
Abstract
Lysosomal dysfunction has been implicated both pathologically and genetically in neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease (PD). Lysosomal gene deficiencies cause lysosomal storage disorders, many of which involve neurodegeneration. Heterozygous mutations of some of these genes, such as GBA1, are associated with PD. CTSD is the gene encoding Cathepsin D (CTSD), a lysosomal protein hydrolase, and homozygous CTSD deficiency results in neuronal ceroid-lipofuscinosis, which is characterized by the early onset, progressive neurodegeneration. CTSD deficiency was also associated with deposition of α-synuclein aggregates, the hallmark of PD. However, whether partial deficiency of CTSD has a role in the late onset progressive neurodegenerative disorders, including PD, remains unknown. Here, we generated cell lines harboring heterozygous nonsense mutations in CTSD with genomic editing using the zinc finger nucleases. Heterozygous mutation in CTSD resulted in partial loss of CTSD activity, leading to reduced lysosomal activity. The CTSD mutation also resulted in increased accumulation of intracellular α-synuclein aggregates and the secretion of the aggregates. When α-synuclein was introduced in the media, internalized α-synuclein aggregates accumulated at higher levels in CTSD+/− cells than in the wild-type cells. Consistent with these results, transcellular transmission of α-synuclein aggregates was increased in CTSD+/− cells. The increased transmission of α-synuclein aggregates sustained during the successive passages of CTSD+/− cells. These results suggest that partial loss of CTSD activity is sufficient to cause a reduction in lysosomal function, which in turn leads to α-synuclein aggregation and propagation of the aggregates.
Similar content being viewed by others


Main
Maintaining protein homeostasis (proteostasis) is crucial in not only maintenance of physiological functions of cells, but survival of cells. Proteostasis is a particularly important issue for the survival of post-mitotic cells, such as neurons, while dividing cells can dilute aged and misfolded proteins during the mitosis process.1, 2 For the clearance of protein burden, cells utilize two major protein degradation systems, ubiquitin proteasome system and lysosomal degradation, the latter degrades endosomal and autophagosomal cargos.3, 4, 5, 6 Dysregulation of ubiquitin proteasome system and lysosome has been shown to cause protein conformational diseases, including neurodegenerative disorders and metabolic disorders.7, 8 Genetic studies have suggested that impairment of lysosomal functions has important roles in the pathogenesis of neurodegenerative diseases. Mutations in ATP13A2, GBA1 and VPS35 have been associated with PD.9, 10, 11, 12 Mutations in progranulin and charged multivesicular body protein 2B (CHMP2B) have been identified as genetic causes of amyotrophic lateral sclerosis and frontotemporal dementia.13, 14, 15 Postmortem brain tissues of neurodegenerative diseases have exhibited deposition of endosomal and autophagic vesicles.16 Therefore, neurodegenerative proteinopathies might be attributed to lysosomal dysfunction.
Pathological examinations of patient tissues have exhibited that protein aggregates, such as amyloid beta (Aβ), tau and α-synuclein aggregates, spread to larger brain regions as disease progresses.17 In animal models, intracerebrally injected α-synuclein aggregates could spread into larger brain regions both in α-synuclein transgenic and non-transgenic mice.18, 19, 20, 21 Inoculation of Aβ or tau aggregates into either non-transgenic or transgenic models of AD also exhibited propagation of those aggregates.22, 23, 24, 25, 26, 27, 28 Studies have suggested that cell-to-cell transmission of protein aggregates is the underlying mechanism of the pathological propagation.29, 30
Mounting evidence have suggested that lysosomal function is important for the clearance of the transferred aggregates in recipient neurons during cell-to-cell aggregate transmission.31 This has been extensively studied in cell culture models for α-synuclein transmission. Previous studies showed α-synculein aggregates can be internalized and transported through the endolysosomal pathway.32 Lyososomal dysfunction led to increased accumulation of the internalized α-synuclein aggregates, suggesting that the lysosomal activity in recipient cells is critical in the clearance of the transmitted α-synuclein aggregates.32, 33
Lysosomal storage diseases (LSDs) are caused by defects in the lysosomal degradation process. Mutations in genes encoding lysosomal catabolic enzymes and transporters manifest excessive deposition of the enzyme substrates in various organs.34 Though different LSDs show different symptoms, most of LSD patients exhibit neurological symptoms such as mental retardation, motor dysfunction and progressive neurodegeneration, as well as specific pathological changes in the nervous system.35, 36 In addition, some of progressive neurodegenerative disorders such as AD, PD and Huntington’s disease also show similar pathological features with LSD: accumulations of endosomal and autophagosomal vesicles and undegraded macromolecules, and inflammatory responses in brain.16
Gaucher’s disease (GD) is the most common LSD, which is inherited in an autosomal recessive manner. Homozygous mutations of GBA1 gene, encoding β-glucocerebrosidase 1 (GCase 1), a lysosomal hydrolase, is responsible for GD.37 Evidence has suggested that GD is closely related to PD. Patients with type-1 GD, the most common form of GD, frequently develop parkinsonism.38 Heterozygous carriers of GBA1 mutations are at a higher risk for PD.39, 40 It has been shown that about 75% of Lewy bodies, a pathological hallmark of PD, colocalized with GCase 1 in brains of PD and DLB patients with heterozygous GBA1 mutations.41 These results suggest that lysosomal enzyme deficiency is associated with the development of PD.
Cathepsin D (CTSD) is a major lysosomal endopeptidase, which is critical in the degradation of long-lived proteins.42 Genetic and clinical studies have shown that the homozygous deficiency of CTSD results in the early onset, progressive neurodegeneration, such as congenital neuronal ceroid-lipofuscinosis.43 The heterozygous missense mutations in CTSD have been known to cause the early onset motor and visual problems, brain atrophy, and progressive psychomotor symptoms.44 However, the effects of CTSD deficiency on the late onset progressive neurodegenerative disorders, including AD and PD, remain unclear. Nevertheless, it has become clear that CTSD activity is crucial in the degradation of pathogenic protein aggregates.45, 46
Herein, we generated a cell line with a heterozygous nonsense mutation in CTSD and investigated the roles of the CTSD activity in lysosomal function, α-synuclein aggregation and transcellular transmission of α-synuclein aggregates.
Results
Lysosomal dysfunction by reduced CTSD activity
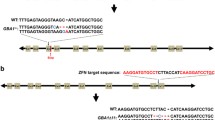
To investigate the roles of CTSD activity on lysosomal function and α-synuclein aggregation, we have generated SH-SY5Y cell lines with nonsense mutations in CTSD gene using zinc finger nucleases technology. We designed the zinc finger nucleases that introduce mutations in exon 4 of CTSD and obtained six clones with mutations in CTSD gene. The presence of nonsense mutations were confirmed by DNA sequencing (Figure 1a). All the clones we selected carried mutations only in a single copy. We were not able to generate a clone with mutations in both copies. This is consistent with the previous transgenic mouse study, in which CTSD-null mice were lethal. Figure 1b showed that the nonsense mutation in CTSD (CTSD+/−) resulted in a reduced expression of CTSD (Figure 1b). We measured the intracellular activity of CTSD both in the WT and CTSD+/− cell lines in the presence or absence of pepstatin A, an inhibitor of CTSD. As expected, pepstatin A-sensitive CTSD activity was decreased in CTSD+/− cells (Figure 1c).

Generation of CTSD-deficient SH-SY5Y cell line by using zinc finger nucleases. (a) To generate nonsense mutations in the CTSD gene, SH-SY5Y human neuroblastoma cells were transfected with zinc finger nucleases targeting exon 4. The nonsense mutations in single copy of CTSD exon 4 were confirmed by DNA sequencing. (b) Western blot analysis of CTSD in SH-SY5Ycell lysates. (c) The significant reduction of intracellular CTSD activity. n=4, *P<0.05 by paired, two-tailed Student’s t test
To address the effects of CTSD activity on lysosomal clearance, we measured intracellular accumulation of lysosomal substrates, such as polyubiquitinated proteins and p62, a polyubiquitin binding protein. CTSD+/− cells exhibited higher steady state levels of p62 and polyubiquitinated proteins than WT cells (Figures 2a and b). Next, we examined the lysosomal degradation activity using fluorescence-labeled dextran. CTSD+/− cells exhibited significantly reduced degradation rate of ectopically introduced dextran than WT cells (Figure 2c). We also analyzed the morphological changes in CTSD+/− cells using transmission electron microscopy and found accumulation of autophagic vesicles and damaged cellular organelles (Figures 2d and e). These results suggested that the reduction in CTSD activity caused lysosomal impairment.

Loss of CTSD activity led to the lysosomal dysfunction. (a and b) The accumulation of p62 (a) and polyubiquitinated proteins (b) were analyzed in Triton X-100-insoluble fraction. For quantification of polyubiquitinated proteins, the quantified size range is indicated by the line to the right of the blot. n=4, * P<0.05 by paired, two-tailed Student’s t test. (c) To test lysosomal degradation rate, degradation rate of internalized dextran–fluorescein isothiocyanate (FITC) was calculated. Scale bars: 20 μm, n=4, *P<0.05 by paired, two-tailed Student’s t test. One hundred cells were analyzed per experiment. (d and e) Electron microscopy of WT (d) and CTSD+/− (e). The boxed areas in the upper images are magnified in the lower panels. Scale bars: 2 μm
Alterations in α-synuclein metabolism by CTSD deficiency
Previous studies have shown that the lysosomal function is critical for degradation of protein aggregates.47 Therefore, we tested whether the loss of CTSD activity might accelerate the accumulation of α-synuclein aggregates. First, we analyzed the intracellular amount of the endogenous α-synuclein both in WT and CTSD+/− cells. The loss of CTSD activity did not change the levels of the endogenous α-synuclein (Figures 3a and b). This result might be because of the low-level expression of the endogenous α-synuclein. In other words, even half the normal level of CTSD might be enough to handle the endogenous α-synuclein. We then investigated the effects of CTSD mutation after overexpression of α-synuclein. Both WT and CTSD+/− cells were infected with the adenoviral vector for human α-synuclein, and the intracellular levels of α-synuclein were measured. α-Synuclein aggregates accumulated at higher levels in the Triton X-100-insoluble fractions of CTSD+/− cells than in those of WT cells, whereas the CTSD deficiency did not alter the levels of monomeric α-synuclein (Figures 3c and d).

Alterations in accumulation and secretion of α-synuclein aggregates by CTSD deficiency. (a and b) Western blot analysis of α-synuclein both in the WT and CTSD+/− SH-SY5Y cells. After differentiation, the levels of α-synuclein was measured in Triton X-100-soluble and -insoluble fraction. Owing to low expression level, α-synuclein was not detected in the Triton-insoluble fraction. The amount of α-synuclein in the Triton-soluble fraction was analyzed in (b). n=3. (c and d) Western blot analysis of α-synuclein. Human α-synuclein was overexpressed both in the WT and CTSD+/− SH-SY5Y cells. On day 3 after infection, the levels of α-synuclein was measured in Triton X-100-soluble and -insoluble fraction. For quantification of α-synuclein aggregates, the quantified size range is indicated by the line to the right of the blot. Loss of CTSD activity increased the accumulation of α-synuclein aggregates in the Triton-insoluble fraction (d). n=3, *P<0.05 by paired, two-tailed Student’s t test. (e) The amount of secreted α-synuclein aggregates in cell culture media were measured using α-synuclein aggregate-specific ELISA. n=4, **P<0.01 by paired, two-tailed Student’s t test
Previous studies showed that neuronal cells can release small amounts of both monomeric and aggregate forms of α-synuclein to extracellular space.48 The secretion of α-synuclein aggregates was increased under certain stress conditions, such as oxidative stress and proteosomal stress.49 We examined alterations in the secretion of α-synuclein aggregates in CTSD+/− cells using α-synuclein aggregate-specific enzyme-linked immunosorbent assay. The amounts of secreted α-synuclein aggregates from CTSD+/− cells was higher than those from WT cells (Figure 3e). These results showed that the reduction in CTSD activity causes an increase in the accumulation and secretion of α-synuclein aggregates.
We then examined the role of CTSD in degradation of internalized α-synuclein aggregates when the protein was introduced in the culture media. We compared the accumulation kinetics of α-synuclein aggregates both in WT cells and CTSD+/− cells. When α-synuclein fibrils were administered in the media, the time required for reaching the maximum level was 24 h in WT cells, while the maximum level was reached at 8 h in CTSD+/− cells (Figures 4a and b). When the cell-released α-synuclein was introduced to the culture, the time to reach the maximum level was reduced from 5 to 2 min in CTSD+/− cells compared with the WT cells (Figures 4c and d). These results indicate that CTSD+/− cells have reduced ability to degrade the internalized exogenous α-synuclein aggregates.

Increased accumulation of exogenous α-synuclein aggregates in CTSD+/− cells. (a and b) Increased accumulation of α-synuclein fibrils in CTSD+/− cells. Internalized α-synuclein fibrils were analyzed by western blotting at the indicated times. The levels of α-synuclein aggregates in (a) was quantified and presented in (b). The relative values to the maximum level of internalized α-synuclein in WT cells were presented in the graph in (b); n=3. (c and d) Accumulation of cell-derived α-synuclein aggregates. Internalized α-synuclein aggregates were analyzed by western blotting at the indicated times. The relative levels to the maximum level of internalized α-synuclein in WT cells were presented in (d); n=3
CTSD deficiency accelerates the transmission of α-synuclein aggregates
Partial deficiency of CTSD caused both increase in secretion of α-synuclein aggregates and reduction in degradation of internalized exogenous α-synuclein aggregates. These results strongly implicate increased cell-to-cell transmission of α-synuclein aggregates with CTSD defects. To assess the roles of CTSD in transcellular transmission of α-synuclein aggregates, we utilized the bimolecular fluorescence complementation co-culture system (BiFC), which has been described previously as a cellular model for the study of cell-to-cell transfer and seeded aggregation of α-synuclein.50 This model system is composed of two SH-SY5Y cell lines that stably express α-synuclein conjugated with either the N-terminal (V1S) or C-terminal (SV2) fragments of the Venus fluorescence protein (Figure 5a). When these stable cell lines are co-cultured, α-synuclein proteins were secreted from each cell line and transferred to another stable lines, resulting in co-aggregation between the transferred α-synuclein and the intrinsic α-synuclein. This co-aggregation leads to reconstitution of Venus fragments and can be monitored by fluorescence emission.

Lysosomal dysfunction in CTSD+/− SV2 cells. (a) Scheme of the dual cell BiFC co-culture system. The system consists of two stable cell lines expressing α-synuclein conjugated with either the amino terminus (V1S) or carboxy terminus (SV2) fragment of Venus fluorescence protein. When the cells were co-cultured, cell-to-cell transmission of α-synuclein was detected by using BiFC fluorescence resulting from dimerization or oligomerization of the V1S and SV2 fusion proteins. (b) Nonsense mutation in single copy of CTSD gene in SV2 cells. (c and d) Western blot analysis showing reduced CTSD expression in SV2 CTSD+/− cells. n=4, **P<0.01 by paired, two-tailed Student’s t test. (e) Reduction of the intracellular CTSD activity. n=4, *P<0.05 by paired, two-tailed Student’s t test. (f) Levels of p62 in the Triton-insoluble fractions. n=3, *P<0.05 by paired, two-tailed Student’s t test. (g) Levels of polyubiquitinated proteins. Quantified regions in the blots are indicated by the line on the right. n=3, *P<0.05 by paired, two-tailed Student’s t test. (h) Degradation efficiency of imported dextran-fluorescein isothiocyanate (FITC). Scale bars: 20 μm. n=4, ***P<0.005 by paired, two-tailed Student’s t test. One hundred cells were analyzed per experiment
Using the zinc finger nucleases targeting exon 4 in the CTSD gene, we have generated the nonsense mutations in the CTSD gene in the SV2 cell line (Figure 5b). Like the naive SH-SY5Y cells, we only obtained heterozygous mutant cell lines (SV2 CTSD+/−), and the nonsense mutations in single copy of CTSD decreased both the expression and activity of CTSD in SV2 cells (Figures 5c and e). Lysosomal substrates, such as p62 and polyubiquitinated proteins, accumulated at higher levels in SV2 CTSD+/− cells than in SV2 cells (Figures 5f and g). Degradation rates of ectopically introduced dextran were reduced in SV2 CTSD+/− cells compared with SV2 cells (Figure 5h).
Next, we tested alterations in the accumulation and secretion of α-synuclein aggregates in SV2 CTSD+/− cells. Unlike the CTSD+/− cells, the SV2 CTSD+/− did not exhibit increased deposition of α-synuclein aggregates compared with the SV2 cells. This is probably due to lower expression levels of α-synuclein in SV2 cells than in SH-SY5Y cells infected with the α-synuclein adenoviral vector. Nevertheless, secretion of α-synuclein aggregates was increased in SV2 CTSD+/− compared with SV2 cells (Figures 6a and b).

CTSD deficiency increased the secretion of α-synuclein aggregates in SV2 cells. (a) Western blot analysis of α-synuclein. The levels of α-synuclein conjugated with C-terminus of Venus fluorescence protein fragment was measured in Triton X-100 soluble and insoluble fraction. (b) The amount of secreted α-synuclein aggregates in cell culture media were measured using α-synuclein aggregate specific enzyme-linked immunosorbent assay. n=4, *P<0.05 by paired, two-tailed Student’s t test
To directly address alterations in the transmission of α-synuclein aggregates, either SV2 or SV2 CTSD+/− cells were co-cultured with V1S cells. Then, we measured BiFC fluorescence, an indicator of co-aggregation between the α-synuclein proteins derived from V1S and SV2 cells (Figure 7a).50 When V1S cells were co-cultured with SV2 CTSD+/− cells, the number of BiFC fluorescence-positive cells was significantly increased compared with the V1S/SV2 co-culture (Figures 7a and b), indicating that reduction in CTSD expression leads to increased transmission of α-synuclein aggregates. In a previous study, we showed that transmission of α-synuclein aggregates perpetuates through multiple cells.50 To determine the effects of CTSD mutation on the perpetual α-synuclein transmission, we continuously sub-cultured both V1S/SV2 and V1S/SV2 CTSD+/− co-cultures through several passages. The number of BiFC-positive cells was elevated with increasing passage numbers in both co-cultures, and the percentage of BiFC-positive cells was consistently higher in the V1S/SV2 CTSD+/− co-culture than the V1S/SV2 culture throughout the passages (Figures 7c and d). Consistent with this result, the amounts of secreted α-synuclein aggregates were higher in V1S/SV2 CTSD+/− co-culture than in V1S/SV2 co-culture (Figure 7e). Collectively, these results suggest that the reduction in CTSD expression/activity potentiates the transcellular transmission of α-synuclein aggregates by accelerating secretion and impairing the clearance of the transferred α-synuclein aggregates.

CTSD deficiency increased cell-to-cell transmission of α-synuclein aggregates. (a and b) Increased cell-to-cell transmission of α-synuclein aggregates in V1S/SV2 CTSD+/− co-culture. BiFC-positive aggregates are indicated by arrowheads. Scale bars: 20 μm. The number of BiFC-positive cells in (a) was quantified in (b); n=4, 500 cells per experiment, *P<0.05 by paired, two-tailed Student’s t test. (c and d) Increased continuous transcellular transmission of α-synuclein aggregates in V1S/SV2 CTSD+/− co-culture. BiFC-positive aggregates are indicated by arrowheads. Scale bars: 20 μm. The number of BiFC-positive cells in (c) was quantified in (d); n=4, 500 cells per experiment, *P<0.05, ****P<0.001 by paired, two-tailed Student’s t test. (e) α-Synuclein aggregate-specific enzyme-linked immunosorbent assay analysis of culture media from the indicated subcultures. n=3, *P<0.05, ***P<0.005 by paired, two-tailed Student’s t test
Discussion
In this study, we showed that experimental haploinsufficiency of CTSD resulted in the reduction of lysosomal functions, impaired α-synuclein catabolism, increased α-synuclein secretion and decreased degradation of internalized α-synuclein, leading to increased cell-to-cell transmission of α-synuclein aggregates. The current study suggests that reduction in CTSD activity is sufficient to cause a significant decline of lysosomal functions and accelerate the propagation of Lewy body pathology.
In humans, homozygous mutations of CTSD causes neuronal ceroid-lipofuscinosis, an early onset lysosomal storage disease, while heterozygous missense mutations results in early-onset motor and visual disturbances, brain atrophy, and progressive psychomotor problems.44 However, the effects of partial decline of CTSD activity on development and progression of late-onset neurodegenerative disease, such as PD, has not been addressed. CTSD is a lysosomal hydrolase, which has important roles in the degradation of long-lived proteins. Ectopic expression of CTSD in cells led to a reduction of α-synuclein aggregation and neurodegeneration, whereas neither transduction of cathepsin B nor L was protective against α-synuclein aggregates-induced toxicity.45, 46 Studies with CTSD-deficient mice showed accumulation of insoluble α-synuclein aggregates in the brain, whereas these were absent in wild-type littermates. Cullen et al.46 also reported the deposition of phosphorylated α-synuclein aggregates only in CTSD-mutant sheep, not in the normal subjects. Human postmortem studies with brains from early-onset neuronal ceroid-lipofuscinosis patients showed the accumulation of α-synuclein aggregates.46 Furthermore, in an α-synuclein transgenic fly model, depletion of CTSD accelerated the formation of α-synuclein aggregates and the associated pathogenic phenotypes, such as accumulation of numerous vacuoles and disruption of the retina structure.46 These results indicate that CTSD is critical for maintaining the levels of α-synuclein in check, and therefore, reduced expression of CTSD might increase the risk of synucleinopathies.
In the current study, we examined the effects of partial deficiency of CTSD not only on lysosomal functions and α-synuclein catabolism but also on secretion and cell-to-cell transmission of α-synuclein aggregates. Our study showed that the reduction in CTSD expression caused a decline in lysosomal function, which in turn led to an increase in the accumulation of intracellular α-synuclein aggregates and the secretion of the aggregates. We further demonstrated that partial loss of CTSD resulted in a reduction in clearance of internalized α-synuclein. As a combined effect of increased secretion and reduced clearance of α-synuclein, CTSD partial deficiency resulted in accelerated cell-to-cell transmission of α-synuclein aggregates. The observed effects on cell-to-cell transmission was not limited to a single transmission event, rather continued on to subsequent rounds of transmission. These results provide significant implications in the role of CTSD in metabolism of α-synuclein aggregates and in the propagation of α-synuclein pathology during the progression of PD and potentially other synucleinopathies.
The mechanism by which lysosomal dysfunction led to the increase in α-synuclein secretion is not understood yet. Our earlier studies suggested that α-synuclein is secreted through the ER-Golgi-independent unconventional exocytosis.48, 49 Exosome-associated secretion and exophagy (exocytosis through the fusion of autophagosome/amphisome with the plasma membrane) have been suggested as the mechanisms of unconventional exocytosis of α-synuclein.51, 52 Although it has not been experimentally demonstrated, one should also consider lysosomal exocytosis as one of the mechanisms.53 The relative contribution and pathophysiological significance of these mechanisms are not clear. It is worth noting that all the exocytosis mechanisms discussed above can be activated by lysosomal dysfunction as alternative pathways to degradation.
Heterozygous mutations in GBA1, homozygous mutations of which cause GD, are known as strong genetic risk factors of PD and perhaps DLB.40 Individuals carrying GBA1 mutations are five times more likely to have PD than the normal control. And, compared with the healthy control, an incidence of parkinsonism is 6 to 17 times higher in patients with type 1 GD.38 Furthermore, a large multicenter study reported that PD patients with GBA1 mutations not only had a higher incidence of dementia, but also showed earlier onset and faster progress of disease.54 Recently, we have shown that the depletion of GBA1 resulted in lysosomal defects and accelerated the cell-to-cell transmission of α-synuclein aggregates.50 These data might explain why carriers of GBA1 mutations exhibit higher incidence and faster progression of the disease.50 In the current study, we show that partial deficiency of CTSD results in the similar phenotypes as GBA1 deficiency, exhibiting decline in lysosomal functions and increased secretion and cell-to-cell transmission of α-synuclein aggregates. Given the similarities between CTSD and GBA1 phenotypes in cells, it might be worth investigating the relationships among CTSD genotypes, PD incidence, and the rate of disease progression.
Even though most of lysosomal hydrolase activities are redundant, it has been repeatedly shown that a single lysosomal mutation can cause lysosomal dysfunction.55, 56 Likewise, only a partial loss of CTSD activity is sufficient to cause lysosomal defects. CTSD heterozygous deficient mice exhibited an increase in LAMP1 levels and were more sensitive to MPTP-induced neurotoxicity than the wild-type controls.57 This indicates that a half of the activity of CTSD is not sufficient for maintaining the normal lysosomal functions. These results suggest that at least for some hydrolases, keeping the proper quantities is critical for maintaining the normal lysosomal activity. The CTSD protein levels and activities have indeed been shown to be reduced in PD patients,58, 59 although it has not been unanimously agreed upon yet.39, 58 We speculate that the proper control of expression of lysosomal hydrolases, perhaps through the activation of transcription factor EB, is important for maintenance of proteostasis, and therefore, for the therapy for neurodegenerative diseases.60 Furthermore, genetic and epigenetic changes in genes encoding lysosomal proteins might represent common risks for PD and other neurodegenerative diseases.
In conclusion, haploinsufficiency of CTSD resulted in lysosomal dysfunction and abnormal α-synuclein metabolism and secretion, thereby promoting cell-to-cell transmission of α-synuclein aggregates. Given the similarities to the GBA1 studies, our current results suggest that the reduced expression of CTSD leads to accelerated disease progression in Lewy body diseases. Our study shows that lysosomal functions could be sensitive to quantities of individual hydrolases. We propose a hypothesis that quantitative changes in certain lysosomal gene expression is the determining factor for development and/or progression rate of PD and other synucleinopathies.
Materials and Methods
Materials
The following antibodies were used in this study: CTSD monoclonal antibody CTD-19 (ab6313; Abcam, Cambridge, MA, USA; 1 : 1000 dilution), β-actin monoclonal antibody AC-15 (A5441; Sigma-Aldrich, St. Louis, MO, USA; 1 : 10 000 dilution), p62 monoclonal antibody p62 LCK ligand (610833; BD Transduction Laboratories, Swampscott, MA, USA; 1 : 1000 dilution), ubiquitin polyclonal antibodies (Dako, Glostrup, Denmark, and Chemicon, Temecula, CA, USA; 1 : 1000 dilution), α-synuclein monoclonal antibody (610787; BD Biosciences, San Diego, CA, USA; 1 : 1500 dilution), α-synuclein monoclonal antibody Ab274 (1 : 1500 dilution), α-synuclein monoclonal antibody Ab62 (1 : 1000 dilution), HRP-conjugated goat anti-mouse IgG (H+L) (172-1011; Bio-Rad Laboratories, Hercules, CA, USA; 1 : 3000 dilution) and HRP-conjugated goat anti-rabbit IgG (H+L) (Bio-Rad Laboratories; 1 : 3,000 dilution).
Fluorescein-conjugated dextran (10 000 molecular weight; D-1821), TO-PRO-3 iodide (T3605) and LysoTracker Red DND-99 (L-7528) were purchased from Invitrogen (Carlsbad, CA, USA).
Generation of CTSD knockout cell lines
Plasmids encoding zinc finger nuclease and a magnetic reporter (ToolGen, Seoul, Korea) was transfected to SH-SY5Y cells (CRL-2266; ATCC, Manassas, VA, USA) by using electroporation. After incubation for 48 h, transfected cells were enriched by magnetic separation. Briefly, cells were mixed with magnetic bead-conjugated antibody against H-2Kk (MACSelect Kk microbeads; Miltenyi Biotech, Gladbach, Germany), and the mixture was applied to a MACS LS column (Miltenyi Biotech). Single cells were isolated from the eluates and maintained until the clonal colonies were picked from the culture dish. Nonsense mutations in the CTSD gene were confirmed by DNA sequencing. Six clones with non-sense mutations in exon 4 were generated. Among these clones, two clones were further analyzed.
Cell culture
SH-SY5Y human neuroblastoma cell lines were cultured as described previously.50 Cells were split every 2 days at 37 °C in humidified air with 5% CO2 in Dulbecco’s modified eagle’s medium (SH30243.01, HyClone, Logan, UT, USA) containing 10% fetal bovine serum (SH30396.03, HyClone), 100 units/ml penicillin and 100 units/ml streptomycin (15140-122, Gibco, Grand Island, NY, USA). To differentiate SH-SY5Y cells, cells were cultured in the presence of 50 μM all-trans retinoic acid (R2625, Sigma-Aldrich). For overexpression of human α-synuclein, differentiated SH-SY5Y cells were infected with a recombinant adenoviral vector (serotype Ad5, CMV promoter) containing human α-synuclein cDNA at a multiplicity of infection of 33.3.
For co-culture, V1S and SV2 (or SV2 CTSD+/−) stable cells (180 000 cells each) were mixed in a coverslip and cultured for 3 days. In order to investigate the continuous transmission of α-synuclein, the mixture of V1S and SV2 (or SV2 CTSD+/−) cells was sub-cultured every 2 days (48 h).
Preparation of conditioned medium
Differentiated SH-SY5Y cells were infected with a recombinant adenoviral vector as described above. On day 2 post infection, cells were washed three times with fresh Dulbecco’s modified eagle’s medium and cultured in serum-free Dulbecco’s modified eagle’s medium. After 18-h incubation at 37 °C, conditioned medium was collected and centrifuged at 4 °C at 250 × g for 10 min, followed by centrifugation at 10 000 × g for 10 min at 4 °C to remove cell debris. The conditioned medium was concentrated using an Amicon 10 K MWCO filters (Millipore, Tullagreen, Ireland).
Preparation of cell extracts
After washing twice with ice-cold PBS, cells were lysed in extraction buffer (1% Triton X-100, 1% (v/v) protease inhibitor cocktail (Sigma, St. Louis, MO, USA) in PBS). Cell lysates were incubated on ice for 10 min and centrifuged at 16 000 × g for 10 min. The Triton X-100 insoluble fraction was resuspended in 1 × Laemmli sample buffer and sonicated briefly.
Western blotting
Western blotting was performed as previously described.50 Images were obtained using the FUJIFILM Luminescent Image Analyzer LAS-3000 and analyzed with Multi Gauge (v3.0) software (FUJIFILM, Tokyo, Japan).
CTSD activity assay
Cellular CTSD activity was determined using a CTSD assay kit purchased from Sigma (CS0500). The assay was carried out as described by the manufacturer’s protocol. Briefly, CTSD activity was determined using an internally quenched fluorescent substrate, MCA-Gly-Lys-Pro-Ile-Leu-Phe-Phe-Arg-Leu-Lys(DNP)-D-Arg-NH2 trifluoroacetate salt. Cells were lysed in lysis buffer (0.5% CHAPS in PBS). Ten micrograms of cell lysates were mixed with internally quenched fluorescent substrate either in the presence or absence of 0.2 mg/ml pepstatin A solution, a CTSD inhibitor. After incubation at 37 °C for 10 min, the fluorescence was measured at 10-min intervals (excitation at 328 nm, emission at 393 nm). The activity unit was calculated by using standard reaction curve of recombinant CTSD enzyme.
Immunofluorescence staining
The procedure for immunofluorescence staining was performed as previously described.50 Briefly, cells grown on poly-L-Lysine-coated coverslips were fixed in 4% paraformaldehyde in PBS and permeabilized in 0.1% Triton X-100 in PBS. After incubation with the blocking solution (5% bovine serum albumin/3% goat serum in PBS), cells were incubated with primary antibodies diluted in blocking solution. After washing with ice-cold PBS, cells were incubated with fluorescent dye-conjugated secondary antibodies diluted in blocking solution. Nuclei were stained with TOPRO-3 iodide (Invitrogen). Cells were mounted onto slide glasses in the presence of Prolong Gold Antifade Reagent (Invitrogen). Images were obtained by using Olympus FV1000 confocal laser scanning microscopy (Olympus, Tokyo, Japan).
Dextran degradation assay
To determine the degradation ratio of ectopically transduced dextran, cells were incubated with 20 μg/ml of fluorescein isothiocyanate-labeled dextran (Invitrogen) for 2 h. After washing with Dulbecco’s modified eagle’s medium, cells were incubated with fresh growth medium for 30 min. Cells were fixed with a 4% PFA solution. The fluorescence intensity was measured using Olympus FV1000 software. The extent of degradation of internalized dextran–fluorescein isothiocyanate was calculated using the following equation: (Ftime0 − Ftime30)/Ftime0. Ftime0 and Ftime30 are the integrated fluorescence intensities at 0 and 30 min, respectively.
Electron microscopy
After fixation with Karnovsky’s fixative solution (2% glutaraldehyde, 2% paraformaldehyde and 0.5% CaCl2), cells were immersed in 1% osmium tetraoxide for 1.5 h. After dehydrating with 50, 60, 70, 80, 90, 95 and 100% absolute ethanol, cells were infiltrated with propylene oxide and EPON mixture (EPON 812, MNA, DDSA, DMP30) for 10 min before being embedded in EPON mixture. After embedding, cells were sectioned with an EM UC-7 Ultramicrotome (Leica Microsystems, Vienna, Austria) and stained with 6% uranyl acetate and lead citrate. Grids were observed using a transmission electron microscope (JEM-1011, JEOL, Tokyo, Japan) and analyzed using MegaView III software (Soft Imaging System, Münster, Germany).
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assay was performed as previously described.61 Briefly, 96-well enzyme-linked immunosorbent assay plates (Nalge Nunc International, Rochester, NY, USA) were coated with 1 μg/ml of capture antibody (Ab62) in 50 mM carbonate buffer (pH 9.6) at 4 °C overnight. After washing with PBS with 0.05% Tween 20 (PBST), SuperBlock T20 (PBS) Blocking Buffer (Thermo Scientific, Rockford, IL, USA) was added to each well. After incubation for 1 h at room temperature with shaking, plates were washed five times in PBST. Samples and standards were incubated at room temperature for 2.5 h with shaking. After washing with PBST, 1 μg/ml of biotinylated Ab62 in blocking buffer was added to each well. After incubation for 1.5 h at room temperature, the plates were washed with PBST. Avidin-conjugated peroxidase (ExtrAvidin, Sigma) was added to each well. After incubation for 1 h at room temperature, plates were washed with PBST. One hundred microliters of 3,3′,5,5′-tetramethylbenzidine solution (Sigma) were added to each well and incubated for 15 min with shaking. To stop the reaction, 50 μl of 2 N H2SO4 was added to each well. The absorbance was measured at 450 nm.
Statistical analysis
Values shown in the figures are means±S.E.M. To analyze the statistical significance, P values were calculated by means of paired, two-tailed Student’s t tests by using InStat version 3.05 software (GraphPad Software, San Diego, CA, USA).
Abbreviations
- AD:
-
Alzheimer’s disease
- BiFC:
-
Bimolecular fluorescence complementation
- CHMP2B:
-
Charged multivesicular body protein 2B
- CTSD:
-
Cathepsin D
- GCase 1:
-
Glucocerebrosidase 1
- GD:
-
Gaucher’s disease
- LSD:
-
Lysosomal storage disease
- PD:
-
Parkinson’s disease
References
Rujano MA, Bosveld F, Salomons FA, Dijk F, van Waarde MA, van der Want JJ et al. Polarised asymmetric inheritance of accumulated protein damage in higher eukaryotes. PLoS Biol 2006; 4: e417.
Taylor RC, Dillin A . Aging as an event of proteostasis collapse. Cold Spring Harb Perspect Biol 2011; 3: 5.
Korolchuk VI, Menzies FM, Rubinsztein DC . Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett 2010; 584: 1393–1398.
Lecker SH, Goldberg AL, Mitch WE . Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 2006; 17: 1807–1819.
Ciechanover A . Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 2005; 6: 79–87.
Menzies FM, Ravikumar B, Rubinsztein DC . Protective roles for induction of autophagy in multiple proteinopathies. Autophagy 2006; 2: 224–225.
Matsuda N, Tanaka K . Does impairment of the ubiquitin-proteasome system or the autophagy-lysosome pathway predispose individuals to neurodegenerative disorders such as Parkinson's disease? J Alzheimers Dis 2010; 19: 1–9.
Pan T, Kondo S, Le W, Jankovic J . The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson's disease. Brain 2008; 131: 1969–1978.
Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5P-type ATPase. Nat Genet 2006; 38: 1184–1191.
Sidransky E, Lopez G . The link between the GBA gene and parkinsonism. Lancet Neurol 2012; 11: 986–998.
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011; 89: 162–167.
Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 2011; 89: 168–175.
Schymick JC, Yang Y, Andersen PM, Vonsattel JP, Greenway M, Momeni P et al. Progranulin mutations and amyotrophic lateral sclerosis or amyotrophic lateral sclerosis-frontotemporal dementia phenotypes. J Neurol Neurosurg Psychiatry 2007; 78: 754–756.
Talbot K, Ansorge O . Recent advances in the genetics of amyotrophic lateral sclerosis and frontotemporal dementia: common pathways in neurodegenerative disease. Hum Mol Genet 2006; 2: R182–R187.
Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 2005; 37: 806–808.
Bahr BA, Bendiske J . The neuropathogenic contributions of lysosomal dysfunction. J Neurochem 2002; 83: 481–489.
Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ . Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 2015; 16: 109–120.
Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H et al. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun 2014; 2: 88.
Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012; 338: 949–953.
Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM . Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 2012; 209: 975–986.
Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H et al. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013; 136: 1128–1138.
Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD et al. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci 2000; 20: 3606–3611.
Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006; 313: 1781–1784.
Rosen RF, Fritz JJ, Dooyema J, Cintron AF, Hamaguchi T, Lah JJ et al. Exogenous seeding of cerebral beta-amyloid deposition in betaAPP-transgenic rats. J Neurochem 2012; 120: 660–666.
Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ et al. Purified and synthetic Alzheimer's amyloid beta (Abeta) prions. Proc Natl Acad Sci USA 2012; 109: 11025–11030.
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 2009; 11: 909–913.
Iba M, Guo JL, McBride JD, Zhang B, Trojanowski JQ, Lee VM . Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci 2013; 33: 1024–1037.
Peeraer E, Bottelbergs A, Van Kolen K, Stancu IC, Vasconcelos B, Mahieu M et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis 2015; 73: 83–95.
Lee SJ, Desplats P, Sigurdson C, Tsigelny I, Masliah E . Cell-to-cell transmission of non-prion protein aggregates. Nat Rev Neurol 2010; 6: 702–706.
Guo JL, Lee VM . Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 2014; 20: 130–138.
Lee HJ, Bae EJ, Lee SJ . Extracellular alpha—synuclein-a novel and crucial factor in Lewy body diseases. Nat Rev Neurol 2014; 10: 92–98.
Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ . Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol 2008; 40: 1835–1849.
Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA 2009; 106: 13010–13015.
Futerman AH, van Meer G . The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol 2004; 5: 554–565.
Vitner EB, Platt FM, Futerman AH . Common and uncommon pathogenic cascades in lysosomal storage diseases. J Biol Chem 2010; 285: 20423–20427.
Platt FM, Boland B, van der Spoel AC . The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol 2012; 199: 723–734.
Brady RO, Kanfer J, Shapiro D . The metabolism of glucocerebrosides. I. Purification and properties of a glucocerebroside-cleaving enzyme from spleen tissue. J Biol Chem 1965; 240: 39–43.
Rosenbloom B, Balwani M, Bronstein JM, Kolodny E, Sathe S, Gwosdow AR et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells Mol Dis 2011; 46: 95–102.
Gegg ME, Burke D, Heales SJ, Cooper JM, Hardy J, Wood NW et al. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 2012; 72: 455–463.
Yang NY, Lee YN, Lee HJ, Kim YS, Lee SJ . Glucocerebrosidase, a new player changing the old rules in Lewy body diseases. Biol Chem 2013; 394: 807–818.
Goker-Alpan O, Stubblefield BK, Giasson BI, Sidransky E . Glucocerebrosidase is present in alpha-synuclein inclusions in Lewy body disorders. Acta Neuropathol 2010; 120: 641–649.
Dean RT . Lysosomes and protein degradation. Ciba Found Symp 1979; 75: 139–149.
Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J et al. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain 2006; 129: 1438–1445.
Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Bruck W et al. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet 2006; 78: 988–998.
Qiao L, Hamamichi S, Caldwell KA, Caldwell GA, Yacoubian TA, Wilson S et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 2008; 1: 17.
Cullen V, Lindfors M, Ng J, Paetau A, Swinton E, Kolodziej P et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2009; 2: 5.
Cuervo AM, Dice JF . When lysosomes get old. Exp Gerontol 2000; 35: 119–131.
Lee HJ, Patel S, Lee SJ . Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 2005; 25: 6016–6024.
Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ . Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 2010; 113: 1263–1274.
Bae EJ, Yang NY, Song M, Lee CS, Lee JS, Jung BC et al. Glucocerebrosidase depletion enhances cell-to-cell transmission of alpha-synuclein. Nat Commun 2014; 5: 4755.
Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010; 30: 6838–6851.
Ejlerskov P, Rasmussen I, Nielsen TT, Bergstrom AL, Tohyama Y, Jensen PH et al. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem 2013; 288: 17313–17335.
Stinchcombe J, Bossi G, Griffiths GM . Linking albinism and immunity: the secrets of secretory lysosomes. Science 2004; 305: 55–59.
Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 2013; 70: 727–735.
Neufeld EF . Lysosomal storage diseases. Annu Rev Biochem 1991; 60: 257–280.
Ballabio A, Gieselmann V . Lysosomal disorders: from storage to cellular damage. Biochim Biophys Acta 2009; 1793: 684–696.
Crabtree D, Boyer-Guittaut M, Ouyang X, Fineberg N, Zhang J . Dopamine and its metabolites in cathepsin D heterozygous mice before and after MPTP administration. Neurosci Lett 2013; 538: 3–8.
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH . Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 2009; 35: 385–398.
Kurzawa-Akanbi M, Hanson PS, Blain PG, Lett DJ, McKeith IG, Chinnery PF et al. Glucocerebrosidase mutations alter the endoplasmic reticulum and lysosomes in Lewy body disease. J Neurochem 2012; 123: 298–309.
Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A . TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci USA 2013; 110: E1817–E1826.
Lee HJ, Bae EJ, Jang A, Ho DH, Cho ED, Suk JE et al. Enzyme-linked immunosorbent assays for alpha-synuclein with species and multimeric state specificities. J Neurosci Methods 2011; 199: 249–257.
Acknowledgements
This work was supported by the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI14C0093).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhrasky
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bae, EJ., Yang, N., Lee, C. et al. Haploinsufficiency of cathepsin D leads to lysosomal dysfunction and promotes cell-to-cell transmission of α-synuclein aggregates. Cell Death Dis 6, e1901 (2015). https://doi.org/10.1038/cddis.2015.283
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2015.283
- Springer Nature Limited
This article is cited by
-
Thioredoxin-1 decreases alpha-synuclein induced by MPTP through promoting autophagy-lysosome pathway
Cell Death Discovery (2024)
-
Disease-, region- and cell type specific diversity of α-synuclein carboxy terminal truncations in synucleinopathies
Acta Neuropathologica Communications (2021)
-
Putative second hit rare genetic variants in families with seemingly GBA-associated Parkinson’s disease
npj Genomic Medicine (2021)
-
Transmission of α-synuclein seeds in neurodegenerative disease: recent developments
Laboratory Investigation (2019)
-
LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation
Nature Communications (2018)