes the cleavage of K63-linked but not K48-linked ubiquitin chains. This deubiquitinase activity was reduced by LPS treatment in HLMVECs (Fig. 3b). Furthermore, the USP40 abundance was decreased in a time-dependent manner in response to LPS exposure (Fig. 3c). USP40 was highly expressed in vessels in lungs from control mice, but its expression was decreased in LPS-challenged lungs (Fig. 3d). USP40 activity and expression are reduced in response to LPS exposure in cells and murine lungs, suggesting that these reductions contribute to inflammatory responses by increasing EC permeability and inflammation.

a HLMVECs were transfected with empty vector or the Flag-USP40 plasmid for 48 h, and Flag-USP40 was immunoprecipitated with an anti-Flag tag antibody, followed by incubation with di-K48-linked ubiquitin (di-K48Ubi) or di-K63-linked ubiquitin (di-K63Ubi). Immunoblot analysis was performed to detect USP40 DUB activity. b HLMVECs were transfected with empty vector or the Flag-USP40 plasmid for 48 h, and Flag-USP40 was immunoprecipitated with an anti-Flag tag antibody, followed by incubation with AMC-conjugated ubiquitin (Ub-AMC). USP40 DUB activity was measured by Ub-AMC analysis (n = 3). c HLMVECs were treated with LPS for 3–24 h. Immunoblot analysis was performed with the indicated antibodies. Fold changes in the USP40/β-actin ratio were analyzed (n = 3). ICAM1 expression was used as a positive control for LPS treatment. d USP40 immunohistochemical staining in lung tissues from mice with i.t. instillation of PBS or LPS. Scale bars = 50 µm.
USP40 protects against LPS-induced acute lung injury
To further investigate whether the reductions in USP40 expression and activity contribute to the pathogenesis of acute lung injury, mice with EC-specific USP40 deficiency (USP40EC-KO) were generated by crossbreeding USP40floxp/floxp mice with Tek-Cre1Ywa/J or Cdh5-Cre7Mlia/J transgenic mice (Supplementary Figs. 4, 5). Intratracheal administration of LPS increased neutrophil influx into alveolar spaces, and this effect was enhanced in USP40−/− and USP40EC-KO mice (Fig. 4a). Furthermore, lung endothelial cell expression of ICAM1 (CD54) and neutrophil infiltration in LPS-challenged mice were assessed by flow cytometric analysis (Fig. 4b). Depletion of USP40 (USP40−/− and USP40EC-KO) significantly increased ICAM1 expression in endothelial cells and neutrophil infiltration in the lungs of mice treated with LPS (i.t. instillation, 2 mg/kg, 24 h). Increased ICAM-1 expression on inflamed endothelial cells enhances leukocyte adhesion and promotes neutrophil transendothelial migration12. LPS challenge increased protein levels in bronchoalveolar lavage (BAL) fluid and Evans blue extravasation in lung tissues, indicating that LPS induces microvascular leakage in the lungs. These effects were enhanced in USP40−/− and USP40EC-KO mice (Fig. 4c, d). Depletion of USP40 promoted the LPS-induced increases in the levels of cytokines, including IL-6, KC, and TNFα, in BAL fluid (Fig. 4e–g). Histological staining indicated that both types of USP40 depletion in mice worsened lung injury (Fig. 4h). Considering that Tek-Cre mice exhibit substantial hematopoietic deletion, USP40cdh5-ECKO mice were generated, and the role of endothelial USP40 in LPS-induced acute lung injury was confirmed (Supplementary Fig. 6). Furthermore, we evaluated the survival rate of USP40−/− mice compared with that of WT mice after CLP. As shown in Fig. 4i, USP40−/− mice exhibited a significantly lower survival rate. To investigate whether overexpression of USP40 mitigates LPS-induced acute lung injury, a lentiviral vector containing the USP40 gene was administered to mice. Both lung epithelial and endothelial cells exhibited ectopic expression of USP40, but infiltrated neutrophils did not, as shown in Supplementary Fig. 7. Introduction of lenti-USP40 significantly reduced the lung wet/dry ratio (Fig. 5a), BAL protein levels (Fig. 5b), Evans blue extravasation (Fig. 5c), BAL cytokine levels (Fig. 5d–f), the MPO level (Fig. 5g), and neutrophil influx (Fig. 5h) in mouse lungs after i.t. LPS administration. Taken together, these data demonstrate that depletion of USP40 worsens lung injury, while induction of USP40 expression by lentiviral delivery ameliorates lung injury.

USP40+/+, USP40−/-, and USP40EC-KO mice were challenged by i.t. instillation of LPS (2 mg/kg) for 24 h. BAL cells were analyzed by cytological staining (a), and flow cytometric analysis of cells from LPS-exposed lung tissue was performed (b). A gating strategy was used to identify populations expressing the endothelial cell surface marker CD31 (CD45−/CD326−/CD31+) and the neutrophil marker Ly6G (CD45+/Ly6G+). Representative plots showing the percentages of CD54+ endothelial cells and Ly6G+ neutrophils in the lungs of mice treated with PBS or LPS (i.t. instillation, 2 mg/kg, 24 h). Differences among the three groups were compared using one-way ANOVA with Tukey’s multiple comparison test. c BAL protein levels were measured. d USP40+/+, USP40−/−, and USP40EC-KO mice were challenged by i.t. instillation of LPS (2 mg/kg), 24 h. Evans blue (EB) was injected into the tail vein 30 min before the mice were sacrificed. Evans blue extravasation in lung tissues was measured. e–g mIL-6, KC and TNFα concentrations in BAL fluid were measured by ELISA. h Lung tissues were subjected to H&E staining. Scale bars = 50 µm. i Mortality curve for CLP mice in the USP40−/− (n = 16) and wild-type control (Wt, n = 15) groups.

C57/BL6J mice injected intravenously with lentiviral vector (LentiCont) and lentiviral USP40 (LentiUSP40) were challenged by i.t. instillation of LPS (2 mg/kg) for 24 h. a. The wet/dry ratio of lung tissues was measured. b BAL protein levels were measured. c Evans blue was injected into the tail vein 30 min before the mice were sacrificed. Lung tissues were imaged, and Evans blue extravasation in the lung tissues was measured. d–f mIL-6, KC and TNFα concentrations in BAL fluid were measured by ELISA. g The MPO content in lung tissues was analyzed. h Lung tissues and BAL cells were subjected to H&E staining and cytological staining. Scale bars = 50 µm.
USP40 deubiquitinates and inactivates HSP90β
To further investigate the molecular mechanisms by which USP40 preserves endothelial biological functions, we used an unbiased strategy to identify USP40-interacting proteins. Coimmunoprecipitation (Co-IP) followed by proteomic analysis showed that USP40 interacts with multiple functional proteins (Fig. 6a, Table 1). Among the potential USP40 substrates, HSP90β plays a central role in the regulation of EC dysfunction through inhibition of the NF-κB pathway and Rho-mediated EC stress fiber formation (Fig. 6b). Consistent with previous studies, inhibition of HSP90β attenuated LPS-induced ICAM1 expression on HLMVECs (Fig. 6c). The co-IP and coimmunofluorescence staining experiments confirmed that USP40 interacted with HSP90β (Fig. 6d, e), while this interaction was diminished after LPS or TNFα treatment (Fig. 6d, i, j). Ubiquitination of HSP90β has been reported23,24. We found that USP40 reduced the polyubiquitination and increased the lysine acetylation of HSP90β (Fig. 6f). Consistent with data showing that USP40 catalyzes the cleavage of K63-linked ubiquitin chains (Fig. 3a), we found that USP40 catalyzed the cleavage of K63-linked polyubiquitin chains on HSP90β (Fig. 6g). Furthermore, USP40 reduced HSP90β ATPase activity (Fig. 6h). It has been shown that lysine acetylation of HSP90β negatively regulates HSP90β activity19. USP40 overexpression increased lysine acetylation of HSP90β (Fig. 6i, j), suggesting that USP40 deubiquitinates HSP90β, resulting in an increase in lysine acetylation of HSP90β, thereby leading to inactivation of HSP90β and preservation of EC integrity. It has been reported that USP40 knockdown disrupts the permeability of glomerular endothelial cells in zebrafish by reducing the nestin level25. We also investigated whether nestin plays a role in USP40-mediated biological functions of lung ECs and found that nestin expression was not reduced in HLMVECs with USP40 downregulation or the lungs of USP40−/− mice (Fig. 2g and Supplementary Fig. 2a). Thus, USP40 preserves the biological functions of lung endothelial cells by targeting HSP90β but not nestin.

a Proteomic profiling of USP40interacting protein complexes in HLMVECs. b Schematic diagram showing the role of HSP90 in EC inflammation and hyperpermeability. c HLMVECs were treated with 17-AAG prior to stimulation with LPS (0.2 μg/ml) for 6 h. ICAM1 expression was analyzed by immunoblotting. Quantification of ICAM1 expression relative to β-actin expression was performed. d HLMVECs were treated with LPS (0.2 μg/ml) for 30 min, and cell lysates were subjected to IP with an anti-USP40 antibody or an anti-HSP90β antibody, followed by immunoblot analysis of USP40 or HSP90β. e HLMVECs were transfected with the USP40-V5 plasmid for 48 h. The cells were subjected to immunofluorescence staining with anti-V5 (green) and anti-HSP90β (red) antibodies. Scale bars = 50 µm. f HLMVECs were transfected with empty vector or the Flag-USP40 plasmid for 48 h. Denatured proteins in cell lysates were subjected to IP with an anti-HSP90β antibody followed by immunoblotting with anti-ubiquitin or anti-acetylated lysine (AcK) antibodies. g HLMVECs were transfected with empty vector or the USP40-V5 plasmid for 48 h, and denatured proteins in cell lysates were subjected to IP with an anti-HSP90β antibody followed by immunoblotting with an anti-K63-linked ubiquitin antibody. h HLMVECs were cotransfected with the HSP90β-HA plasmid and empty vector or the Flag-USP40 plasmid for 48 h. HSP90β-HA was immunoprecipitated, and its ATPase activity was measured (n = 4). i HLMVECs were transfected with empty vector or the Flag-USP40 plasmid for 48 h, followed by LPS treatment for 1 h. Cell lysates were subjected to IP with an antibody against HSP90β followed by immunoblotting with the indicated antibodies. j HLMVECs were treated with TNFα (10 ng/ml) for 30 min. Cell lysates were subjected to IP with an antibody against HSP90β followed by immunoblotting with the indicated antibodies.
USP40 docking site and acetylation site in HSP90β
To identify a specific HSP90 motif interacting with USP40, HSP90 C-terminal and N-terminal deletion mutants were generated by mutagenesis, and the corresponding plasmids were constructed (Fig. 7a). Only the N-terminal fragment (1-325 aa) of HSP90β was unable to bind to USP40, suggesting that the USP40 binding site is located between residues 325 and 425 of HSP90β (Fig. 7b). Independent of HSP90 activity, wild-type HSP90 and the dominant-negative mutant (HSP90βD88N) had similar binding affinities for USP40 (Fig. 7c). To determine the mechanism of USP40-mediated deubiquitination of Hsp90β, several constructs with mutations in the region between aa 300 and 425 were generated by site-specific mutagenesis. HSP90β∆402-406 was found to demonstrate enhanced binding to USP40 (Fig. 7d), suggesting that the region between residues 402 and 406 is most likely also involved in the interaction but inhibits binding. Furthermore, expression of HSP90∆402-406 significantly reduced the monoubiquitination of HSP90β (Fig. 7e), possibly due to the high binding affinity of this mutant for USP40. These data indicate that targeting the 402KVIRK406 sequence with a small molecule may attenuate proinflammatory responses and pulmonary EC barrier disruption by enhancing the association of Hsp90β with USP40. Furthermore, we confirmed that K284 is an acetylation site in Hsp90β (Fig. 7f), consistent with the previously reported K294 in Hsp90α34.

a Schematic diagram of HSP90β deletion mutants. The structure of HSP90β consists of an N-terminal ATPase domain, a ribosomal protein S5 domain 2, and a C-terminal domain. The HSP90β C-terminal and N-terminal deletion mutants were generated by site-specific mutagenesis, and the corresponding plasmids were constructed. b HEK293 cells were cotransfected with USP40-V5 and HA-tagged N- or C-terminal deletion mutants of HSP90β for 48 h. The USP40 binding site in HSP90β was identified by co-IP of USP40-V5 using an anti-V5 antibody followed by immunoblotting with an antibody against the HA tag. c HEK293 cells were cotransfected with Flag-USP40 and vector or HA-tagged wild-type HSP90β or the dominant-negative (D88N) HSP90β mutant for 48 h. Cell lysates were subjected to IP with an antibody against HA followed by immunoblotting with the indicated antibodies. d HEK293 cells were cotransfected with Flag-USP40 and vector or HA-tagged wild type (wt) HSP90β or HSP90β mutants (S307A, S391A, ∆350–354, ∆402–406) for 48 h. Cell lysates were subjected to IP with an antibody against the HA tag followed by immunoblotting with the indicated antibodies. e HEK293 cells were transfected with HA-tagged wild type (wt) Hsp90β or Hsp90β mutants (S307A, S391A, ∆350–354, ∆402–406) for 48 h. The ubiquitination of HSP90 was examined by modified IP (under denaturing conditions) with an anti-HA antibody, followed by immunoblotting with antibodies against ubiquitin (Ubi) and HA. f HEK293 cells were transfected with HA-tagged wild-type (wt) HSP90β or HSP90β mutants (K275R, K284R, and K354R) for 48 h. Cell lysates were subjected to IP with an antibody against acetylated lysine (AcK) followed by immunoblotting with the indicated antibodies. g USP40 preserves EC integrity by deubiquitinating and inactivating HSP90β, resulting in reductions in inflammatory stimulus-induced EC inflammation and hyperpermeability. Schematic diagram showing HSP90β-mediated NF-κB pathway activation and cytoskeletal rearrangement in ECs.
Discussion
Ubiquitination is involved in inflammatory responses and cell‒cell junctions22. Our previous studies showed that E3 ligases such as FBXL19, Nedd4L, and FBXL2 exhibit anti-inflammatory properties35,36,37,38, while DUBs, such as USP11 and USP14, play proinflammatory roles38,39. In this study, we focused on the role of a DUB in EC inflammation and barrier dysfunction. We revealed that USP40 preserves EC function by deubiquitinating and inactivating HSP90β. USP40, a newly recognized DUB, was reported to regulate glomerular permeability in zebrafish by mediating nestin expression25. A study from the same group further demonstrated that depletion of Usp40 in zebrafish disrupted glomerulogenesis, but surprisingly, Usp40KO mice showed a normal kidney phenotype. Additionally, this group showed that in contrast to the reduction in nestin expression in Usp40-depleted zebrafish, nestin was upregulated in the glomeruli of Usp40KO mice26. We confirmed that the nestin level was not changed in USP40-deficient HLMVECs or the lungs of USP40KO mice (Fig. 2g and Supplementary Fig. 2a), indicating that the mechanism by which USP40 preserves lung EC barrier integrity is not mediated through nestin. Here, we reveal a new mechanism of regulation of HSP90β deubiquitination and inactivation by USP40.
HSP90 is a critical modulator of cell signaling and has been shown to play a detrimental role in EC integrity16. Inhibition of HSP90 protects lung EC integrity by attenuating NF-κB pathway activity and cytoskeletal rearrangement17,18. USP40 deubiquitinates HSP90β, resulting in the inactivation of HSP90β in ECs, suggesting that USP40 protects EC integrity by blocking HSP90β-mediated EC dysfunction. This is the first study to identify a DUB that regulates both the inflammation and barrier function of ECs. USP40 has been shown to cleave K48-linked polyubiquitin chains on c-FLIPL in lung cancer cells40. In ECs, USP40 does not catalyze the cleavage of K48-linked polyubiquitin chains. However, USP40 catalyzes the cleavage of K63-linked polyubiquitin chains on HSP90β. Unlike other reported substrates, USP40 regulates HSP90β deubiquitination without altering HSP90β stability. In general, K63-linked polyubiquitination regulates protein trafficking, protein‒protein interactions, and enzyme activity, while K48-linked polyubiquitination promotes proteasomal protein degradation21,22. HSP90β can undergo K48- and K63-linked polyubiquitination mediated by a ubiquitin E3 ligase, CHIP23. Hectd1, another ubiquitin E3 ligase, mediates K63-linked polyubiquitination, leading to the cytosolic localization of HSP90 and a reduction in HSP90 secretion24. Hyperacetylation of HSP90β reduces its chaperone activity19. In this study, we reveal that USP40 increases the acetylation and decreases the activation of HSP90β. Consistent with a previous report, lysine 284 is an acetylation site in Hsp90β34. Of particular interest is determining whether deubiquitination by USP40 modulates the interaction between HSP90 and its lysine acetyltransferase or deacetylase. Taken together with findings showing that USP40 deubiquitinates HSP90β, these data suggest that USP40 inactivates HSP90β by modulating its ubiquitination and acetylation.
The detrimental effect of HSP90 activity in ECs has been well reported17,41. Inhibition of HSP90 suppresses NF-κB signaling and prevents the nuclear translocation of NF-κB, thereby diminishing inflammation17,42. In ECs, HSP90 inhibitors prevent LPS-induced endothelial barrier dysfunction by attenuating RhoA-mediated phosphorylation of MLC16. HSP90 inhibition protects the brain microvascular endothelium against oxidative stress43. A large amount of evidence indicates that inhibition of HSP90 is a potential therapeutic strategy for EC dysfunction-related inflammatory diseases16,18,41,43. HSP90 has been shown to associate with DUBs, such as USP19 and USP5044,45. HSP90 regulates the DUB activity of USP19 and promotes USP50-mediated Wee1 stability45; however, a DUB for HSP90 has not been reported. This is the first report to reveal that USP40 deubiquitinates and inactivates HSP90. Consistent with findings indicating that inhibition of HSP90 diminishes EC inflammation and barrier dysfunction, USP40 protects lung ECs against inflammatory stimulus-induced inflammatory responses and barrier disruption in vitro and in vivo. Nestin is a type VI intermediate filament protein and is a developmentally regulated protein46. It seems that nestin is not involved in EC inflammation and barrier function. Nestin is considered to play a role in the formation of the cytoskeleton in newly formed ECs46; thus, USP40 may play a role in vascular regeneration during lung repair and remodeling.
We showed that the USP40 level was reduced in response to endotoxin exposure. USP40 (USP40−/− and USP40EC-KO) deficiency significantly exacerbated lung injury. Consistent with a previous study40, no apparent abnormal phenotypes were observed in USP40KO or USP40 EC-KO mice. We believe that the pathological phenotypes resulting from USP40 deletion in this study are not secondary to preexisting vascular and nonvascular changes during embryogenesis and adulthood. We employed mice with endothelium-specific deletion of USP40 to determine the role of endothelial USP40. Although Tek-Cre mice have been reported to show at least partial Cre recombinase activity in cells of the hematopoietic lineage, via the TgTek-cre1Ywa/J allele, a small number of circulating cells have been reported to be Cre positive in adult mice47. Thus, a portion of immune cells may also lack USP40 expression in the Tek-Cre system. This might be due to Cre leakiness in the Tek-Cre system, as there were no differences in BAL cell counts, vascular leakage, or BAL cytokine levels between USP40KO (global knockout) mice and USP40EC-KO mice with USP40 deficiency driven by Tek-Cre (Fig. 4). USP40cdh5-ECKO mice were generated by breeding USP40floxp/floxp mice with Cdh5-cre7Mlia/J transgenic mice. As shown in Supplementary Fig. 5c and 5d, USP40 excision events in blood cells were not detected in the Cdh5-Cre system. The increased lung injury in mice with EC-specific USP40 deficiency (USP40cdh5-ECKO) confirmed the protective effects of USP40 in ECs (Supplementary Fig. 6).
Taken together, this study shows that USP40 deubiquitinates HSP90β, thereby inhibiting its activity by increasing its acetylation (Fig. 7g). We reveal a new molecular mechanism for the regulation of posttranslational modification and activation of HSP90β. HSP90 inhibitors have emerged as potential anti-inflammatory drugs. Understanding the molecular regulation of HSP90β activity by USP40 is expected to lead to the development of new drugs to target HSP90. USP40 activity and protein expression are suppressed under inflammatory conditions, suggesting that these reductions contribute to the pathogenesis of acute lung injury. The molecular regulation of USP40 expression and activity is anticipated to be a new focus for understanding the processes of inflammatory diseases and developing new therapeutic strategies for acute lung injury and sepsis.
Data availability
All data and study materials that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Rao, R. M., Yang, L., Garcia-Cardena, G. & Luscinskas, F. W. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ. Res. 101, 234–247 (2007).
Gill, S. E., Taneja, R., Rohan, M., Wang, L. & Mehta, S. Pulmonary microvascular albumin leak is associated with endothelial cell death in murine sepsis-induced lung injury in vivo. PloS One. 9, e88501 (2014).
Goldenberg, N. M., Steinberg, B. E., Slutsky, A. S. & Lee, W. L. Broken barriers: a new take on sepsis pathogenesis. Sci. Transl. Med. 3, 88ps25 (2011).
Mackow, E. R., Gorbunova, E. E. & Gavrilovskaya, I. N. Endothelial cell dysfunction in viral hemorrhage and edema. Front. Microbiol. 5, 733 (2014).
Hiyoshi, M. et al. Influenza A virus infection of vascular endothelial cells induces GSK-3beta-mediated beta-catenin degradation in adherens junctions, with a resultant increase in membrane permeability. Arch. Virol. 160, 225–234 (2015).
Sukriti, S., Tauseef, M., Yazbeck, P. & Mehta, D. Mechanisms regulating endothelial permeability. Pulmonary Circ. 4, 535–551 (2014).
Birukova, A. A. et al. Endothelial permeability is controlled by spatially defined cytoskeletal mechanics: atomic force microscopy force mapping of pulmonary endothelial monolayer. Nanomed.: Nanotechnol. Biol. Med. 5, 30–41 (2009).
Schnoor, M. et al. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell Mol. Life Sci. 74, 1985–1997 (2017).
Lin, T. et al. Rho-ROCK-LIMK-cofilin pathway regulates shear stress activation of sterol regulatory element binding proteins. Circ. Res. 92, 1296–1304 (2003).
Gorovoy, M. et al. LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. J. Biol. Chem. 280, 26533–26542 (2005).
Owen-Woods, C. et al. Local microvascular leakage promotes trafficking of activated neutrophils to remote organs. J. Clin. Invest. 130, 2301–2318 (2020).
Yang, L. et al. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood 106, 584–592 (2005).
Kilpatrick, L. E. & Kiani, M. F. Experimental approaches to evaluate leukocyte-endothelial cell interactions in sepsis and inflammation. Shock 53, 585–595 (2020).
Ledebur, H. C. & Parks, T. P. Transcriptional regulation of the intercellular adhesion molecule-1 gene by inflammatory cytokines in human endothelial cells. Essential roles of a variant NF-kappa B site and p65 homodimers. J. Biol. Chem. 270, 933–943 (1995).
Kim, I. et al. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J. Biol. Chem. 276, 7614–7620 (2001).
Joshi, A. D. et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am. J Respir. Cell Mol. Biol. 50, 170–179 (2014).
Thangjam, G. S. et al. Hsp90 inhibition suppresses NF-kappaB transcriptional activation via Sirt-2 in human lung microvascular endothelial cells. Am. J Physiol. Lung Cell. Mol. Physiol. 310, L964–L974 (2016).
Chatterjee, A. et al. Heat shock protein 90 inhibitors prolong survival, attenuate inflammation, and reduce lung injury in murine sepsis. Am. J Respir. Crit. Care Med. 176, 667–675 (2007).
Scroggins, B. T. et al. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25, 151–159 (2007).
Mollapour, M. & Neckers, L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 1823, 648–655 (2012).
Schnell, J. D. & Hicke, L. Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J Biol. Chem. 278, 35857–35860 (2003).
Mukhopadhyay, D. & Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315, 201–205 (2007).
Kundrat, L. & Regan, L. Identification of residues on Hsp70 and Hsp90 ubiquitinated by the cochaperone CHIP. J. Mol. Biol. 395, 587–594 (2010).
Sarkar, A. A. & Zohn, I. E. Hectd1 regulates intracellular localization and secretion of Hsp90 to control cellular behavior of the cranial mesenchyme. J. Cell Biol. 196, 789–800 (2012).
Takagi, H. et al. USP40 gene knockdown disrupts glomerular permeability in zebrafish. Am. J. Phys. Renal Phys. 312, F702–F715 (2017).
Takahashi, S. et al. USP40 deubiquitinates HINT1 and stabilizes p53 in podocyte damage. Biochem. Biophys. Res. Commun. 614, 198–206 (2022).
Pelletier, S., Gingras, S. & Green, D. R. Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity 42, 18–27 (2015).
Roy, N. et al. Dysregulation of lipid and glucose homeostasis in hepatocyte-specific SLC25A34 knockout mice. Am. J. Pathol. 192, 1259–1281 (2022).
Li, L., Wei, J., Mallampalli, R. K., Zhao, Y. & Zhao, J. TRIM21 mitigates human lung microvascular endothelial cells’ inflammatory responses to LPS. Am. J. Respir. Cell Mol. Biol. 61, 776–785 (2019).
Free, R. B., Hazelwood, L. A. & Sibley, D. R. Identifying novel protein-protein interactions using co-immunoprecipitation and mass spectroscopy. Curr. Protoc. Neurosci. Chapter 5, Unit 5 28 (2009).
Yang, N. et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 393, 809–812 (1998).
Chen, C. C., Rosenbloom, C. L., Anderson, D. C. & Manning, A. M. Selective inhibition of E-selectin, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 expression by inhibitors of I kappa B-alpha phosphorylation. J. Immunol. 155, 3538–3545 (1995).
Muller, W. A. Mechanisms of transendothelial migration of leukocytes. Circ. Res. 105, 223–230 (2009).
Kekatpure, V. D., Dannenberg, A. J. & Subbaramaiah, K. HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling. J. Biol. Chem. 284, 7436–7445 (2009).
Zhao, J. et al. F-box protein FBXL19-mediated ubiquitination and degradation of the receptor for IL-33 limits pulmonary inflammation. Nat. Immunol. 13, 651–658 (2012).
Wei, J. et al. Regulation of the ubiquitylation and deubiquitylation of CREB-binding protein modulates histone acetylation and lung inflammation. Sci. Signal 10, eaak9660 (2017).
Chen, B. B. et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat. Immunol. 14, 470–479 (2013).
Zhao, J. et al. Destabilization of lysophosphatidic acid receptor 1 reduces cytokine release and protects against lung injury. EBioMedicine 10, 195–203 (2016).
Mialki, R. K., Zhao, J., Wei, J., Mallampalli, D. F. & Zhao, Y. Overexpression of USP14 protease reduces I-kappaB protein levels and increases cytokine release in lung epithelial cells. J. Biol. Chem. 288, 15437–15441 (2013).
An, W. et al. Glucocorticoid modulatory element-binding protein 1 (GMEB1) interacts with the de-ubiquitinase USP40 to stabilize CFLARL and inhibit apoptosis in human non-small cell lung cancer cells. J. Exp. Clin. Cancer Res. 38, 181 (2019).
Barabutis, N., Uddin, M. A. & Catravas, J. D. Hsp90 inhibitors suppress P53 phosphorylation in LPS - induced endothelial inflammation. Cytokine 113, 427–432 (2019).
Thangjam, G. S. et al. Novel mechanism of attenuation of LPS-induced NF-kappaB activation by the heat shock protein 90 inhibitor, 17-N-allylamino-17-demethoxygeldanamycin, in human lung microvascular endothelial cells. Am. J. Respir. Cell Mol. Biol. 50, 942–952 (2014).
Uddin, M. A. et al. Hsp90 inhibition protects the brain microvascular endothelium against oxidative stress. Brain Disord. 1, 100001 (2021).
Lee, J. G., Kim, W., Gygi, S. & Ye, Y. Characterization of the deubiquitinating activity of USP19 and its role in endoplasmic reticulum-associated degradation. J. Biol. Chem. 289, 3510–3517 (2014).
Aressy, B. et al. A screen for deubiquitinating enzymes involved in the G(2)/M checkpoint identifies USP50 as a regulator of HSP90-dependent Wee1 stability. Cell Cycle 9, 3815–3822 (2010).
Wagner, N., Wagner, K. D., Scholz, H., Kirschner, K. M. & Schedl, A. Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms’ tumor suppressor Wt1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R779–R787 (2006).
Kisanuki, Y. Y. et al. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230–242 (2001).
Acknowledgements
This research was supported by the National Institutes of Health, Nos. R01HL131665, R01HL136294, and R01HL157164 to Y.Z.; R01GM115389 and R01HL151513 to J.Z.; and P01 HL114453, R01 HL081784, and R01HL097376 to R.K.M. All the authors have read the journal’s authorship agreement, and the manuscript has been reviewed by and approved by all named authors.
Author information
Authors and Affiliations
Contributions
J.M., L.L., N.S., A.M.J., S.J.T., P.S., J.Z., and Y.Z. performed the experiments and data analysis. J.Z. supervised the project. J.Z. and Y.Z. finalized the manuscript. R.K.M. reviewed the data, provided suggestions on the studies, and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Miao, J., Li, L., Shaheen, N. et al. The deubiquitinase USP40 preserves endothelial integrity by targeting the heat shock protein HSP90β. Exp Mol Med 56, 395–407 (2024). https://doi.org/10.1038/s12276-024-01160-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-024-01160-y
- Springer Nature Limited