
Abstract
Mycobacterium tuberculosis, the causative agent of tuberculosis, induces apoptosis in infected macrophages in vitro and in vivo. However, the molecular mechanism controlling this process is not known. In order to study the involvement of the mitochondrial apoptotic pathway in M. tuberculosis-induced apoptosis, we analysed cell death in M. tuberculosis-infected embryonic fibroblasts (MEFs) derived from different knockout mice for genes involved in this route. We found that apoptosis induced by M. tuberculosis is abrogated in the absence of Bak and Bax, caspase 9 or the executioner caspases 3 and 7. Notably, we show that MEF deficient in the BH3-only BCL-2-interacting mediator of cell death (Bim) protein were also resistant to this process. The relevance of these results has been confirmed in the mouse macrophage cell line J774, where cell transfection with siRNA targeting Bim impaired apoptosis induced by virulent mycobacteria. Notably, only infection with a virulent strain, but not with attenuated ESX-1-defective strains, such as Bacillus Calmette-Guerin and live-attenuated M. tuberculosis vaccine strain MTBVAC, induced Bim upregulation and apoptosis, probably implicating virulence factor early secreted antigenic target 6-kDa protein in this process. Our results suggest that Bim upregulation and apoptosis is mediated by the p38MAPK-dependent pathway. Our findings show that Bim is a master regulator of apoptosis induced by M. tuberculosis.
Similar content being viewed by others

Main
Mycobacterium tuberculosis, the causative agent of tuberculosis, is primarily an intracellular pathogen that has successfully developed strategies to colonise host alveolar macrophages and overcome their bactericidal defence mechanisms.1
Apoptosis is a physiological type of cell death characterised by the preservation of the plasma membrane integrity, which prevents local inflammatory reactions and tissue damage. Intracellular pathogens have co-evolved with the host to develop strategies for modulation of host cell apoptosis to favour infection.2 During M. tuberculosis infection, presence of apoptotic cells has been detected in lungs from both infected humans and mice.3, 4, 5 ESX-1 secretion system, which regulates early secreted antigenic target 6-kDa protein (ESAT-6) secretion, seems to play a crucial role in apoptosis induction and virulence during mycobacterial infection.3, 6 It has been shown that attenuated strains, like Bacillus Calmette-Guerin (BCG) and the live-attenuated M. tuberculosis vaccine Mycobacterium tuberculosis vaccine strain (MTBVAC),7 which lack a functional ESX-1 secretion system, have lost their ability to induce apoptosis and cell death.3, 8 Altogether, these results suggest that the ability to induce apoptotic cell death is a feature characteristic of virulent strains. Indeed, similarly to other authors, we have shown that apoptosis triggered by virulent mycobacteria is required for bacterial spread.3, 9
The activation of the mitochondrial cell death pathway is regulated by the Bcl-2 family of proteins consisting of pro-apoptotic (Bak, Bax, Bim, Bid and so on) and anti-apoptotic (Bcl-2, Bcl-XL, Mcl-1 and so on) members, whose activity is reciprocally modulated.10 BH3-only pro-apoptotic proteins (i.e., Bid, BCL-2-interacting mediator of cell death (Bim), Puma and Noxa) interfere with anti-apoptotic proteins Bcl-2, Bcl-XL or Mcl-1, and induce Bak and Bax activation by conformational change, leading to mitochondrial permeabilization.11 Pore formation on mitochondrial membrane leads to the release of pro-apoptotic factors to cytosol. One of these molecules, cytochrome c, is necessary to activate caspase 9,12 which activates the effector caspases 3 and 7 by cleavage. These are ultimately responsible for the appearance of the apoptotic phenotype.
The intracellular mediators of apoptosis induced by M. tuberculosis are poorly understood. Previous works have shown that virulent M. tuberculosis strains are able to activate the mitochondrial cell death pathway including cytochrome c release and caspase activation.4, 13 However, the molecular mechanism including the involvement of the Bcl-2 family in this process remains unknown. In this work, we conducted an in-depth analysis of the implication of different pro-apoptotic members of the Bcl-2 family during apoptosis induced by the clinical isolate MT103 in different cell lines. We have identified the BH3-only protein Bim as a key modulator of apoptosis induction and bacterial spread.
Results
M. tuberculosis induces apoptosis through the mitochondrial cell death pathway
It has been previously described that the mitochondrial apoptotic pathway is activated in M. tuberculosis-infected macrophages.14 However, the role of this signalling route has not been studied in detail and the molecular players involved are still elusive. Thus, we used MEF cells from mice knockout for different proteins of the intrinsic cell death pathway in order to dissect the molecular mechanism of apoptosis induced by M. tuberculosis. MEF cells were infected with the virulent M. tuberculosis clinical isolate MT103, and apoptosis was analysed by monitoring phosphatidylserine (PS) translocation and membrane integrity. We analysed apoptosis at day 7 post infection because at this time point we observed the highest rate of apoptotic cells (Supplementary Figure S1). As shown in Figure 1a, wild-type MEF (MEF.wt) cells showed a characteristic apoptotic-like phenotype, staining with Annexin V and maintaining cellular impermeability to 7-actinomycin D (7-AAD). In contrast, MEF deficient for Bax and Bak (MEF.Bak/Bax DKO), caspase 9 (MEF.Casp9−/−), or the executioner caspases 3 and 7 (MEF.Casp3/7 DKO) were profoundly resistant to MT103-induced apoptosis. Single Bak- or Bax-deficient MEF cells were as susceptible to apoptosis as MEF.wt (Figure 1a), indicating that presence of either Bak or Bax is sufficient to activate the mitochondrial cell death pathway during MT103 infection. Results obtained with MEF.Casp9−/− and MEF.Casp3/7 DKO cells confirmed the implication of the mitochondrial apoptotic route. Both cell lines were resistant to apoptosis, indicating that MT103 activates the classical mitochondrial route including the activation of caspase 9 and the executioner caspases 3 and 7. We also noticed a residual cell death of about 25% in all MEF-resistant cell lines, suggesting that MT103 may exert some cytotoxicity in host cells in a mitochondria- and caspases 3/7-independent manner.

MT103 induces apoptosis on MEF by activation of the mitochondrial apoptotic route. Wild-type MEF (WT) and MEF knockouts for Bax, Bak, caspases 3 and 7(C3/7 DKO), caspase 9 (C9), Bak and Bax (Bax/Bak DKO), Bim, Bid were infected with MT103 (MOI 30 : 1) during seven days. (a and b) Cells were stained with annexinV and 7-AAD, and analysed by flow cytometry. Representative dot plots are shown in the right panels. Data in the graphs (left panels) are represented as mean±S.E.M. of four independent experiments. (c) Cells were stained with an anti-active caspase 3 antibody and analysed by flow cytometry. Representative histograms are shown in the upper panel. Black and red lines represent infected-C3/7 DKO MEF and the indicated MEF types, respectively. Data in the graph (lower panel) correspond with the ratio of mean fluorescent intensities (MFI) of the different infected MEF types versus the C3/7 DKO MEF control and are shown as mean±S.E.M. of three independent experiments. (d) Seven days post infection, 150 cells per well were seeded and incubated in fresh medium during eight additional days. Survival was calculated as percentage of colonies relative to the number of colonies in the non-infected controls. Data in the graphs are represented as mean±S.E.M. of two independent experiments. Statistical analysis was done using one-way ANOVA and Bonferroni’s post hoc test comparing with MT103-infected wild-type cells. *P<0.05; **P<0.01; ***P<0.001
Apoptosis induced by MT103 in MEF cells is regulated by the BH3-only protein Bim
We studied the possible role of the BH3-only proteins, Bim and Bid, as activators of the intrinsic route in MT103-infected MEF cells. Bid has been identified as the BH3-only protein that links the extrinsic and the intrinsic apoptotic pathways.15 Bim has being identified to respond to cellular stress stimuli, being a key regulator of apoptosis induced by endoplasmic reticulum (ER) stress response.16 As shown in Figure 1b, Bid-deficient (MEF.Bid−/−) cells underwent apoptosis when infected with MT103, suggesting that this BH3-only protein is not triggering the mitochondrial route in our model. On the contrary, MEF.Bim−/− cells were completely resistant to apoptosis, similar to MEF.Bax/Bak DKO, MEF.Casp9−/− and MEF.Casp3/7 DKO cells. These data indicate that Bim could result important for M. tuberculosis-induced apoptosis, at least in MEF cells.
Next, we analysed caspase 3 activation by flow cytometry. As shown in Figure 1c, active caspase 3 was only detected in MEF.wt and MEF.Bid−/− cells infected with MT103. Fluorescence intensity in MT103-infected MEF.Bax/Bak DKO, MEF.Casp9−/− or MEF.Bim−/− cells was similar to MEF.Casp3/7 DKO-negative controls. This result corroborates that caspase 3 activation occurs downstream of the mitochondria.
Bim deficiency recovers the clonogeneic potential of MEF cells infected with virulent MT103 strain
Results above indicated that M. tuberculosis could not induce cell death in Bax/Bak, Bim, Casp9 or Casp3/7 knockout MEF cells up to 7 days post infection. In order to elucidate whether cell death would take place at later time points, we performed a clonogenic assay. This kind of assays represents a gold standard test in the field of cell death research used to discern if a specific stimulus is able to kill a target cell at long term, and is based on the ability of treated cells to proliferate and to form colonies. Our previous work showed that M. tuberculosis-infected MEF cells lose clonogenic capacity.4 Clonogenic assay studies performed in this work showed that MEF.Bax/Bak DKO, MEF.Casp9−/−, MEF.Bim−/− or MEF.Casp3/7 DKO cells were totally resistant to M. tuberculosis cytotoxic effect, whereas MEF.Bid−/− or single MEF.Bax−/− and MEF.Bak−/− cells were as susceptible as MEF.wt to cell death induced by MT103 strain (Figure 1d).
Bim is upregulated in M. tuberculosis-infected macrophages
Our results strongly point to Bim as a key factor for triggering apoptosis in infected cells. To assess the possible physiological relevance of this finding, we studied the implication of Bim during M. tuberculosis-induced apoptosis in macrophages, the main target cell for this pathogen.
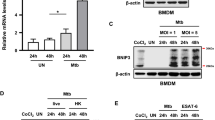
We analysed whether Bim expression was altered by M. tuberculosis infection in the murine macrophage cell line J774. As shown in Figure 2, Bim appeared strongly upregulated in infected cells, both at transcriptional (Figure 2a) and at protein (Figure 2b) levels. Remarkably, the three main Bim isoforms, BimEL, BimL and BimS,17 were augmented in infected cells (Figure 2b).

MT103 upregulates Bim in J774 cells. J774 cells were infected with MT103 (MOI 10 : 1) up to 72 h. Bim expression was analysed by RT-qPCR (a), or by western blot (b) at 24 h, 48 h and 72 h post infection. (a) Data in the graph are represented as mean±S.E.M. of three independent experiments. (b) Quantification of the western blot is represented as the ratio of Bim/Actin densities. A representative blot of three independent is shown in the figure
Bim upregulation in macrophages is induced only by virulent M. tuberculosis and correlates with apoptosis induction
Our and other authors’ works have previously described that ESX-1-deficient strains have lost the ability to induce apoptosis resulting in an attenuated phenotype,3, 9, 18, 19 as confirmed in MEF cells infected with attenuated strains BCG or MTBVAC (Supplementary Figure S2). In the present study, we analysed whether ESX-1-deficient strains failed to upregulate Bim in infected J774 cells. As shown in Figures 3a and b, attenuated MTBVAC presented impaired capacity to induce Bim expression as well as to trigger apoptosis in infected cells. MTBVAC principally failed to induce the shorter Bim isoform, BimS, whereas BimEL and BimL isoforms were upregulated when compared with non-infected controls, although to a lesser extent than in cells infected with MT103. In agreement with these data, capacity of MT103 to kill host cells and to induce Bim was abolished when we infected with heat-killed MT103, comparably as observed with non-virulent live MTBVAC. This result strongly implies that M. tuberculosis has developed active mechanisms to trigger the pro-apoptotic Bim cascade that leads to host cell death.

Bim expression is induced by virulent M. tuberculosis. (a and b) J774 cells were infected with MT103 or the live-attenuated vaccine candidate MTBVAC (MOI 10 : 1) for 72 h. (a) Cells were stained with annexinV and 7-AAD, and analysed by flow cytometry. Data in the graph are represented as mean±S.E.M. of three independent experiments. (b) A representative blot of two independent experiments is shown. Quantification of the western blot is represented as the ratio of Bim/Actin densities. (c and d) J774 cells were infected with live or heat-killed (HK) MT103 or MTBVAC bacteria. (c) Cells were stained with annexinV and 7-AAD, and analysed by flow cytometry. Data represent the mean±S.E.M. of two independent experiments. (d) A representative blot of two independent experiments is shown. Quantification of the western blot is represented as the ratio of Bim/Actin densities
Additionally, direct implication of ESX-1 in this process was confirmed infecting J774 cells with BCG and BCG:RD1 strains. BCG:RD1 contains a functional ESX-1 system, which restores both virulence6 and pro-apoptotic capacity.3 Comparing both strains, we assessed a strong correlation between apoptosis induction and Bim level expression (Supplementary Figure S3). Finally, we studied the involvement of phoP virulence factor in Bim upregulation. As shown in Supplementary Figure S4, phoP-deficient strain SO220 failed to increase Bim expression in infected J774 cells. Remarkably, complementation of SO2 strain with a functional copy of phoP restored the wild-type phenotype.
Bim is involved in M. tuberculosis-induced apoptosis in macrophages
Our results show a correlation between Bim upregulation and apoptosis induction during M. tuberculosis infection of J774 cells, suggesting a role for Bim in this process. To test whether Bim is required for MT103-induced cell death, we transfected J774 cells with four different siRNA specific for Bim, and subsequently infected cells with MT103. First, we tested the capacity of four different siRNAs to downregulate Bim levels (Figure 4a). The different siRNAs showed variable efficiency to modulate Bim expression when compared with control cells, with siRNA4 resulting the least effective of the four. Importantly, MT103-induced apoptosis was significantly prevented by siRNA1, siRNA2 and siRNA3, implicating Bim in the pro-apoptotic mechanism triggered by M. tuberculosis in these cells (Figure 4b). Confirming the specificity of the siRNA approach, siRNA4 did not protect infected cells, which correlated with its limited capacity to downmodulate Bim.

siRNAs targeting Bim impair MT103-induced apoptosis. (a) Bim knockdown induced by specific siRNAs was confirmed by western blot. (b and c) Non-transfected and Bim-specific siRNAs-transfected J774 cells were infected with MT103 (MOI 5 : 1 and 10 : 1) during 72 h. (b) Cells were stained with annexinV and 7-AAD, and analysed by flow cytometry. Data in the graphs from four independent experiments are represented as mean±S.E.M. of the percentage of dead cells. (c) Percentage of GFP-positive cells at 0 and 72 h post infection was determined by flow cytometry. Data are shown as mean±S.E.M. of three independent experiments. Statistical analysis was done using one-way ANOVA and Bonferroni’s post hoc test. *P<0.05; **P<0.01; ***P<0.001
We have previously described that apoptosis inhibition impaired the capacity of M. tuberculosis to spread cell to cell and colonise fresh macrophages.3 Thus, using a GFP-expressing MT103 strain to monitor host cell infection, we observed that infection at 72 h was less efficient in cells transfected with siRNA1, siRNA2 and siRNA3, as compared with non-transfected controls. Importantly, MT103 cell-to-cell spread was not prevented in siRNA4-transfected cells (Figure 4c).
p38MAPK activation mediates M. tuberculosis-induced Bim upregulation
In agreement with previous works,21, 22 we recently demonstrated that chemical inhibition of p38MAPK resulted in abrogation of M. tuberculosis-induced apoptosis.3 Consequently, we hypothesised that p38MAPK could be involved in M. tuberculosis-induced Bim upregulation.
First, we analysed whether p38MAPK was activated in J774 cells infected with MT103 virulent strain. As shown in Figure 5a, MT103 triggered p38MAPK phosphorylation mainly at early time points (4 h) post infection. Upon infection with MTBVAC, p38MAPK activation at 4 h resulted much less pronounced than observed with the virulent mycobacteria. Looking at later time points (72 h) post infection, we detected that only MT103-infected cells showed sustained p38MAPK activation. Next, we confirmed that the specific p38MAPK inhibitor SB203580 impaired M. tuberculosis-induced apoptosis. Correlatively, incubation with the inhibitor reduced notably Bim expression. Data revealed that SB203580 abrogates expression of the three isoforms of Bim, even though this reduction was much more evident in the case of BimS.

Bim induction triggered by MT103 is mediated by p38MAPK. (a) J774 cells were infected with MT103 or the live-attenuated vaccine candidate MTBVAC (MOI 10 : 1) for up to 72 h. A time course of p38MAPK phosphorylation (P-p38) was analysed by western blot at 1, 4 and 72 h post infection. A representative blot of two independent experiments is shown. Quantification of the western blot is represented as the ratio of phospho-p38MAPK/actin densities. (b and c) Cells were infected with MT103 for 72 h in the presence or absence of SB203580 10 μM. (b) Cells were stained with annexinV and 7-AAD, and analysed by flow cytometry. Data are represented as mean±S.E.M. of two independent experiments. (c) A representative blot of two independent experiments is shown. Quantification of the western blot is represented as the ratio of Bim/actin densities
Discussion
The mitochondrion is a widely used target by many pathogens for modulating cell death.23 In the present work, we show that the pro-apoptotic member of the Bcl-2 family, the BH3-only protein Bim, is a key regulator of apoptosis induced by virulent M. tuberculosis clinical isolate MT103. To our knowledge, this is the first report demonstrating that a BH3-only protein is responsible for apoptosis induced by a virulent M. tuberculosis strain. Nevertheless, Bim has a central role in the control of host cell death by other pathogens, such as Neisseria gonorrhoeae, which is able to promote or inhibit apoptosis upregulating or downregulating Bim levels, respectively.24, 25 Chlamydia trachomatis also favours Bim degradation to inhibit host cell death.26 Altogether, these data suggest that Bim modulation could be a general strategy exploited by different types of bacteria.
Resistance to programmed cell death by infected Bak- and Bax-deficient cells indicates pore formation in the extracellular mitochondrial membrane, which would allow release of apoptogenic factors including cytochrome c from the intermembrane space. Caspase-9-knockout MEF cells resistance suggests the likely formation of apoptosome structure. Apoptosis resistance of caspases 3- and 7-knockout cells indicates that, ultimately, cell death depends on activation of the executioner caspases. In the absence of Bim, the apoptotic phenotype is highly impaired, and importantly, most of the infected cells recover their clonogeneic potential indicating that these cells are resistant to any type of cell death. Translating these results to macrophages, the main target cell of M. tuberculosis, we demonstrate that infection with M. tuberculosis significantly increases Bim expression, and more importantly, downmodulation of Bim levels after transfection with specific siRNA markedly abrogates MT103-induced apoptosis. Moreover, Bim downmodulation also reduces cell-to-cell spread of M. tuberculosis indicating that Bim-induced apoptosis could be a principal factor that contributes to colonisation of new host cells during M. tuberculosis infection.
During the preparation of this manuscript, a report was published describing a role of Bim in BCG-triggered apoptosis, in an ERK1/2-dependent process.27 Our data are discrepant with this work as we maintain that mycobacteria-induced apoptosis is a feature exclusive to virulent strains. Using our experimental settings, and in agreement with other works8, we do not find cell death induced by BCG infection, or other attenuated strains, such as MTBVAC vaccine candidate. Nevertheless, we tested whether ERK1/2 could be involved in M. tuberculosis-induced apoptosis and Bim induction, and data shown in Supplementary Figure S5 clearly differ from this hypothesis. J774 incubation with MEK1 inhibitor, U0126, did not prevent MT103-induced apoptosis and importantly, Bim upregulation.
Our data indicate that virulent M. tuberculosis activates the canonical intrinsic apoptotic route to kill the host cell in a process that likely involves the ESX-1 secretion system, as shown by the differences found in this work between BCG and BCG:RD1 strains. However, we cannot discard the implication of other cytotoxic molecules apart from ESX-1, because MTBVAC and SO2 attenuated strains are deficient in phoP, a virulence factor that regulates many other mechanisms apart from the ESAT-6 secretion machinery.
The observation that the absence of Bak and Bax prevents apoptosis and cell death after M. tuberculosis infection is also novel. Recently, a study analysed cell death induced by M. tuberculosis in macrophages from Bak- and Bax-deficient mice and found that loss of mitochondrial membrane potential following infection was independent of Bak and Bax.28 However, these experiments were performed with a high multiplicity of infection (MOI), which accounts for necrosis-like cell death,29 making it difficult to conclude the role of Bak and Bax in M. tuberculosis-induced apoptosis.
Bim activity can be regulated transcriptionally by post-translational modifications, such as phosphorylation,30 or by its interaction with other proteins, including the anti-apoptotic partner Mcl-131 or with DLC1 or DLC2 dynenin light chains, in the case of BimEL and BimL isoforms.32 In this context, a previous work shows that Mcl-1 downregulation leads to an increment of H37Rv-induced apoptosis,33 which could be explained by the release of Bim after Mcl-1 downmodulation. Our data indicate that in J774 macrophages Bim expression is upregulated at both mRNA and protein levels following infection with M. tuberculosis. Moreover, MT103 and MTBVAC differentially induced Bim in infected cells, with the major difference being the induction of BimS predominantly by MT103. Of note, BimS has been described as the most cytotoxic isoform of Bim,34 with a strong capacity to directly translocate Bax to mitochondria.35 BimS activity is mainly regulated by transcriptional activity,36 whereas BimEL and BimL expression does not necessarily lead to apoptosis as they can be sequestered by the cytoskeleton.32
Our data suggest that p38MAPK has a crucial role in Bim-dependent apoptosis following MT103 infection. We detected p38MAPK phosphorylation in MT103-infected cells mainly at early time points post infection, suggesting that this is an early event necessary to trigger the subsequent apoptotic machinery. Of note, MTBVAC transiently induced p38MAPK activation at 4 h post infection, although to a lesser extent than MT103. At longer time points, only MT103 was shown to sustain p38MAPK phosphorylation. Importantly, duration of p38MAPK activation has been suggested to be crucial in the role of this kinase, promoting death or survival. Transient and rapid activation has been associated with survival and proliferation, whereas sustained activation favours cells to undergo apoptosis.37, 38
Two main pathways have been described to control Bim expression: ER stress,16 and the transcription factor FOXO3.39 p38MAPK can modulate both routes. It directly phosphorylates FOXO3, promoting its translocation to the nucleus.40 Similarly, it phosphorylates CHOP, a transcription factor induced during ER stress, enhancing its transcriptional activity.41 It has been previously found that ESAT-6 induces different ER stress markers, including CHOP expression both in vitro and in vivo.5, 13, 42 Moreover, Bim has been reported to be induced in a CHOP-dependent fashion.16 Consequently, we analysed CHOP expression in MT103- and MTBVAC-infected J774 cells and surprisingly CHOP was similarly upregulated by both strains (data not shown), indicating that CHOP induction did not correlate with cell death nor Bim increase in our model. Nevertheless, this result does not discard a role of ER stress signalling in M. tuberculosis-induced apoptosis, as ER stress-induced cell death can occur in a CHOP-independent way.43, 44 Remarkably, a recent paper describes a role of FOXO3 in mycobacteria-induced cell death, which could suggest that this transcription factor might be responsible for Bim induction during mycobacterial infection but more work is needed to elucidate in detail this mechanism.
Our results show that a virulent clinical isolate of M. tuberculosis upregulates the expression of the BH3-only protein Bim, leading to apoptosis in both fibroblast and macrophage cells through activation of the mitochondrial cell death pathway. Moreover, we show that Bim inhibition reduces cell-to-cell spread of M. tuberculosis, suggesting that Bim may be a critical host factor that promotes M. tuberculosis spread and virulence. This finding could be a potential basis for the design of new treatments targeting the molecular regulators of Bim-mediated apoptosis.
Materials and Methods
Cell culture
For this study, we used mouse embryonic fibroblasts (MEFs) immortalised with SV40 virus or using a 3T9 protocol and the mouse bone marrow-derived cell line J774 (Health Protection Agency, Cat No.85011428). All cell lines were cultured in DMEM medium supplemented with 10% inactivated fetal bovine serum (Biological industries, Reactiva, Barcelona, Spain) and 2 mM glutamine (Biological industries) at 37 °C and 5% CO2. MEF used in this study were:
SV40-transformed WT MEF (Health Protection Agency, Cat No.98061101).
SV40-transformed MEF.Caspase 9−/− (MEF.Casp 9−/−) and MEF.Caspases 3/7−/− (MEF.Casp 3/7 DKO), were kindly provided by Richard A Flavell.45
SV40-transformed MEF.Bak−/−, MEF.Bax−/−, MEF.Bid−/−, MEF.Bak/Bax DKO and 3T9-transformed MEF.Bak/Bax DKO were kindly provided by Christoph Borner.
3T9 WT or MEF.Bim−/− cells were kindly provided by Andreas Strasser.46
Bacterial strains and growth conditions
M. bovis BCG Pasteur 1173P2,47 live M. tuberculosis attenuated vaccine MTBVAC,7 M. tuberculosis clinical isolate MT103, its phoP-mutant derivative SO2 (kanamycin resistant) and the phoP-complemented SO2 strain (SO2:phoP)20 were used in this study. MT103 strain was rendered fluorescent by the transfer of plasmid pMV361H gfp (green fluorescent protein).48 Mycobacteria were grown at 37 °C in Middlebrook 7H9 broth (BD Biosciences, Madrid, Spain) supplemented with 0.05% Tween 80 and 10% Middlebrook albumin dextrose catalase enrichment (ADC; BD Biosciences), and when required the medium was supplemented with 20 μg/ml of kanamycin or hygromycin. The virulence and quality of our stocks are always confirmed in vivo in mice. All virulent stocks used to perform these experiments showed high virulence in vivo.
Cell culture and infections
MEF or J774 cells were cultured at 37 °C and 5% CO2 in DMEM medium supplemented with 10% inactivated fetal bovine serum (Biological industries) and 2 mM glutamine (Biological industries). Cells were seeded in 24-plate wells and allowed to attach to the plastic overnight. After clumps removal by low-speed centrifugation of a log-phase culture, bacterial concentration was determined by optical density. For some experiments, bacteria were inactivated by heat treatment for 20 min at 100 °C. Bacterial suspension for indicated MOIs was prepared in DMEM complete medium and put in contact with cells for 4 h. Afterward, cells were washed three times with PBS to remove extracellular bacteria and fresh DMEM complete medium was added. SB203580 or U0126 (Calbiochem, Madrid, Spain) inhibitors were added when corresponds at the indicated concentrations.
GeneSolution siRNA (QIAGEN, IZASA, Barcelona, Spain) was used to knockdown Bim expression in J774 cells. siRNAS were transfected with HiPerfect Transfection Reagent (QIAGEN), using the protocol supplied by the manufacturer for J774 cells transfection. siRNA1: 5′-TCCGCTTATTTAAATGTCTTA-3′; siRNA2: 5′-TGGGTAGGCCTTTGTACTTAA-3′; siRNA3: 5′-CAAGTTGTAATAAACATACAA-3′; siRNA4: 5′-CACCCTCAAATGGTTATCTTA-3′.
Apoptosis analysis in vitro
PS exposure and plasma membrane integrity were evaluated by AnnexinV-APC (AnnV) and 7-AAD (BD Biosciences) staining according to the manufacturer’s instructions, and analysed by flow cytometry. Briefly, cells were washed and incubated with AnnV and 7-AAD in Annexin-binding buffer (ABB) for 15 min in dark at room temperature. Afterward, cells were washed with ABB and fixed with 4% paraformaldehyde (PFA) containing CaCl2.
Caspase 3 activation was analysed by flow cytometry using an antibody against active form of caspase 3 (BD Biosciences), as previously described.49
Clonogenic assay
MEF cells were seeded in 24-well plates at approximately 5 × 103 cells per well and infected with the MT103 at MOI of 30 : 1. Seven days post infection, cells were trypsinized and 150 cells per well were seeded in a final volume of 3 ml in a 6-well plate. Cells were then allowed to grow during eight additional days at 37 °C, following which the medium was removed and cell colonies were counted following 20-min fixation with a mixture of glutaraldehyde (6.0% v/v) and crystal violet (0.5% w/v) at room temperature.
Analysis of Bim expression
RNA from control and infected cells was extracted with the QIAGEN RNAeasy kit (QIAGEN), according to the manufacturer’s instructions. Bim expression was analysed by RT-qPCR using the following primers: forward 5′-TGATTACCGCGAGGCTGAA-3′ and reverse 5′-ACCAGACGGAAGATAAAGCGTAAC-3′. Primers for b-actin expression analysis were forward 5′-TGTTACCAACTGGGACGACA-3′ and reverse 5′-CTGGGTCATCTTTTCACGGT-3′.
Cell homogenates were obtained lysing the cells with a buffer containing 0.1% Triton X-100 and protease and phosphatase inhibitors (Roche, Madrid, Spain). Protein concentration was determined by the Bradford method (BioRad, Madrid, Spain) and 10 μg of total protein was loaded in a 15% polyacrylamide gel, separated by SDS-PAGE and transferred to PVDF membrane (GE Healthcare, Madrid, Spain). Membranes were incubated with anti-β-actin (Sigma, Madrid, Spain), anti-Bim (Cell Signalling, IZASA), anti-CHOP (Cell Signalling) or anti-phospho (Thr180/Tyr182)-p38MAPK (Cell Signalling) primary antibodies according to dilutions indicated by the manufacturer. After corresponding secondary antibodies incubation, membranes were revealed using ECL plus Western Blotting system (GE Healthcare). Western blot quantification was performed by densitometry analysis using the ImageJ64 software.
Abbreviations
- MEF:
-
mouse embryonic fibroblast
- Wt:
-
wild type
- DKO:
-
double knockout
- MTBVAC:
-
Mycobacterium tuberculosis vaccine strain
- ESAT-6:
-
early secreted antigenic target 6-kDa protein
- 7-AAD:
-
7-actinomycin D
- annV:
-
annexin V
- PS:
-
phosphatidylserine
- Bim:
-
BCL-2-interacting mediator of cell death
- ER:
-
endoplasmic reticulum
- BCG:
-
Bacillus Calmette-Guerin
References
Pieters J . Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe 2008; 3: 399–407.
Finlay BB, McFadden G . Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 2006; 124: 767–782.
Aguilo JI, Alonso H, Uranga S, Marinova D, Arbues A, de Martino A et al. ESX-1-induced apoptosis is involved in cell-to-cell spread of Mycobacterium tuberculosis. Cell Microbiol 2013; 15: 1994–2005.
Aporta A, Arbues A, Aguilo JI, Monzon M, Badiola JJ, de Martino A et al. Attenuated Mycobacterium tuberculosis SO2 vaccine candidate is unable to induce cell death. PLoS One 2012; 7: e45213.
Seimon TA, Kim MJ, Blumenthal A, Koo J, Ehrt S, Wainwright H et al. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS One 2010; 5: e12772.
Pym AS, Brodin P, Brosch R, Huerre M, Cole ST . Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 2002; 46: 709–717.
Arbues A, Aguilo JI, Gonzalo-Asensio J, Marinova D, Uranga S, Puentes E et al. Construction, characterization and preclinical evaluation of MTBVAC, the first live-attenuated M. tuberculosis-based vaccine to enter clinical trials. Vaccine 2013; 31: 4867–4873.
Schaible UE, Winau F, Sieling PA, Fischer K, Collins HL, Hagens K et al. Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat Med 2003; 9: 1039–1046.
Davis JM, Ramakrishnan L . The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 2009; 136: 37–49.
Cory S, Adams JM . The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002; 2: 647–656.
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997; 91: 479–489.
Lim YJ, Choi JA, Choi HH, Cho SN, Kim HJ, Jo EK et al. Endoplasmic reticulum stress pathway-mediated apoptosis in macrophages contributes to the survival of Mycobacterium tuberculosis. PLoS One 2011; 6: e28531.
Danelishvili L, McGarvey J, Li YJ, Bermudez LE . Mycobacterium tuberculosis infection causes different levels of apoptosis and necrosis in human macrophages and alveolar epithelial cells. Cell Microbiol 2003; 5: 649–660.
Li H, Zhu H, Xu CJ, Yuan J . Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998; 94: 491–501.
Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007; 129: 1337–1349.
O'Reilly LA, Cullen L, Visvader J, Lindeman GJ, Print C, Bath ML et al. The proapoptotic BH3-only protein bim is expressed in hematopoietic, epithelial, neuronal, and germ cells. Am J Pathol 2000; 157: 449–461.
Derrick SC, Morris SL . The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol 2007; 9: 1547–1555.
Grover A, Izzo AA . BAT3 regulates Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis of macrophages. PLoS One 2012; 7: e40836.
Perez E, Samper S, Bordas Y, Guilhot C, Gicquel B, Martin C . An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol 2001; 41: 179–187.
Aleman M, Schierloh P, de la Barrera SS, Musella RM, Saab MA, Baldini M et al. Mycobacterium tuberculosis triggers apoptosis in peripheral neutrophils involving toll-like receptor 2 and p38 mitogen protein kinase in tuberculosis patients. Infect Immun 2004; 72: 5150–5158.
Kundu M, Pathak SK, Kumawat K, Basu S, Chatterjee G, Pathak S et al. A TNF- and c-Cbl-dependent FLIP(S)-degradation pathway and its function in Mycobacterium tuberculosis-induced macrophage apoptosis. Nat Immunol 2009; 10: 918–926.
Rudel T, Kepp O, Kozjak-Pavlovic V . Interactions between bacterial pathogens and mitochondrial cell death pathways. Nat Rev Microbiol 2010; 8: 693–705.
Howie HL, Shiflett SL, So M . Extracellular signal-regulated kinase activation by Neisseria gonorrhoeae downregulates epithelial cell proapoptotic proteins Bad and Bim. Infect Immun 2008; 76: 2715–2721.
Kepp O, Gottschalk K, Churin Y, Rajalingam K, Brinkmann V, Machuy N et al. Bim and Bmf synergize to induce apoptosis in Neisseria gonorrhoeae infection. PLoS Pathog 2009; 5: e1000348.
Dong F, Pirbhai M, Xiao Y, Zhong Y, Wu Y, Zhong G . Degradation of the proapoptotic proteins Bik, Puma, and Bim with Bcl-2 domain 3 homology in Chlamydia trachomatis-infected cells. Infect Immun 2005; 73: 1861–1864.
Iyoda T, Takada M, Fukatsu Y, Kumokoshi S, Fujisawa T, Shimada T et al. A novel mechanism underlying the basic defensive response of macrophages against Mycobacterium infection. J Immunol 2014; 192: 4254–4262.
Lee J, Repasy T, Papavinasasundaram K, Sassetti C, Kornfeld H . Mycobacterium tuberculosis induces an atypical cell death mode to escape from infected macrophages. PLoS One 2011; 6: e18367.
Lee J, Remold HG, Ieong MH, Kornfeld H . Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J Immunol 2006; 176: 4267–4274.
Puthalakath H, Strasser A . Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ 2002; 9: 505–512.
Hsu SY, Lin P, Hsueh AJ . BOD (Bcl-2-related ovarian death gene) is an ovarian BH3 domain-containing proapoptotic Bcl-2 protein capable of dimerization with diverse antiapoptotic Bcl-2 members. Mol Endocrinol 1998; 12: 1432–1440.
Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A . The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol Cell 1999; 3: 287–296.
Sly LM, Hingley-Wilson SM, Reiner NE, McMaster WR . Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J Immunol 2003; 170: 430–437.
O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J 1998; 17: 384–395.
Weber A, Paschen SA, Heger K, Wilfling F, Frankenberg T, Bauerschmitt H et al. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2 proteins. J Cell Biol 2007; 177: 625–636.
Ley R, Ewings KE, Hadfield K, Cook SJ . Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ 2005; 12: 1008–1014.
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME . Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995; 270: 1326–1331.
Frasch SC, Nick JA, Fadok VA, Bratton DL, Worthen GS, Henson PM . p38 mitogen-activated protein kinase-dependent and -independent intracellular signal transduction pathways leading to apoptosis in human neutrophils. J Biol Chem 1998; 273: 8389–8397.
Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol Cell Biol 2000; 20: 9138–9148.
Ho KK, McGuire VA, Koo CY, Muir KW, de Olano N, Maifoshie E et al. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J Biol Chem 2012; 287: 1545–1555.
Wang XZ, Ron D . Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science 1996; 272: 1347–1349.
Choi HH, Shin DM, Kang G, Kim KH, Park JB, Hur GM et al. Endoplasmic reticulum stress response is involved in Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis. FEBS Lett 2010; 584: 2445–2454.
Hiramatsu N, Messah C, Han J, Lavail MM, Kaufman RJ, Lin JH . Translational and posttranslational regulation of XIAP by eIF2alpha and ATF4 promotes ER stress-induced cell death during the unfolded protein response. Mol Biol Cell 2014; 25: 1411–1420.
Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005; 307: 935–939.
Lakhani SA, Masud A, Kuida K, Porter Jr GA, Booth CJ, Mehal WZ et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 2006; 311: 847–851.
Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 1999; 286: 1735–1738.
Brosch R, Gordon SV, Garnier T, Eiglmeier K, Frigui W, Valenti P et al. Genome plasticity of BCG and impact on vaccine efficacy. Proc Natl Acad Sci USA 2007; 104: 5596–5601.
Astarie-Dequeker C, Le Guyader L, Malaga W, Seaphanh FK, Chalut C, Lopez A et al. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog 2009; 5: e1000289.
Pardo J, Wallich R, Martin P, Urban C, Rongvaux A, Flavell RA et al. Granzyme B-induced cell death exerted by ex vivo CTL: discriminating requirements for cell death and some of its signs. Cell Death Differ 2008; 15: 567–579.
Acknowledgements
This work was supported by grant BIO2011-23555 and SAF2011-25390 from Spanish Ministry of Economy and Competitiveness, European FP7 grant NEWTBVAC 241745 and Gobierno de Aragón/Fondo Social Europeo. JP was supported by Aragón I+D (ARAID). BCG-RD1 strain was kindly provided by Roland Brosch (Institut Pasteur, Paris, France). We acknowledge the use of the Scientific and Technical Services at CIBA (Instituto Aragonés de Ciencias de la Salud–SAI, Universidad de Zaragoza, Spain).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Munoz-Pinedo
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Aguiló, N., Uranga, S., Marinova, D. et al. Bim is a crucial regulator of apoptosis induced by Mycobacterium tuberculosis . Cell Death Dis 5, e1343 (2014). https://doi.org/10.1038/cddis.2014.313
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2014.313
- Springer Nature Limited
This article is cited by
-
C-terminal region of Rv1039c (PPE15) protein of Mycobacterium tuberculosis targets host mitochondria to induce macrophage apoptosis
Apoptosis (2024)
-
Apoptosis and its pathways as targets for intracellular pathogens to persist in cells
Parasitology Research (2024)
-
Absence of Bim sensitizes mice to experimental Trypanosoma cruzi infection
Cell Death & Disease (2021)
-
New insights into the evasion of host innate immunity by Mycobacterium tuberculosis
Cellular & Molecular Immunology (2020)
-
Corticosteroids inhibit Mycobacterium tuberculosis-induced necrotic host cell death by abrogating mitochondrial membrane permeability transition
Nature Communications (2019)