
Abstract
It is known that human papillomavirus (HPV) infection can cause squamous cell neoplasms at several sites, such as cervix uteri carcinoma and oral squamous carcinoma. There is little information on the expression of HPV and its predictive markers in tumours of the major and minor salivary glands of the head and neck. We therefore assessed oral salivary gland neoplasms to identify associations between HPV and infection-related epidermal growth factor receptor (EGFR), cyclin-dependent kinase inhibitor 2A (CDKN2A/p16) and tumour protein p53 (TP53). Formalin-fixed, paraffin-embedded tissue samples from oral salivary gland carcinomas (n=51) and benign tumours (n=26) were analysed by polymerase chain reaction (PCR) analysis for several HPV species, including high-risk types 16 and 18. Evaluation of EGFR, CDKN2A, TP53 and cytomegalovirus (CMV) was performed by immunohistochemistry. Epstein–Barr virus (EBV) was evaluated by EBV-encoded RNA in situ hybridisation. We demonstrated that salivary gland tumours are not associated with HPV infection. The expression of EGFR, CDKN2A and TP53 may be associated with tumour pathology but is not induced by HPV. CMV and EBV were not detectable. In contrast to oral squamous cell carcinomas, HPV, CMV and EBV infections are not associated with malignant or benign neoplastic lesions of the salivary glands.
Similar content being viewed by others

Introduction
Several viruses are associated with neoplastic diseases of the head and neck, particularly human papillomavirus (HPV), human herpesvirus-4/Epstein–Barr virus (EBV) and human herpesvirus-5/cytomegalovirus (CMV). HPV is frequently found in squamous cell carcinomas,1,2,3 while EBV is detectable in nasopharyngeal carcinomas, lymphomas and post-transplant lymphoproliferative disease.4,5 CMV, which is associated with sialadenitis, has recently been shown to be detectable in mucoepidermoid carcinomas.6,7
HPV consists of several subtypes, which are classified according to their carcinogenic potential, including low-risk HPV (e.g., types 6, 11, 40, 42, 43, 44, 54, 61, 70, 72) and high-risk HPV (types 16, 18, 31, 26, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 73, 82).1,2 After cellular infection, HPV manipulates the intracellular signalling associated with cell cycle regulation and differentiation via several viral proteins (e.g., E5, E6 and E7). These proteins interact with host cell factors such as cyclin-dependent kinase inhibitor 2A (CDKN2A/p16), tumour protein p53 (TP53/p53) and epidermal growth factor receptor (EGFR). Stimulation of EGFR by extracellular cytokines or by intracellular HPV-derived protein E5 induces activation of intracellular signalling cascades that lead to proliferation. Cyclin-dependent kinase 4 (CDK4) is a component of the protein kinase complex that regulates the G1–S cell cycle phase. CDK4 inactivates retinoblastoma 1 (RB1) by phosphorylation, preventing the active (hypophosphorylated) RB1 protein from binding to E2F transcription factor 1 (E2F1). CDKN2A inhibits CDK4 and thereby regulates RB1–E2F1 binding. HPV protein E7 interferes with this regulatory cascade by binding to RB1 (thus, E2F1 can no longer bind to RB1). In addition, E7 binds to the E2F1–DNA complex and induces RB1-independent gene transcription. The binding of E7 to RB1 results in a compensatory increase in CDKN2A expression, but this accumulation of CDK4-inhibiting protein is ineffective due to the presence of E7. CDKN2A also protects transcription factor TP53 from degradation, but HPV protein E6 counteracts this function by binding to TP53 and inducing increased degradation of the protein. Through these mechanisms, viral proteins mediate uncontrolled activation of cell division and inhibition of apoptosis, which can ultimately result in neoplastic transformation of the infected cell population.1,2
It has been reported that HPV can be detected in benign salivary gland tumours and adenocarcinomas.8,9,10,11,12 CDKN2A expression has been observed in salivary gland tumours without detection of HPV.13,14 We hypothesised that screening a large number of HPV types by a sensitive and specific polymerase chain reaction (PCR) assay may reveal an association between viral infection and CDKN2A, TP53 and EGFR expression in salivary gland tumours.
Materials and methods
Patient cohort and tumour samples
We selected tissue samples from salivary gland neoplasms (n=77) and non-neoplastic salivary gland tissues (n=65). Non-neoplastic controls consisted of 58 normal glands in the tumour margins and 7 unrelated cases without adenocarcinomas. Tumour types were as follows: adenoid cystic carcinomas (n=20), adenocarcinomas not otherwise specified (NOS) (n=17), mucoepidermoid carcinomas (n=11), carcinomas ex pleomorphic adenomas (n=3), pleomorphic adenomas (n=4) and Warthin’s tumours (n=22). Surgical resection and subsequent routine pathological examination were performed between 2000 and 2006.
The retrospective analysis of archived tissue was approved by our local ethics committee.
Analysis of HPV infection in salivary gland tumours
HPV was analysed in all tumours by PCR analysis (Chipron, Berlin, Germany) as previously described.15,16 Briefly, DNA was extracted from tumour blocks (>80% tumour cells) in formalin fixed-paraffin embedded (FFPE) tissue sections with the DNEasy Blood & Tissue Kit (Qiagen GmbH, Hilden, Germany). A total of 50 ng of DNA (determined with a Micro-Volume UV-Vis Nanodrop 2000 spectrophotometer; Thermo Scientific, Wilmington, DE, USA) was used for multiplex PCR for 32 high- and low-risk HPV samples (types 6, 11, 16, 18, 31, 33, 35, 39, 42, 44, 45, 51–54, 56, 58, 59, 61, 62, 66–68, 70, 72, 73, 81–84, 90, 91). The sensitivity of the PCR for each of the above-mentioned HPV types is reported by the manufacturer to be 20 virus copies per µg of extracted DNA (Chipron, Berlin, Germany). The controls were FFPE tissue-derived DNA from HPV-positive cervix samples and DNA-free water.
For the evaluation of EBV and CMV, two tissue microarrays (each with 60 samples) were used, which contained all the carcinoma, pleomorphic adenoma and control samples. Deparaffinised and rehydrated FFPE tissue sections (approximately 2–3 µm) were processed in an automated staining system (Benchmark ULTRA; Ventana Medical Systems, Tucson, AZ, USA). Infection of tumour cells by EBV and/or CMV was tested by in situ hybridisation (EBV-encoded RNA; Ventana Medical Systems, Tucson, AZ, USA) and immunohistochemistry (CMV; Thermo Fisher Scientific, Schwerte, Germany), respectively. Controls were FFPE samples from an EBV+lymphoma patient and a CMV+colitis patient.
Protein expression analysis of HPV-associated human genes
Carcinoma and pleomorphic adenoma samples underwent cell cycle and receptor protein expression analysis using a tissue microarray. Sections (approximately 2–3 µm) were used for automated immunohistochemistry (Benchmark ULTRA, Ventana Medical Systems, Tucson, AZ, USA) of CDKN2A (Santa Cruz Biotechnology, Santa Cruz, CA, USA), TP53 (Dako, Glostrup, Denmark) and EGFR (Zytomed, Berlin, Germany).
Immunohistochemistry was scored semi-quantitatively as described previously17: 0 (no apparent reaction), 1+ (positivity in <30% of tumour cells), 2+ (positivity in ≥30% but <60%) and 3+ (positivity in ≥60%). Immunostaining intensity was scored as 0 (absent), 1+ (weak), 2+ (intermediate) or 3+ (strong).
Statistical analysis
Overall survival was obtained from the medical records. Survival analysis was performed with Prism 5.0 (GraphPad Software, San Diego, CA, USA) by applying the log-rank (Mantel–Cox) test. P values<0.05 were considered statistically significant.
Results
Clinical characteristics of patients with salivary gland tumours
The median patient age was 63 years (24 females/53 males). All patients were of European descent. Tumours were located in the major salivary glands of the head and neck (parotid n=45, submandibular n=3 and sublingual gland n=1) or in minor salivary glands of the oral cavity (n=28). Carcinoma grades were G1 in 7/51 (14%), G2 in 30/51 (59%) and G3 in 14/51 (27%) of the cases. The median follow-up period in carcinoma cases was 111 months, and the tumour-free survival rate was 71%. In this small patient cohort, patients with mucoepidermoid carcinoma had a significantly better survival rate than patients with other types of salivary gland tumours (P<0.05). The characteristics of the study group are summarised in Table 1.
Salivary gland tumours are not associated with HPV infection
HPV was tested by PCR in carcinomas and benign salivary gland tumours but could not be detected in any of these samples. None of the tumour samples under investigation showed infection with EBV (no EBV-encoded RNA hybridisation signals) or CMV (no immunostaining).
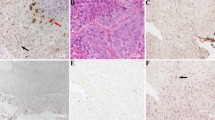
A subfraction of tumour samples exhibited expression of cell cycle and receptor proteins (Figure 1). The most frequent tumour types with positive expression were adenoid cystic carcinomas for CDKN2A/p16 and adenocarcinomas, NOS for TP53/p53 (Table 2). EGFR was mainly expressed in mucoepidermoid carcinomas (Table 2). A detailed description of the percentage of positive tumour cells and the intensity of immunostaining is provided in Supplymentary Table 1.

Expression of cell cycle factors and EGFR in salivary gland carcinomas. (a) Representative histomorphology of an HPV-negative adenoid cystic carcinoma expressing CDKN2A/p16 in a subfraction of tumour cells. (b) Adenoid cystic carcinoma with TP53/p53 expression. (c) Adenoid cystic carcinoma showing EGFR expression. (d) Mucoepidermoid carcinoma with partial EGFR positivity. Original magnification of ×200 in a, c and d and ×400 in b, as well as in inserts in a, c and d. Microscopic images were produced with a BZ-9000 slide scanner (Keyence, Neu-Isenburg, Germany). CDKN2A, cyclin-dependent kinase inhibitor 2A; EGFR, epidermal growth factor receptor; HPV, human papillomavirus; TP53, tumour protein p53.
Discussion
Viruses have been demonstrated to be the mediators of neoplastic proliferation in squamous tumour types of the head and neck,1,2 while adenocarcinomas are rarely associated with viruses.10,11,12 Recently, CMV was analysed by immunohistochemistry in mucoepidermoid carcinomas; remarkably, it was detected in these carcinoma cells.7 If CMV has a major causal role in neoplastic proliferation, it would be expected that the majority of carcinoma cells would be positive. In contrast, it has been shown that the luminal carcinoma cells were particularly positive for immunostaining,7 and in our cohort, none of the carcinomas, including the mucoepidermoid carcinomas, showed positivity for CMV. In another study, PCR analysis did not reveal CMV in salivary tumours.18 EGFR and other cell cycle factors were expressed in these tumour samples,7 but it is not clear whether these factors are directly related to CMV detection or are a result of aberrantly increased proliferation. Because CMV results have not yet been confirmed by in situ hybridisation or PCR, CMV infection cannot be regarded as a major pathogenic driver in adenocarcinomas of the head and neck.
EBV can be found in nasopharyngeal carcinomas and lymphoepithelial carcinomas.19 Indolent EBV infection of B cells is very common among adults, and the oro-nasal cavity is the main anatomical site of virus entry. EBV persists in ductal epithelial cells of salivary glands and can be involved in aberrant proliferation, such as in sporadic B-cell lymphoma or Hodgkin lymphoma and post-transplant lymphoproliferative disease.4,5 Although EBV is localised within ductal epithelial cells and can mediate neoplastic proliferation, we and others have shown that salivary gland tumours in the Caucasian population are negative for this virus.20 Regarding the role of ethnic background, the Inuit are known to have a higher risk for developing EBV-positive high-grade salivary gland carcinomas and have higher frequencies of EBV-positive nasopharyngeal carcinomas, similar to Asians and North Africans.21 The pathobiology of this ethnic predisposition is currently unknown, and it is also not known why nasopharyngeal carcinomas are usually associated with EBV, while oropharyngeal carcinomas are associated with HPV, despite their close anatomical relationship. HPV has been analysed by in situ hybridisation and PCR in oral, nasal and parotid gland tumours.8,10,11,12 In one study, 7% of cases (n=2/27) were found to be positive for HPV subtype 16 and revealed a concomitant strong and diffuse expression of CDKN2A.10 These two tumours were high-grade adenoid cystic carcinomas of the nasal cavity, and the ethnic background of these two patients was not specified.10 The correlation of HPV and CDKN2A presence in oropharyngeal carcinoma and cervical carcinoma has shown that strong and diffuse CDKN2A positivity is an indicator of underlying HPV infection.22,23 However, CDKN2A was also strongly and diffusely detectable in HPV-negative, high-grade adenoid cystic carcinomas and other salivary gland tumours, indicating that this kinase inhibitor can be increased independent of HPV status.10,13 Corresponding to the lack of HPV infection in our cohort, tumour expression of CDKN2A, TP53 and EGFR was mainly weak to moderate and often patchy. Previous studies have demonstrated similar expression of these three factors in salivary gland carcinomas and the frequency of positive tumour types in our analysis is similar to findings in other cohorts.24,25,26 For example, the frequency of EGFR-positive mucoepidermoid carcinomas was 91% in our cohort and 82% in the analysis of Shang et al.25 Taken together, these findings show an aberrant expression of cell cycle factors and EGFR in a subfraction of tumour cells refers to the deregulated cell homeostasis and not to virus infection.
In another recently published study, PCR analyses of mucoepidermoid carcinomas from American patients revealed that 36% (n=35/98) of samples were HPV-positive, mainly HPV subtype 16 and, less frequently, HPV 18 or HPV 16+18.11 In our cohort of salivary gland carcinomas (European patients), we used a sensitive and specific PCR method15 but did not identify any HPV-positive cases among a variety of different tumours types, including 20 adenoid cystic carcinomas and eleven mucoepidermoid carcinomas. Another study demonstrated only occasional HPV-positive parotid gland carcinomas (n=1/39, 3%).12 Similar to our results, in three other studies which included more than 100 salivary gland carcinoma cases, no HPV-positive case could be identified.27,28,29
While salivary gland carcinomas show no general association with HPV infection, some reports have indicated that benign salivary gland neoplasms are frequently positive. In particular, greater than 50% of HPV-positive Warthin’s tumours were identified in small cohorts.8,9 In contrast, another study that included a larger number of benign parotid tumours reported the identification of less than 10% of HPV-positive cases (n=3/49, 7.5%).12 We analysed a set of Warthin’s tumours and pleomorphic adenomas and expected that a subfraction would be HPV-positive according to previous results,8,9 but no association was found. Therefore, similar to carcinomas, HPV is not a general mediator of aberrant proliferation in benign salivary gland neoplasms.
In the minority of salivary gland tumours that are HPV-positive, the transmission route of HPV infection is not fully understood. Extrapolating from studies on squamous tumours,1,2 sexual transmission could be one risk factor. Viraemia after HPV entry in an anatomical site other than the salivary glands is an alternative hypothesis. Immunosuppression, particularly by co-infection with human immunodeficiency virus, could be another factor that increases the potential of HPV to infect non-squamous cells.30 In contrast to salivary gland neoplasms, cervical cancer data show that squamous carcinomas are often infected by HPV, as are a subfraction of adenocarcinomas.31 Similar to adenocarcinomas of the cervix, HPV can also be detected in adenocarcinomas of the lung, but the frequency of HPV-associated lung carcinoma is lower and appears to be related to ethnicity (higher in Asia, lower in parts of Europe and America).32,33 The exact entry mechanism that allows HPV to enter glandular but not squamous cells is still uncertain. Recent experiments in squamous cells suggest that HPV binds nonspecifically to glycoproteins (particularly heparan-sulfonated proteoglycans) and that this complex activates EGFR.34,35 The EGFR-activated mTOR/PI3K signalling cascade leads to translocation of intracellular annexin A2 to the extracellular leaflet of the plasma membrane. At the site of HPV/EGFR/annexin A2 interaction, endosome formation is induced and ultimately leads to internalisation of the virus.34 Chronic stress with (transient) squamous metaplasia of salivary duct cells could increase the likelihood of HPV entry. In situ precursor lesions of HPV-positive cervical adenocarcinomas are often located adjacent to cervical squamous intraepithelial dysplasia/neoplasms.34 Therefore, infection of a common adenosquamous precursor cell at the site of transition between squamous and glandular epithelial cells is possible. Furthermore, a HPV-positive adenosquamous carcinoma of the tongue has been described.36
In summary, HPV can be found in a few nasal-type high-grade adenocarcinomas,10 in a few benign and malignant parotid gland tumours12 and in some mucoepidermoid carcinomas.11 In contrast to oropharyngeal carcinomas, HPV is not generally associated with salivary gland tumours. Similar to HPV findings, infection with either EBV or CMV is not a major driver of aberrant cell proliferation in salivary gland carcinomas.
References
Riechelmann H . [Human papilloma virus in head and neck cancer]. Laryngorhinootologie 2010; 89( 1): 43–48, quiz 49–51. German.
Allen CT, Lewis JS Jr, El-Mofty SK et al. Human papillomavirus and oropharynx cancer: biology, detection and clinical implications. Laryngoscope 2010; 120( 9): 1756–1772.
Isayeva T, Li Y, Maswahu D et al. Human papillomavirus in non-oropharyngeal head and neck cancers: a systematic literature review. Head Neck Pathol 2012; 6( Suppl 1): S104–S120.
Kaneda A, Matsusaka K, Aburatani H et al. Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res 2012; 72( 14): 3445–3450.
Hussein K, Maecker-Kolhoff B, Klein C et al. [Transplant-associated lymphoproliferation]. Pathologe 2011; 32( 2): 152–158. German.
Wax TD, Layfield LJ, Zaleski S et al. Cytomegalovirus sialadenitis in patients with the acquired immunodeficiency syndrome: a potential diagnostic pitfall with fine-needle aspiration cytology. Diagn Cytopathol 1994; 10( 2): 169–174.
Melnick M, Sedghizadeh PP, Allen CM et al. Human cytomegalovirus and mucoepidermoid carcinoma of salivary glands: cell-specific localization of active viral and oncogenic signaling proteins is confirmatory of a causal relationship. Exp Mol Pathol 2012; 92( 1): 118–125.
Hafed L, Farag H, Shaker O et al. Is human papilloma virus associated with salivary gland neoplasms? An in situ-hybridization study. Arch Oral Biol 2012; 57( 9): 1194–1199.
Vageli D, Sourvinos G, Ioannou M et al. High-risk human papillomavirus (HPV) in parotid lesions. Int J Biol Markers 2007; 22( 4): 239–244.
Boland JM, McPhail ED, García JJ et al. Detection of human papilloma virus and p16 expression in high-grade adenoid cystic carcinoma of the head and neck. Mod Pathol 2012; 25( 4): 529–536.
Isayeva T, Said-Al-Naief N, Ren Z et al. Salivary mucoepidermoid carcinoma: demonstration of transcriptionally active human papillomavirus 16/18. Head Neck Pathol 2013; 7( 2): 135–148.
Descamps G, Duray A, Rodriguez A et al. Detection and quantification of human papillomavirus in benign and malignant parotid lesions. Anticancer Res 2012; 32( 9): 3929–3932.
Brunner M, Koperek O, Wrba F et al. HPV infection and p16 expression in carcinomas of the minor salivary glands. Eur Arch Otorhinolaryngol 2012; 269( 10): 2265–2269.
Jour G, West K, Ghali V et al. Differential expression of p16(INK4A) and cyclin D1 in benign and malignant salivary gland tumors: a study of 44 Cases. Head Neck Pathol 2013; 7( 3): 224–231.
Pilatz A, Altinkilic B, Rusz A et al. Role of human papillomaviruses in persistent and glucocorticoid-resistant juvenile phimosis. J Eur Acad Dermatol Venereol 2013; 27( 6): 716–721.
Scharenberg C, Eckardt A, Tiede C et al. Expression of caspase 14 and filaggrin in oral squamous carcinoma. Head Neck Pathol 2013; 7( 4): 327–333.
Bockmeyer CL, Maegel L, Janciauskiene S et al. Plexiform vasculopathy of severe pulmonary arterial hypertension and microRNA expression. J Heart Lung Transplant 2012; 31( 7): 764–772.
Atula T, Grénman R, Klemi P et al. Human papillomavirus, Epstein–Barr virus, human herpesvirus 8 and human cytomegalovirus involvement in salivary gland tumours. Oral Oncol 1998; 34( 5): 391–395.
Rytkönen AE, Hirvikoski PP, Salo TA . Lymphoepithelial carcinoma: two case reports and a systematic review of oral and sinonasal cases. Head Neck Pathol 2011; 5( 4): 327–334.
Pollock AM, Toner M, McMenamin M et al. Absence of Epstein–Barr virus encoded RNA and latent membrane protein (LMP1) in salivary gland neoplasms. J Laryngol Otol 1999; 113( 10): 906–908.
Boysen T, Friborg J, Andersen A et al. The Inuit cancer pattern—the influence of migration. Int J Cancer 2008; 122( 11): 2568–2572.
van Monsjou HS, van Velthuysen ML, van den Brekel MW et al. Human papillomavirus status in young patients with head and neck squamous cell carcinoma. Int J Cancer 2012; 130( 8): 1806–1812.
von Knebel Doeberitz M, Reuschenbach M, Schmidt D et al. Biomarkers for cervical cancer screening: the role of p16(INK4a) to highlight transforming HPV infections. Expert Rev Proteomics 2012; 9( 2): 149–163.
Etges A, Nunes FD, Ribeiro KC et al. Immunohistochemical expression of retinoblastoma pathway proteins in normal salivary glands and in salivary gland tumours. Oral Oncol 2004; 40( 3): 326–331.
Shang J, Shui Y, Sheng L et al. Epidermal growth factor receptor and human epidermal growth receptor 2 expression in parotid mucoepidermoid carcinoma: possible implications for targeted therapy. Oncol Rep 2008; 19( 2): 435–440.
Ettl T, Schwarz S, Kleinsasser N et al. Overexpression of EGFR and absence of C-KIT expression correlate with poor prognosis in salivary gland carcinomas. Histopathology 2008; 53( 5): 567–577.
Bishop JA, Yonescu R, Batista D et al. Mucoepidermoid carcinoma does not harbor transcriptionally active high risk human papillomavirus even in the absence of the MAML2 translocation. Head Neck Pathol 2014; DOI: 201410.1007/s12105-014-0541-9. [Epub ahead of print].
Skálová A, Kašpírková J, Andrle P et al. Human papillomaviruses are not involved in the etiopathogenesis of salivary gland tumors. Cesk Patol 2013; 49( 2): 72–75.
Jour G, West K, Ghali V et al. Differential expression of p16 (INK4A) and cyclin D1 in benign and malignant salivary gland tumors: a study of 44 Cases. Head Neck Pathol 2013; 7( 3): 224–231.
Fatahzadeh M, Schlecht NF, Chen Z et al. Oral human papillomavirus detection in older adults who have human immunodeficiency virus infection. Oral Surg Oral Med Oral Pathol Oral Radiol 2013; 115( 4): 505–514.
Andersson S, Mints M, Wilander E . Results of cytology and high-risk human papillomavirus testing in females with cervical adenocarcinoma in situ. Oncol Lett 2013; 6( 1): 215–219.
Wang JL, Fang CL, Wang M et al. Human papillomavirus infections as a marker to predict overall survival in lung adenocarcinoma. Int J Cancer 2014; 134( 1): 65–71.
Chen YC, Chen JH, Richard K et al. Lung adenocarcinoma and human papillomavirus infection. Cancer 2004; 101( 6): 1428–1436.
Dziduszko A, Ozbun MA . Annexin A2 and S100A10 regulate human papillomavirus type 16 entry and intracellular trafficking in human keratinocytes. J Virol 2013; 87( 13): 7502–7515.
Surviladze Z, Sterk RT, DeHaro SA et al. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. J Virol 2013; 87( 5): 2508–2517.
Hanna J, Reimann JD, Haddad RI et al. Human papillomavirus-associated adenocarcinoma of the base of the tongue. Hum Pathol 2013; 44( 8): 1516–1523.
Author information
Authors and Affiliations
Corresponding author
Additional information
Note: Supplementary information for this article can be found on International Journal of Oral Science's website .
Supplementary information
supplementary Table 1
Summary of the percentage of positive tumour cells and the staining intensity. (DOC 68 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permissing from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Senft, E., Lemound, J., Stucki-Koch, A. et al. Expression of cyclin-dependent kinase inhibitor 2A 16, tumour protein 53 and epidermal growth factor receptor in salivary gland carcinomas is not associated with oncogenic virus infection. Int J Oral Sci 7, 18–22 (2015). https://doi.org/10.1038/ijos.2014.28
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijos.2014.28
- Springer Nature Limited