
Abstract
The treatment of HIV presents significant challenges due to the low solubility and bioavailability of antiretroviral drugs. These limitations impact the absorption of the drugs in the gastrointestinal tract, leading to reduced effectiveness. Patients often exhibit varying responses due to inconsistent levels of the drug in their bloodstream. Achieving desired plasma concentrations is challenging and often requires higher doses that increase the risk of side effects. Developing drugs with low solubility is a complex and costly process, slowing down overall drug development. Traditional formulation strategies have proven ineffective in addressing these issues, necessitating exploration of innovative techniques. Nanocrystallization has emerged as a transformative approach that enhances solubility and dissolution rates by reducing drug particles to the nanometer scale. This increased surface area improves drug solubility and bioavailability. Nanocrystallized drugs demonstrate a faster onset of action, higher drug load, and enhanced safety and efficiency, offering a potential solution to the challenges posed by the poor water solubility of many antiretroviral drugs. This article delves into nanocrystal formulations of HIV drugs and examines the effectiveness of commonly used stabilizers and surfactants in nanocrystal production. The aim is to determine whether the popularization of nanocrystallization could potentially overcome the prevalent issues of low solubility and bioavailability in existing antiretroviral therapies.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The human immunodeficiency virus (HIV) belongs to the Lentivirus genus in the Retroviridae family. There are two strains of HIV: HIV-1 and HIV-2. HIV-1 is the globally spread type of HIV and is a significant public health issue [1]. HIV infection is transmitted through the exchange of bodily fluids. This can occur through blood transfusion, organ transplantation, physical contact, or from an infected parent to their children [2]. No single prevention or treatment method will be sufficient to control the HIV epidemic. However, there is a growing number of treatments that have shown promise in partially preventing the acquisition and transmission of HIV [3]. Some measures to combat HIV include the use of antiretroviral drugs by infected individuals, raising awareness about viral load levels, practicing safe behaviors such as condom use and male circumcision, and treating curable sexually transmitted diseases [4]. HIV infects the CD4 cells of the host, which play a crucial role in the immune system’s defense against infection. HIV-1 relies on the host’s CD4 cells to reproduce and sustain its existence; without them, it cannot replicate independently. Reverse transcriptase, integrase, and protease are the three essential enzymes of HIV. Without these enzymes, the virus is unable to infect CD4 cells [5]. The HIV genome consists of three structural genes—gag, pol, and env—as well as six regulatory genes, which include tat, rev, nef, vif, vpr, and vpu [6].
As shown in the Fig. 1, the HIV life cycle can be divided into several main stages after the virus enters the host’s body. These stages are as follows:
-
1.
Binding and fusion: During these initial steps, HIV targets and enters the host’s CD4 cells.
-
2.
Reverse transcription: This stage involves the conversion of the viral RNA into DNA.
-
3.
Integration: The newly formed viral DNA is integrated into the host cell’s DNA.
-
4.
Replication: In this stage, the virus reproduces its genetic material.
-
5.
Assembly and budding: During the final two stages, the virus reassembles and matures before being released from CD4 cells into the bloodstream, allowing it to infect new cells [7].

HIV Life Cycle and Drug Targets
The treatment of HIV with medication is known as antiretroviral therapy (ART). These drugs work by inhibiting the multiplication of the virus, and they are categorized into six groups based on the specific stages of the virus's life cycle that they target [8].
As listed in Table 1, HIV drugs can be divided in to six group. The following description provides separate information on each groups of HIV drugs.
-
Nunucleoside Reverse Transcriptase Inhibitors (NRTIs):
NRTIs were the first group of drugs that were developed for the treatment of HIV and approved by the FDA. During the process of reverse transcription, RNA, the genetic material of HIV, is converted into DNA after entering the host's body. By inhibiting the reverse transcriptase enzyme, NRTIs stop the reverse transcription process and prevent the replication of HIV.[9] NRTIs are phosphorylated and activated by cellular kinases once they enter the host cell. Because NRTIs lack a 3-hydroxyl group in the 2-deoxyribosyl part, they inhibit the formation of 3ʹ-5ʹ-phosphodiester bonds in the DNA chain, ultimately preventing DNA replication [10].
-
Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs):
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) bind to a region away from the active site of the HIV-1 reverse transcriptase enzyme. They cause a structural change that prevents the substrate from binding. However, it is important to note that HIV-2 is often resistant to NNRTIs [11]. The main limitation of NNRTIs is their susceptibility to high resistance within this class of drugs. A single mutation in the binding site of NNRTIs is enough to confer high-level resistance to first-generation NNRTIs. To overcome this limitation, NNRTIs should be used in combination with at least two other antiretroviral drugs [12].
-
Integrase Strand Transfer Inhibitor (INSTIs):
The integrase enzyme plays a crucial role in the replication of HIV-1. It facilitates the integration of the virus’s cDNA into the DNA of the host CD4 cell. Integrase inhibitors, on the other hand, prevent the virus from integrating into the host cell’s DNA. As a result, the virus is unable to replicate [13]. The first-generation drugs in this category include Raltegravir (RAL) and elvitegravir (EVG], which were approved in 2007 and 2012, respectively. These drugs were effective in controlling HIV, but they have a relatively low genetic barrier. As a result, if other active antiviral drugs are not used alongside them, widespread cross-resistance can occur. Due to the emergence of viruses that are resistant to the first-generation INSTIs drugs, research and development of second-generation drugs began [14, 15].
Second-generation drugs, such as dolutegravir (DTG), bictegravir (BIC), and cabotegravir (CAB), are better tolerated and safer compared to the first generation [6].
-
Protease inhibitors (PIs):
HIV protease inhibitors are crucial compounds in combined antiretroviral treatments because they exhibit relatively low levels of drug resistance. In the life cycle of HIV, the role of protease is vital in the maturation process of the virus, as it prevents the virus from multiplying. The HIV genome contains the precursors of gag and gag-pol polyproteins. HIV1 protease cleaves these precursors at nine processing sites to separate the gag and gag-pol polyproteins and generate active proteins. Under normal circumstances, the substrate needs to access the enzyme’s active site by opening the two beta-hairpin flaps that cover it. However, by obstructing these active sites, protease activity can be inhibited [16].
-
Entry inhibitors:
-
Fusion inhibitors: Glycoprotein Gp41 is responsible for the physical fusion of the virus and CD4 cells, enabling HIV to enter the host cell. By binding to the glycoprotein gp41 on the virus envelope, fusion inhibitors prevent the conformational changes required for the fusion of the virus and the cell membrane. This effectively stops HIV from entering the host’s CD4 cells [3].
-
CCR5 antagonists: Among the various receptors found on the surface of CD4 cells, the CCR5 receptor plays a critical role in facilitating the entry of HIV into host cells. CCR5 antagonists work by binding to this receptor, effectively blocking it. By doing so, these antagonists prevent the virus from establishing a strong connection with the target cell, ultimately halting the replication process [17].
-
Attachment inhibitors: GP41 and GP120 are two crucial glycoproteins found on the surface of the virus. CCR5 and CXCR4, on the other hand, are two significant receptors located on the CD4 surface of the host cell. These receptors play a vital role in facilitating the entry of the virus into the host cell. Initially, the interaction between gp120 on the virus's surface and the CD4 cell receptor takes place. Following this connection, structural changes occur on the surface of the CD4 cell, exposing the CCR5 and CXCR4 chemical receptors. Attachment inhibitors refer to a group of drugs that hinder the binding between gp120 and the CD4 cell receptors, thereby preventing the HIV virus from entering the host cell [18].
-
Post-attachment inhibitors: Ibalizumab is the only drug in this category that has been approved by the FDA. It is a humanized IgG4 monoclonal antibody that works by binding to the second (C2) domain of CD4 cells, reducing their flexibility. This binding prevents gp120 from accessing the CCR5 and CXCR4 receptors [19].
-
-
In some cases, people use more than one drug.
-
Pharmacokinetic enhancers: The body’s Cytochrome P450 (CYP) 3A4 enzymes are responsible for breaking down certain drugs and molecules, which prevents these drugs from remaining in the body for extended periods of time. Pharmacokinetic enhancers can inhibit these cytochrome P450 enzymes, increasing the effectiveness and longevity of certain drugs. Two pharmacokinetic enhancers used in HIV treatment are Cobicistat and Ritonavir. Unlike Ritonavir, Cobicistat does not have antiviral properties. Instead, it is a powerful inhibitor of cytochrome P450 3A enzymes, including the important subtype CYP3A4 [20].
-
Combining different antiretroviral medications is a widely used strategy for effectively managing HIV/AIDS. Studies have demonstrated that utilizing multiple drugs from different drug groups is more successful in achieving virus suppression during HIV treatment. This treatment approach, involving the use of a combination of antiretroviral therapies, is referred to as cART [21].
-
The usual HIV treatment for an individual typically involves three HIV drugs from a minimum of two distinct HIV drug categories [25]. Combination antiretroviral therapy (cART) has greatly reduced AIDS-related mortality worldwide over the past 30 years. While these medications are generally well-tolerated, they do have limitations due to factors such as bioavailability, permeability, and a short half-life [2].
Therefore, there is an urgent requirement for novel antiviral categories that possess innovative chemical scaffolds (chemotypes) or mechanisms of action. Additionally, alternative strategies are needed to address these issues effectively [26]. Nanotechnology is an emerging, multidisciplinary field that is revolutionizing the field of medicine in the twenty-first century [27]. The primary concept behind nanotechnology is to alter the way a molecule behaves in the body, with the aim of effectively eradicating HIV [17]. Nanocrystallization is employed to increase the effectiveness of drugs that have limited solubility, particularly those used in HIV treatment [16]. The absorption, distribution, metabolism, and excretion of an anti-HIV drug in nanocrystal form are influenced by the physicochemical properties of the nanosystem. These properties include the surface-exposed molecules, electrical charge, and size of the nanocrystals [28]. Poorly water-soluble drugs, such as certain antiretroviral drugs used in HIV treatment, often face challenges related to low bioavailability. Nanocrystals, which have a smaller particle size, have greater solubility due to their increased surface area. This means they dissolve more easily, leading to improved drug absorption and bioavailability [29]. Nanocrystallized drugs demonstrate faster onset of action, higher drug load, and enhanced safety and efficiency, offering a potential solution to the challenges posed by poor water solubility of many antiretroviral drugs [30]. Nanocrystals can also be engineered to release the drug gradually over time, creating a sustained release profile. This ensures consistent therapeutic levels in the body and reduces the need for frequent dosing. In the case of HIV drugs, maintaining consistent drug retention and concentrations is crucial for effective viral suppression [31]. Nanocrystals can even be incorporated into targeted drug delivery systems. By activating the nanocrystals with specific ligands or antibodies, they can selectively deliver the drug to HIV-infected cells. This targeted approach minimizes side effects and increases drug efficacy [32]. Additionally, nanocrystals are more resistant to degradation and aggregation, providing a longer shelf life for HIV drugs. This reduces wastage and ensures a reliable supply [33]. Nanocrystals can be formulated in various forms, including oral suspensions, injectables, and topical creams. This flexibility allows for customization based on patient preferences and specific treatment needs [34]. Some HIV drugs have bioavailability that depends on whether they are taken with or without food. However, nanocrystals can mitigate this issue by continuously releasing the drug regardless of food intake. In summary, nanocrystallization shows promise for increasing the efficacy, stability, and patient compliance of HIV medications [35, 36].
1.1 Cocrystallization
Cocrystallization is a technique aimed at enhancing the solubility and bioavailability of drugs. Unlike nanocrystallization, which reduces drug particles to the nanometer scale, cocrystallization involves forming crystalline structures composed of the drug and a co-former. This co-former, typically a small molecule, interacts with the drug through non-covalent bonds, creating a stable crystalline lattice [37]. Cocrystals can significantly improve the physical and chemical properties of drugs, such as solubility, stability, and mechanical strength. By forming a cocrystal, the dissolution rate of a poorly soluble drug can be greatly enhanced, leading to better absorption and bioavailability. This approach is particularly beneficial for drugs that are not amenable to other solubility enhancement techniques [38]. Despite its promise, cocrystallization faces challenges such as the selection of suitable coformers, scalability, stability issues, regulatory hurdles, and thorough characterization. Nonetheless, it remains a valuable strategy for drug development due to its potential to improve the solubility, bioavailability, and stability of active pharmaceutical ingredients (APIs) [39].
1.2 The advantages of nanocrystallization over cocrystallization
Nanocrystallization and cocrystallization each have their own advantages, but nanocrystallization can be considered better in certain contexts. For instance, nanocrystals can be used for a wide range of drugs, including those that are not suitable for cocrystallization due to the lack of suitable coformers. Additionally, nanocrystallization techniques, such as wet milling and antisolvent precipitation, have been well-established and can be scaled up relatively easily for industrial production. Nanocrystals can also be stabilized using various surfactants and polymers, which helps in maintaining their stability during storage and handling. However, it’s important to note that the choice between nanocrystallization and cocrystallization depends on the specific drug and its properties. Both techniques have their own set of challenges and benefits, and the best approach is often determined on a case-by-case basis [40, 41].
Several industrially relevant technologies have emerged for the preparation of nanocrystals. We will discuss these technologies below.
1.3 Methods for producing nanocrystals
In the production of medicinal nanocrystals, the solid particles created can be either amorphous or crystalline, depending on the method used to prepare the nanocrystals. Amorphous structures have a faster dissolution rate. Therefore, if these structures are formed during production, the time required for dissolution will be reduced, resulting in a faster-acting drug [42].
Top-down and bottom-up methods are based on the raw material used. The Table 2 summarizes these methods. In top-down methods, larger solid particles are decomposed to obtain the raw material, and mechanical processes are primarily responsible for reducing particle size. High-pressure homogenization and pearl milling or bead milling are some examples of these methods [43].
Bottom-up techniques have emerged as a response to the limitations of top-down methods. One such limitation is the instability of the final product caused by shearing [44].
However, the use of seeds for abrasion in this technique is time-consuming and can potentially lead to an increased risk of microbial contamination in the final product [42].
In bottom-up techniques, nanocrystals are created from molecules using methods such as the deposition method. In this technique, nucleation and precipitation occur in a supersaturated solution through the addition or removal of solvent, depending on the execution method. The particles formed in the final formulation of this method have the potential to grow and cause issues in pharmaceutical products by increasing in size. To prevent this phenomenon, stabilizers can be added to the formulation [42, 45].
A combination of bottom-up and top-down techniques can be used to achieve smaller particles with a more uniform size distribution. However, this approach is only employed when necessary due to the significant increase in costs [43, 44].
2 Top-down technique
The top-down method involves subjecting large drug particles to high pressure, resulting in their reduction to nanosized. This method has the advantage of being simple and applicable to almost all high-volume drugs. However, it also has some disadvantages, such as pollution and impurities caused by shear force, as well as non-uniformity of particle size and chemical composition. The top-down mechanisms involved in the preparation of nanocrystals will be discussed below [47]
The summery is depicted in Fig. 2

Top-down techniques
2.1 Bead milling
This method involves physically grinding drug microparticles with special devices in a milling environment to obtain drug nanoparticles. The particles are subjected to mechanical pressure and shear forces, which cause them to break down into smaller nanoparticles. The milling environment is composed of glass, zirconium oxide, and polyester resin, which facilitate the movement of the mill. To prepare the nanoparticles, the drug is loaded into the chamber along with a suitable stabilizer and either water or a compatible buffer. The milling process is conducted at a constant temperature, typically taking around 30–60 min to produce nanoparticles smaller than 200 nm. This method is particularly effective for drugs that are insoluble in both water and organic solvents. However, one significant drawback is the erosion of grains, which can lead to contamination in the final formulation. To address this issue, network polystyrene resin, which is more resistant to wear and tear, can be utilized. Additionally, excessive milling time may promote microbial growth, further compromising the quality of the final product [48].
2.2 High-pressure homogenization
This technique plays a crucial role in the preparation of nanoparticles with desired properties. High-pressure homogenization (HPH) is a process in which a fluid, such as a drug suspension, is subjected to high pressure through a homogenizing nozzle with a very narrow gap. It utilizes a high-pressure pump and a valve with a diameter of 25 µm. The drug suspension is passed through a cylindrical chamber with a diameter of 3 mm, specifically through the mentioned valve. During homogenization, the drug microparticles are crushed by high shear forces, resulting in intense particle impact and the formation of drug nanoparticles. The particle size is influenced by the pressure of the homogenizer (150–100 bar) and the number of cycles in which the drug suspension is homogenized (3–10 cycles). This method allows for the preparation of nanoparticles with a narrow size distribution. Moreover, it is suitable for drugs that can dissolve in both organic solvents and water. However, it is necessary to first micronize the particles before preparing them with a high-pressure homogenizer [49].
3 Bottom-up technique
The summery is depicted in Fig. 3

Bottom-up techniques
3.1 Solvent-anti-solvent precipitation
In this method, a drug solution in an organic solvent that can mix well with water is thoroughly mixed with an anti-solvent, which can be either water or another aqueous phase. This process forms a supersaturated solution in the aqueous phase, resulting in nucleation and precipitation of the drug molecules. This ultimately leads to the production of medicinal nanocrystals. The uniformity and stability of the nanocrystal product depend on factors such as the type and ratio of solvent and anti-solvent used, the conditions of supersaturation, temperature, and the speed of addition and mixing [50, 51].
3.2 Supercritical fluid technology
Supercritical fluids, which possess properties of both gases and liquids, are not only cost-effective but also environmentally friendly. They allow for precise control of the crystallization process and the production of fine particles with the desired morphology and particle size distribution. Among the various supercritical fluids, CO2 is the most commonly used and popular one. Its critical parameters (TC: 31 °C/PC: 78 bar) can be easily obtained in an industrial device. Different supercritical techniques, such as RESS (Rapid Expansion of Supercritical Solutions], RESOLVE (Rapid Expansion of a Supercritical Solution into a Liquid Solvent), SAS (Supercritical Antisolvent), PGSS (Particles from Gas Saturated Solutions), and DELOS (a process involving depressurization of an expanded liquid organic solution, facilitating the formation of fine particles with controlled properties), are available. For instance, the RESS technique involves introducing high-temperature and high-pressure liquid CO2 to achieve a supercritical fluid state. The supercritical fluid is mixed with the solvent in an autoclave. Next, the mixture is depressurized from high pressure to atmospheric pressure using a nozzle. This sudden pressure decrease induces nucleation by reducing the solvent’s ability to dissolve. As gaseous CO2 comes into contact with the surrounding conditions, solutes form and gather in a reservoir. The CO2 is then released from the chamber through a valve, purified, and ultimately recycled [52, 53].
3.3 Solvent evaporation and spray drying
Spray drying is a widely used method for producing nanocrystals. It involves converting liquid content (a drug dissolved in a solvent) into dry particles by spraying it into a hot drying chamber. The hot air in the chamber causes the liquid droplets to evaporate, leaving behind dried particles in the surrounding environment. There are two main techniques used in spraying: spray drying and spray freezing. In spray drying, the primary action is evaporation, while in spray freezing, the liquid droplets undergo a phase change from liquid to solid. The processes are similar, except for the direction of energy flow. In spray drying, energy is applied to the droplets, leading to solvent evaporation and both energy and mass transfer. In spray freezing, energy is removed from the droplets, causing them to freeze. The spray drying process consists of five main steps: concentration, atomization, droplet contact with air, drying of the droplets, and separation. Variables such as flow rate, atomizer diameter, and equipment design can affect the shape and size of the resulting particles [54].
4 Combination of bottom-up and top-down techniques
The Combination technique utilizes both bottom-up and top-down processes, effectively addressing the limitations associated with each method individually, such as time-consuming processes or equipment requirements. Initially, nanocrystals are formed using a bottom-up approach, followed by further refinement using a top-down technique like high-pressure homogenization or ultrasound. Combining these methods offers the advantage of producing nanocrystals on a smaller and more accessible scale. However, it is important to note that this approach comes with the drawback of increased costs. Consequently, the combined method is typically reserved for situations where it is absolutely necessary [43, 55].
4.1 Aggregation kinetics and colloidal stability
The development of drug nanocrystals requires careful consideration of aggregation kinetics and colloidal stability, especially for applications such as HIV treatment where stability and efficacy are crucial. Aggregation can change pharmacokinetics and decrease drug bioavailability. For this reason, maintaining medication efficacy requires an understanding of and control over its kinetics. A number of variables, including temperature, ionic strength, pH, and the presence of stabilizers, can affect how easily nanocrystals agglomerate. Aggregation kinetics are studied using methods like analytical ultracentrifugation (AUC), nanoparticle tracking analysis (NTA), and dynamic light scattering (DLS). These techniques shed light on how quickly and how much a particle aggregates over time. The ability of nanocrystals to withstand aggregation and hold onto their dispersed state over time and in a variety of environments is referred to as colloidal stability. Several techniques are used to improve colloidal stability, such as using polymers to modify the surface (e.g. g. PEGylation), the application of agents that stabilize (e.g. g. polymers, surfactants), and formulation parameter control [56, 57].
4.2 Key factors in nanocrystal formation and stability
Essential factors that directly impact the formation, stability, and performance of nanocrystals include various driving parameters. The development of drug/nanomaterial nanocrystals is primarily driven by the need to enhance the solubility and bioavailability of poorly water-soluble drugs. One key parameter influencing this process is particle size reduction to the nanometer scale, which increases the surface area-to-volume ratio, leading to an enhanced dissolution rate and improved solubility [29]. Stabilizers play a crucial role by Preventing agglomeration and Ostwald ripening during nanocrystallization, maintaining particle size and ensuring nanocrystal stability. The degree of crystallinity also affects the dissolution rate and stability of the nanocrystals [58]. Techniques such as high-pressure homogenization, wet milling, and antisolvent precipitation impact the quality and characteristics of nanocrystals, influencing particle size distribution, crystallinity, and surface properties [59]. Surface modifiers can alter the surface properties, enhancing drug stability and bioavailability by improving wettability and interaction with biological membranes. Additionally, the pH, temperature, and presence of solvents during formulation affect nanocrystal formation, and optimizing these conditions can enhance nanocrystallization efficiency. By meticulously controlling these parameters, drug/nanomaterial nanocrystals exhibit significantly improved solubility and bioavailability, resulting in better therapeutic efficacy, faster onset of action, and improved patient compliance [58].
4.3 Stabilizers in nanocrystallization of drugs
Stabilizers play a crucial role in the nanocrystallization of drugs. They are necessary for the formation of nanocrystals and for maintaining long-term stability during storage. Stabilizers prevent the aggregation of nanocrystals, ensuring the physical stability of the formulation. Additionally, they can interact with cells and cell layers in the final formulation, affecting the bioavailability of the drug. Stabilizers create a protective layer around nanoparticles, preventing them from clumping together. They also introduce charges that repel nanoparticles, maintaining their dispersion. Some stabilizers even act as cryoprotectants during processes like freeze-drying, Safeguarding the nanocrystals. This is particularly important because the surface tension of drug. Nanocrystals is often high, leading to easy aggregation. By reducing surface tension, a suitable stabilizer can prevent nanocrystal aggregation. For solid nanocrystal drugs, stabilizers are crucial.
In maintaining the redispersibility of the nanocrystals, especially for large-scale production. The selection of an appropriate stabilizer is based on the requirement of physical stability, such as maintaining the nanosized particle size, and its compatibility with the drug to enhance bioavailability. Various types of stabilizers are utilized in the nanocrystallization process to ensure stability and bioavailability of drug nanocrystals. As indicated in the Table 3, Polymers like PVP and HPMC prevent nanocrystal aggregation by forming a physical barrier, while surfactants such as Tween and Poloxamer reduce surface tension to prevent aggregation. Polymers, which are large molecules composed of repeated subunits, provide steric stabilization. Celluloses, derived from plant cell walls, prevent nanocrystal aggregation. Surfactants, which lower the surface tension between liquids and solids, aid in preventing nanocrystal aggregation. Lipids, insoluble in water but soluble in organic solvents, stabilize lipid-based nanocrystals. Ionic surfactants offer electrostatic stabilization, while polymeric stabilizers like poloxamers enhance stabilization through a combination of electrostatic and steric effects [60, 63].
Not every formulation has a unique stabilizer. The choice of stabilizer depends on various factors, including the specific drug, the desired release profile, and the intended route of administration. Formulators select stabilizers based on the unique characteristics of each drug. Some drugs may require specific stabilizers to address solubility challenges, prevent aggregation, or enhance bioavailability. Certain stabilizers, such as Poloxamers or PVP (Polyvinylpyrrolidone), are widely used due to their versatility. They can stabilize a range of drugs, making them popular choices. For targeted drug delivery, specialized stabilizers like Folate-conjugated Poloxamer (FA-P407) may be employed. These allow precise drug localization while minimizing side effects. As mentioned in Table 4, Hydrophilic stabilizers like HPMC K15M and PVP K30 enhance solubility, stability, and drug release, while amphiphilic ones such as Poloxamer 407 and DSPE-PEG 2000 improve cell uptake and reduce toxicity. In complex formulations, stabilizers may work synergistically. For instance, combining mPEG DSPE with other stabilizers can achieve sustained antiretroviral activity. Researchers continually explore novel stabilizers to optimize drug performance. Each formulation is a delicate balance of drug properties and stabilizer characteristics. While not every nanocrystalline formulation has a unique stabilizer, the art lies in selecting the right stabilizer for each drug to unlock its full potential [64, 65].
4.4 Comparison of amorphous and nanocrystal drug systems
When compared to metastable Amorphous forms, nanocrystals provide better physical stability and reduce the risk of re-crystallization and aggregation. Because of their smaller size and larger surface area, nanocrystals offer more uniform and improved drug absorption than amorphous solid dispersions. While amorphous forms need particular storage conditions in order to avoid crystallization, nanocrystals can be stored with less complexity and still maintain stability. Drug development and production may face difficulties if nanocrystal medications become amorphous because they dissolve more quickly and have increased bioavailability, but they also become less stable, more difficult to handle, and may break down more quickly. Itraconazole, a strong antifungal drug that is sold commercially in both amorphous and nanocrystal forms, serves as one example of comparison. Each with a unique therapeutic function. The purpose of Sporanox, which is formulated as nanocrystals, is to increase itraconazole’s solubility and rate of dissolution. By increasing the medication’s bioavailability, sporanox promotes more reliable and effective absorption in the gastrointestinal system [72, 73].
In the following section, we will thoroughly examine existing studies focusing on enhancing the bioavailability and effectiveness of HIV drugs. Additionally, we will explore the production technology for HIV drug nanocrystals. Table 5 refers to a summary of researches in this field.
4.4.1 Abacavir
Abacavir (ABC) is a type of NRTI reverse transcriptase inhibitor. Currently, ABC is used as part of a first-line treatment plan, in combination with two other NRTIs (zidovudine and lamivudine] [88, 89]. One of the problems with Abacavir is its hydrophilicity. In a study aiming to convert Abacavir (ABC) into a hydrophobic compound using myristic acid, it was esterified and transformed into MABC. Myristic acid can inhibit N-myristoyl transferase, an enzyme that catalyzes myristoylation, a process essential to the life cycle of several crucial proteins in HIV. The synthesized MABC was then formulated into a nanosuspension, and the hydrophobic drug crystals of Abacavir were prepared using the high-pressure homogenization (HPH) technique, which is a top-down approach. To cover and enhance the stability of the nanocrystals, the nanocrystals were stabilized with Poloxamer 407 alone in the first form, while in the second form, folate-conjugated P407 (FA-P407) was utilized for coating to enhance targeting. This coating specifically targets FLOR2, a protein expressed on the surface of activated macrophages. As shown in Fig. 4 (A, B and C), Both NMABC and FA-NMABC appeared as cylindrical rods measuring approximately 200 nm, as determined by SEM (scanning electron microscopy) and DLS.

A SEM images of NMABC and B SEM images of FA-NMABC nanoformulations revealed cylindrical monodispersed particles with a diameter of approximately 200 nm. C Particle size at 4 °C and 25 °C for 8 weeks. D FTIR-Peaks at 2915 cm−1 and 2850 cm−1 in MABC FTIR spectrum correspond to alkane (CH2–CH2) stretching of the myrisitic acid alkane
The triplet at 2.36 ppm in the H-NMR spectrum of MABC corresponds to two fatty acid methylene protons adjacent to the carbonyl group linked to ABC. The broad multiple signal in the region of 1.21–1.50 ppm represents 20 hydrogen atoms from the repeating methylene units of the fatty acid chain. Comparing the FTIR spectra of MABC to native ABC and myristic acid, the alkane stretching (CH2–CH2) peaks at 2915 and 2850 cm−1 in the MABC spectrum confirm that the alkane chain of the derivatizing fatty acid is covalently linked to the parent drug. Peaks at 1595 cm−1 in MABC and 1588 cm−1 in the ABC FTIR spectra correspond to CH2 stretching of the native aromatic ring. Peaks at 1673 and 1696 cm−1 in MABC and myristic acid spectra correspond to C=O fatty acid attachments, and the peak at 1033 and 1030 cm−1 in ABC and MABC spectra corresponds to NH2 vibrations of the parent compound. Infusion into a Waters Xevo TQS micro mass spectrometer confirmed a molecular mass ion of 496.2, corresponding to ABC with one myristoyl group. Notably, the size and PDI (polydispersity index) of the nanoparticles remained stable over 8 weeks at temperatures of 4 °C and 25 °C. The drug showed high loading in both forms, with NMABC at 62 ± 1.5% and FA-NMABC at 65 ± 2.4%. FA-NMABC demonstrated an impressive up to 2.5-fold increase in uptake compared to NMABC. The retention of MABC using FA-NMABC was significantly greater than that observed for NMABC. Antiretroviral activity was assessed using the real-time PCR method. The results showed that on Days 1 and 5, FA-NMABC exhibited 100 ± 2.3, while on Days 10 and 15, it showed 88 ± 3.4% and 70 ± 2.5% activity, respectively. In in vivo studies, mice treated with FA-NMABC demonstrated significantly higher blood levels of ABC compared to mice treated with NMABC or native ABC. The increase in ABC levels ranged from 2.6 to 4.9-fold in lymphoreticular tissues (including liver, spleen, and lymph nodes) in mice treated with nanoformulated MABC compared to native ABC. Overall, the nanocrystalline formulation of Abacavir in this study improved absorption, extended the shelf life by 2 weeks, and enhanced the antiretroviral effect of the drug [74].
4.4.2 Atazanavir
Atazanavir sulfate binds to the protease enzyme of the HIV-1 virus, thereby preventing the formation of mature virions. However, it has poor aqueous solubility and is extensively metabolized by the liver, resulting in low bioavailability when eliminated through the bile. To enhance its pharmacokinetic properties, co-administration of atazanavir with other antiretroviral drugs like ritonavir or cobicistat is recommended. This combination not only improves efficacy but also helps prevent resistance. By inhibiting the metabolism of atazanavir in the liver, co-administration can effectively increase its blood levels [75, 90]. Another method to enhance solubility and bioavailability involves the use of nanostructures like nanostructured lipid carriers (NLC) and solid lipid nanoparticles (SLN) for drug encapsulation. In a study conducted to improve solubility and bioavailability, atazanavir was Nanocrystallized using the emulsion solvent diffusion technique, a bottom-up approach. Stabilizers such as HPMC K15M, PVP K30, and PEG 6000 polymers were employed in different ratios. The compatibility between the drug and polymer was examined through Fourier Transform Infrared Spectroscopy (FTIR) within the frequency range of 400–4000 cm−1 and the Differential Scanning Calorimetry (DSC) method. The pure drug’s FT-IR spectrum displayed specific peaks at 3357.84 cm−1, 3060.82 cm−1, and 2873.74 cm−1 for N–H stretching vibrations, Aromatic C–H stretching, and Asymmetric and symmetric aliphatic C–H stretching, respectively. Confirming the presence of an amide group in the structure were the C=O stretching at 1699.17 cm−1 and N–H deformation band at 1650.95 cm−1. When comparing with the Nanocrystals, although the fingerprint region differed due to the presence of polymer in the Nanocrystals, the prominent absorption bands were nearly identical. The FTIR spectrum of the optimized nanocrystals showed slightly lower intensity, indicating no significant interaction between the drug and excipients. As shown in Fig. 5C, D The XRD pattern of atazanavir sulphate exhibited intense, sharp, and well-resolved peaks. These peaks occurred at specific 2θ angles (e.g., 5.77, 10.88, 12.09, etc.), characteristic of a crystalline compound. In contrast, the XRD pattern of the prepared nanocrystals showed less intense and denser peaks compared to atazanavir sulphate. Despite the differences, the characteristic peaks were still preserved, indicating that the crystalline state was not altered.

A SEM of Pure Drug. B SEM of Optimized Formulation. C XRD Study of Pure Drug Atazanavir Sulphate. D XRD Study of Optimized Formulation
The SEM examination of pristine atazanavir sulphate (Fig. 5A, B) revealed significantly smaller particles with a blade-shaped morphology and fines. The thermogram peak of atanazavir sulfate confirmed its crystalline nature, which contributed to its enhanced dissolution. The average optimized particle size was measured at 100 nm, indicating increased solubility of atazanavir sulfate nanocrystals in distilled water and phosphate buffer with pH 6.8. Stability studies conducted over a period of time exhibited minimal changes, suggesting a stable structure. As mentioned in Table 6, the solubility of the optimized batch in distilled water was 40.068 mg/ml (compared to 4.174 mg/ml for the pure drug), and in phosphate buffer it was 160.182 mg/ml (compared to 20.547 mg/ml for the pure drug). The optimal formulation achieved a high drug release percentage of 87.9%, which can be attributed to the long hydrophilic chain of PEG 6000 [75].
4.4.3 Darunavir
Darunavir is an antiviral medication used to treat HIV infection. It works by inhibiting the virus’s ability to multiply in the body. However, darunavir has low solubility in water, which can limit its absorption by the body. To enhance its effectiveness, darunavir is often prescribed in combination with other drugs like ritonavir and cobicistat. These drugs help metabolize the enzyme responsible for breaking down darunavir, leading to increased levels of the medication in the bloodstream. Researchers have also explored various methods to improve the bioavailability of darunavir, such as using mesoporous polymer carriers that dissolve in water. Another solution is to utilize the nanocrystal form of darunavir, which can enhance its solubility and bioavailability due to its smaller particle size [91].
A study was conducted to investigate the coaxial electrospraying method for producing darunavir nanocrystals. Nanocrystals were synthesized using a top-down wet milling technique that employed zirconium oxide beads. Various stabilizers, including Tween 80, Tween 20, HPMC, poloxamer 338 or 188, vitamin E, TPGS 1000 or 400, and SLS, were utilized. The purpose of the study was not to optimize the formulation of the nanocrystals, so no comparison was made between the stabilizers. In the next step, the darunavir nanocrystals were encapsulated using the coaxial electrospraying method with Edoragit L100. This resulted in a core–shell structure, where the outer liquid consisted of Edoragit L100 and the inner liquid consisted of darunavir nanocrystals. As indicated in Fig. 6A–C, the scanning electron microscope (SEM) image showed a narrow particle size distribution, with most particles smaller than 1 μm. Furthermore, SEM analysis of the electrosprayed sample containing 2.5% mannitol showed a uniform dispersion of the electrosprayed DRV particles after 2 h of dissolution in an acidic medium. The results showed that coaxial electrospraying is a unique technique for encapsulating drug nanocrystals within a polymeric shell. The final product of this study holds promise for improving the solubility and bioavailability of darunavir, potentially leading to more effective treatment options [76].

SEM images of a original DRV, b DRV nanocrystals and c DRV electrosprayed particles. d Particle size distribution by DLS of nanosuspensions and electrosprayed samples. e XRD diffractogram of different DRV crystal forms, DRV nanocrystals and DRV electrosprayed sample
M1DRV and M2DRV are lipophilic prodrugs that were created by derivatizing the amine group in DRV with fatty acids of varying carbon chain lengths. On the other hand, M2DRV is a hydrophobic prodrug that was synthesized by modifying the hydroxyl group in DRV. Both prodrugs were prepared using the top-down method, high-pressure homogenization, and stabilized with poloxamer 407 as nanocrystals named NM1DRV and NM2DRV. The nanoformulated prodrugs showed enhanced intracellular prodrug levels, sustained drug retention, and prolonged antiretroviral activities. The drug encapsulation rates for NDRV, NM1DRV, and NM2DRV were 57%, 74%, and 85%, respectively, indicating an increase in encapsulation efficiency in the nanoformulated prodrugs. The particle sizes for NDRV, NM1DRV, and NM2DRV were 384 ± 10 nm, 322 ± 7 nm, and 149 ± 2 nm, respectively, indicating a reduction in particle size in the nanoformulated prodrugs. All nanocrystals remained stable at 4 °C, 25 °C, and 37 °C for a period of 13 weeks. The observed antiviral activity for NM1DRV and NM2DRV formulations is likely due to improved cellular drug uptake and variable prodrug activation rates in HIV target cells. The intracellular prodrug concentrations peaked at 24 h for NM1DRV, reaching 126 nmol/106 cells, and for NM2DRV, reaching 74 nmol/106 cells. These findings demonstrate formulation stability and rapid prodrug uptake in HIV-1 target cells. As shown in Fig. 7, Plasma DRV levels demonstrating sustained drug release for NM1DRV and NM2DRV formulations. Transmission electron microscopy (TEM) images confirmed significant nanoparticle accumulation in macrophages for both NM1DRV and NM2DRV compared to NDRV. The electron micrograph images support previous observations on nanoparticle trafficking and storage in endosomal compartments. Furthermore, when given to mice, both NM1DRV and NM2DRV showed consistent levels of the drug in their blood and tissues for a period of 30 days. This study concludes that it is possible to achieve long-lasting DRV and sustained antiretroviral effects using prodrug nanoformulations [77].

A Plasma DRV levels demonstrating sustained drug release for NM1DRV and NM2DRV formulations. B Plasma M1DRV and M2DRV levels. C Blood M1DRV and M2DRV levels
4.4.4 Doltegravir
Dolutegravir is classified as an integrase strand transfer inhibitor (INSTI). Due to its high permeability and lipophilic nature, this drug has low solubility in water. Consequently, this limited solubility can potentially impact its bioavailability and therapeutic effectiveness [92]. The researchers used a Design of Experiments (DoE) approach to develop a nanosuspension of Doltegravir. This was done in order to enhance the drug's dissolution and improve its performance compared to the pure drug form. The nanosuspension was prepared through top-down methods, high-speed homogenization, and probe sonication techniques. With Soluplus used as a surfactant to stabilize the nanoparticles. Soluplus, being a novel excipient, provides enhanced stability to the formulation by preventing particle aggregation or precipitation. During the characterization process, it was found that the optimized Doltegravir nanosuspension had a particle size of 337.1 nm, a low polydispersity index, and a negative zeta potential. These results indicate that the nanosuspension has good stability. Differential scanning calorimetry and X-ray diffraction analysis confirmed that Doltegravir in the nanosuspension is in its amorphous form. The amorphous form explains the increased solubility; as amorphous drugs tend to dissolve more easily. In an in vitro study, it was found that the nanosuspension exhibited improved drug release and increased solubility. Additionally, an in vivo study conducted on Wistar rats revealed that the nanosuspension had higher bioavailability in comparison to the pure drug. This discovery was further supported by the AUC value (area under the concentration–time curve) [78].
4.4.5 Efavirenz
Efavirenz, a non-nucleoside reverse transcriptase inhibitor, is known for its lipophilic properties and high solubility in lipids. However, it has poor aqueous solubility, measuring only 0.0085 mg/ml. To address this issue, efavirenz can be reduced to nanocrystal size, which enhances its absorption in the body. It’s worth noting that the oral bioavailability of this drug is 40–45%, meaning that only a portion of the prescribed dose reaches the systemic circulation after oral consumption [93].
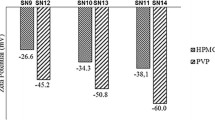
In a study aimed at improving the solubility and bioavailability of this drug, researchers utilized a novel approach. Antisolvent precipitation which is bottom up technique was specifically chosen as the method for nanocrystal preparation. Through this approach, they successfully developed efavirenz nanocrystals. Furthermore, the researchers investigated the dissolution rate and stability of this newly formulated drug. The results highlighted the significance of various factors in optimizing the final formulation, including the type and quantity of stabilizers (HPMC and PVP), the drug load, and the ratio of solvent to anti-solvent [79]. In another study, five different polymers (HPMC, HPC, Lutrol F68, Lutrol F127, and PVP K30) were used to create medicinal nanocrystals of efavirenz. In all preparations (Table 7), SLS was employed as an electrostatic stabilizer at a concentration of 2% (w/w) relative to the mass of EFV.
Only the samples containing HPMC and PVP were able to form uniform suspensions. It was confirmed that the use of HPMC and PVP polymers, at different concentrations, can optimize the production of efavirenz nanocrystals. In XRD results as shown in Fig. 8A, B, all samples exhibit a peak at 6.2° 2θ, which is characteristic of EFV polymorph. Enlargement of peak width suggests a reduction in crystalline domain size, likely associated with smaller particle sizes. The study concluded that HPMC polymer, although slightly hydrophilic, can be used in a lower quantity in the formulation to enhance mixing with the efavirenz drug, prevent crystal clumping, and ultimately generate superior nanocrystals. The solvent antisolvent precipitation method, a bottom-up approach, was employed to produce the nanocrystals. Confirmed that samples remained crystalline. Additionally, the study suggested that, apart from the number of stabilizers, the appropriate selection of stabilizers also plays a crucial role in achieving optimal and effective nanocrystals [81].

A, B X-ray diffraction patterns of the samples. C SEM images based on different concentrations of HPMC and PVP
In another study, 12 batches of Efavirenz nanocrystals were prepared to evaluate the optimal formulation of the drug. The nanocrystals demonstrated a uniform distribution of the drug. According to xrd studies, Pure Efavirenz (EFV) drug has shown crystalline peaks at 14.572° 7.225° 6.692° angles. In three baches crystalline peaks was observed like pure Efavirenz. The scanning electron microscopy was used to examine the surface morphology of these three batches of nanocrystals. It is evident from the SEM images, as shown in Fig. 9C, that the nanocrystals have an irregular shape. Methanol was used as a solvent, and PVP K-30, Poloxamer 188, and SLS polymers were used as stabilizers. The researchers examined the ratio of these polymers to determine the formulation that resulted in the highest release of Efavirenz. The results confirmed that formulations containing PVP K30, Poloxamer F-68, and SLS stabilizers exhibited an increased release of Efavirenz compared to the pure drug. The solubility of pure Efavirenz is 0.017 (practically insoluble). However, by reducing the size and formulating the drug as nanocrystals, the solubility and release of Efavirenz significantly increased, allowing for greater exposure to body fluids. In the FTIR study (Fig. 9D, E), no significant interactions were observed between the drug (Efavirenz) and the polymer stabilizers. This lack of interaction is crucial for maintaining drug stability and effectiveness. The formulations containing PVP K30, Poloxamer F68, and SLS (Sodium Lauryl Sulfate) showed the highest uniform distribution and solubility. The solubility of the drug in water for these formulations was respectively 0.077, 0.069, and 0.061, demonstrating an increase compared to the pure drug. The distribution index of the drug in the nanocrystals was 80.41%, 72.33%, and 69.16%, respectively [80].

A Xrd pattern of pure efavirenze. B XRD pattern of one batch of nanocrystalline efavirenz. C SEM images of three baches of Efavirenz nanocrystals. D FTIR pattern of pure efavirenze. E FTIR pattern of one batch of nanocrystalline efavirenz
4.4.6 Emtricitabine
Emtricitabine, also known as FTC, is a cytidine analog that works by inhibiting the reverse transcriptase enzyme. This inhibitory action hinders the HIV virus from multiplying, resulting in a decrease in the viral load and an increase in the number of CD4 cells [82, 94]. FTC is a drug that is hydrophilic and has a short half-life. In a study, a modified form of FTC called NMFTC was created by esterifying palmitoyl chloride. Nanocrystals of NMFTC were then prepared using high-pressure homogenization, which is a top-down method. And embedded in a surfactant called poloxamer 407 to increase stability. As shown in Fig. 10 XRD analysis of FTC and MFTC demonstrates the crystalline nature of both drugs. The objective of study was to increase the half-life and distribution of the drug in the body. In this study, monocytes were cultured and transformed into macrophages. The researchers examined the uptake of the drug by the cells at different time intervals (1, 2, 4, 8, and 24 h) after incubating the cells with either 100 µM NMFTC or FTC. The results showed that NMFTC was readily taken up by the macrophages, reaching its peak concentration of 25 nmol/106 cells after 4 h. However, the prodrug decayed rapidly over the course of 24 h. On the other hand, minimal levels of the drug were detected inside the cells when exposed to FTC. In a Long-Term Effects study, it was found that after 8 h of NMFTC treatment, intracellular levels of FTC-TP were still detectable for up to 10 days (1.5 pmol/106 cells). Compared to FTC treatment, NMFTC resulted in three times higher levels of FTC-TP on day 5 (3.2 pmol/106 cells). To evaluate the antiretroviral effect, a viral inhibitory study was conducted by measuring reverse transcriptase activity and HIV1 p24 gene expression in flask-attached cells. The results showed that NMFTC completely inhibited viral reverse transcriptase activity in macrophages when the cells were challenged up to 10 days after prior exposure. In contrast, native FTC only showed 30% inhibition after 24 h of exposure. These findings highlight the potential of NMFTC as an effective antiretroviral agent and emphasize its role as a drug carrier. By increasing the cell uptake by macrophages and extending the half-life, NMFTC exhibited better antiviral activity compared to MFTC [82].

A XRD analysis of FTC and MFTC demonstrates the crystalline nature of both drugs. B FTIR spectrum of MFTC showing, highlighted in dashed circles, absorption peaks at 2920 and 2860 cm−1 that correspond to CH2–CH2 stretches of the fatty acid chain and at 1745 cm−1 that corresponds to the carbonyl functional group that is part of the formed ester bond
4.4.7 Lamivudine
Lamivudine, commonly known as 3TC, is a cytidine analog often used in combination with other drugs like zidovudine. It effectively treats both HIV-1 and HIV-2 by inhibiting the reverse transcriptase enzyme. However, due to its short half-life and quick elimination from the body, frequent dosing is necessary, which can be inconvenient for patients. 3TC is typically used in combination with other antiretroviral drugs, such as zidovudine, to create highly active antiretroviral therapy (HAART). Lamivudine is known for its high bioavailability and rapid absorption when taken orally. In terms of its hydrophilicity, lamivudine is a water-soluble compound, which aids in its good absorption and distribution throughout the body [95]. A study developed a new version of the antiretroviral drug lamivudine called M23TC1. This version, modified to be a ProTide, is a long-acting, nano-formulated drug aimed at improving the drug’s effectiveness and pharmacokinetic profile compared to the original. ProTide refers to prodrug nucleotide analogs, which combine the benefits of prodrugs and nucleotide analogs. The production process involves the use of high-pressure homogenization, which is a top-down method to create nanoparticles. The nanoparticles were stabilized using Tween 20 and mPEG DSPE as surfactants. The nano-formulated ProTide is designed to gradually release the active drug over time, making it long-acting and potentially reducing the frequency of dosing.As shown in Fig. 11, In MDMs (monocyte-derived macrophages), M23TC was readily taken up, resulting in a significantly higher intracellular drug concentration compared to native 3TC. In CEM CD4+ T cells, M23TC exhibited rapid prodrug uptake and efficient conversion to 3TC-TP after 8 h, surpassing the levels observed with 3TC treatment. Conversion to 3TC-TP peaked at 8 h. M23TC also facilitated drug retention in MDMs for up to 30 days, whereas native 3TC showed no detectable drug within 24 h. These cells were exposed to 100 μM NM23TC for 8 h. The enhanced intracellular delivery of active metabolites and improved pharmacokinetic profiles suggest that M23TC could be more effective than the native drug, providing better HIV control with potentially fewer side effects. This new formulation can maintain potent antiretroviral activities for at least 30 days in macrophages and improve drug uptake in CD4+ T cells [83].

TEM morphological evaluation in MDM and CEM-CD4+ T cells. Cells were treated with 100 μM NM23TC for 8 h. Control cells (A and C) were given no treatment and collected alongside treated cells (B and D)
4.4.8 Nevirapine
Nevirapine is an NNRTI (non-nucleoside reverse transcriptase inhibitor) that is used to treat HIV-1 infection. Its main function is to block the activity of reverse transcriptase. However, one limitation of nevirapine is its poor water solubility, which may impact its effectiveness [96]. Researchers used high-pressure homogenization and surface modification techniques, which are top-down methods, to produce nevirapine nanosuspensions. The nanosuspensions had a size of approximately 457 nm and were stabilized using serum albumin, polysaccharide, and polyethylene glycol (PEG) 1000. During the homogenization process, nevirapine crystals with a mixed layer of stabilizers were formed, with an average size below 1000 nm. This study aimed to investigate protein adsorption on nevirapine nanosuspensions, as understanding this can help optimize drug delivery systems and enhance targeted drug delivery. The researchers focused on mononuclear phagocytic system (MPS) cells, which are the main reservoir of the virus. In vitro protein adsorption studies were conducted using two-dimensional polyacrylamide gel electrophoresis (2-D PAGE) to compare nevirapine nanosuspension with nevirapine surface-modified nanocrystals. The adsorption patterns were qualitatively identical, but quantitative differences were observed. Immunoglobulins were found to be relatively highly adsorbed, indicating good uptake by the liver and spleen. Overall, by controlling protein adsorption through surface modification, researchers can potentially reduce the need for animal studies and accelerate the development of targeted nanoparticles [84].
4.4.9 Rilpivirine
Rilpivirine (RPV) is a non-nucleoside reverse transcriptase inhibitor (NNRTI) with stronger antiretroviral properties and a longer half-life compared to older NNRTIs like efavirenz, as well as fewer side effects. However, one disadvantage of this drug is its poor solubility in water and oil. An interesting property of RPV is its inherent fluorescence, which was utilized in a study to investigate its release and intracellular tracking after injection. Although RPV drug molecules in dilute solution do not exhibit fluorescence, this property becomes intensified with accumulation. In the study, RPV nanocrystals were synthesized using a combination of methods. Initially, the high-pressure homogenization method, which is a top-down method, was employed for synthesis, followed by the use of solvent-antisolvent precipitation methods to reduce the size and form nanocrystals. To enhance stability and lipophilicity, the synthesized nanoparticles were coated with a lipid film containing DSPE-PEG 2000 and L-α-phosphatidylcholine as stabilizers.
The nanocrystals exhibited a uniform shape with a smooth surface, measuring approximately 200 nm in particle size. XRD analysis was performed on native RPV and RPV crystals before and after homogenization. As shown in Fig. 12B–D, distinct diffraction peaks corresponding to native RPV were retained at positions 12.4°, 17.5°, 20.1°, 21.1°, and 27.9° in RPV crystals before and after homogenization. This consistency indicates that the crystalline nature of native RPV remained intact in the microcrystals and homogenized nanoparticles. For the RPV-NC Localization Study, CD4 and MDM cells (monocyte-derived macrophages) were investigated. CD4+ T cells primarily took up RPV-NC through endocytosis, and confocal microscopy revealed the presence of RPV-NC in the cytoplasm of CD4+ T cells. MDMs were treated with 100 μM RPV-NC for 8 h, and immunostaining was performed to visualize endosomal markers, including Rab5 (early endosomes), Rab7 (late endosomes), and Rab14 (recycling endosomes). Co-localization of RPV-NC was readily observed in stained endosomal compartments, particularly with Rab14 endosomes. Analysis of the in vitro and in vivo results demonstrated an increase in cell uptake by CD4+ and MDM cells, along with a decrease in toxicity. Additionally, the synthesized nanocrystals facilitated visualization of the particles inside cells due to their increased accumulation in tissues such as the liver, owing to the aggregation-induced emission (AIE) feature [85].

A TEM images revealed micron-sized, ‘flower-shaped’ crystals clustered together. B–D XRD patterns of native RPV, RPV-NC before and after homogenization, respectively. E Hydrodynamic size distribution of RPV-NC is shown by DLS (average size ~ 320 nm)
In another study, nanocrystals of the drug RPV were synthesized using the top down method, wet-milling with zirconium beads. The goal was to enhance stability and improve dispersion characteristics. To achieve this, Poloxamer 338 and D-alpha-tocopherol polyethylene glycol 1000 Succinate were used as stabilizers for the synthesized nanocrystals. As shown in Fig. 13, Particle size distribution of the test TMC278 nanosuspensions with differently targeted particle sizes was from 200–800 nm. The results of both in vivo and in vitro studies demonstrated an increase in the 6-month stability of the nanocrystals, along with proper dispersion and monodispersity. Furthermore, after single-dose administration, the plasma concentration profile showed a prolonged release of TMC278 for 3 months in dogs and 3 weeks in mice [86].

Particle size distribution of the test TMC278 nanosuspensions with differently targeted particle sizes: Exp 9005A (200 nm), Exp 10,477 (400 nm) and Exp 10,488 (800 nm)
4.4.10 Tenofovir
Tenofovir disoproxil fumarate (TDF) is a nucleoside reverse transcriptase inhibitor (NRTI) used alongside other antiretroviral drugs to treat patients infected with HIV-1. Tenofovir is similar to deoxyadenosine 5′ monophosphate but lacks the hydroxyl group at the 3′ carbon position of dAMP. This modification causes DNA transcription to end prematurely, effectively preventing virus replication [97]. ProTide technology operates by binding small molecules to pre-activated compounds, aiding in their delivery to target cells. Once inside, these small molecules decompose, allowing the pre-activated compounds to carry out their intended function. Tenofovir sulfonamide (TAF) is another variant of the tenofovir prodrug, which was developed using proTide technology [98]. TAF exhibits greater antiviral activity than TDF and possesses superior distribution within lymphatic tissues. However, despite these advantages, TAF is prone to hydrolysis and is considered an unstable prodrug [99]. A study was conducted to create two lipophilic prodrugs (NM1TFV and NM2TFV) by combining amino acid esters of phenylalanine and alanine with tenofovir. By converting tenofovir into a prodrug, the physical and chemical properties of the drug were altered. This resulted in decreased solubility of the prodrugs in water and increased solubility in octanol, indicating their lipophilicity compared to TAF. The protides and TAF were prepared using a high-pressure homogenizer technique, which is a top-down method. They were then converted into nanocrystals using poloxamer 407 for NM1TFV and NM2TFV, and PEG3350 for NTAF. As shown in Fig. 14B, the absorption bands at 2844 cm−1 and 2912 cm−1 in the FTIR spectrum actually correspond to the asymmetric and symmetrical C–H stretches of long-chain fatty alcohols. These stretches are characteristic of the C–H bonds in alkanes, which are common in long-chain fatty alcohols. The aim of designing the protides and their nanocrystallization was to enhance intracellular transport and prolong the shelf life of the drug. The results showed that NM1TFV protide deposited four times more tenofovir in PBMC and tissue. The IC50 values of M1TFV, M2TFV, and their nanoformulations (NM1TFV and NM2TFV) were assessed in monocyte-derived macrophages (MDM). This was done over a concentration range of 0.00005–5000 nM by measuring the HIV-1 reverse transcriptase (RT) activity after challenging the cells with HIV-1ADA at a multiplicity of infection (MOI) of 0.1. This long-acting form allows for a reduction in the frequency of antiviral drug administration from daily to once every 2 months [87].

A IC50 values of M1TFV, M2TFV, and their nanoformulations (NM1TFV and NM2TFV) in MDM. B FTIR Absorption bands at 2844 and 2912 cm−1
There are no FDA approved HIV medications formulated as nanocrystals as of now. Most HIV drugs, as indicated in Table 8, utilize amorphous solid dispersion technology. This technology helps enhance solubility and bioavailability, crucial for effective treatment.
While nanocrystal technology isn’t currently prominent in HIV drug formulations, ongoing research and development in nanotechnology may lead to advancements in drug delivery systems. Researchers continue to explore how nanocrystals could potentially offer benefits such as improved stability and reduced aggregation in the context of HIV treatment.
5 Conclusion
The future of nanocrystals in HIV treatment holds great promise and potential. Ongoing advancements in nanocrystal development are expected to enhance the effectiveness of HIV drugs by improving their solubility and bioavailability. This, in turn, could lead to more potent formulations that require lower dosages. Furthermore, there is a growing interest in utilizing nanocrystals for targeted drug delivery, which could minimize side effects and improve patient outcomes. One approach involves the use of specialized stabilizers like Folate-conjugated Poloxamer, which enables precise localization of the drug. Additionally, the focus is on developing long-acting formulations that can be administered less frequently, potentially improving adherence to therapy. As HIV can develop resistance to medications, nanocrystals may offer a way to enhance the delivery of combination therapies that can overcome the virus’s resistance. The search for new stabilizers that can work in synergy with nanocrystals to maintain stability and prevent aggregation is an ongoing pursuit. This research holds the potential to unlock new possibilities for drug formulations. Nanocrystal technology is also expected to integrate with other advancements in HIV treatment, such as the use of broadly neutralizing antibodies (bNAbs) and long-acting antiretroviral therapy (ART), to create comprehensive treatment strategies. The application of nanocrystals in HIV treatment could have a significant impact on global health, especially in regions with high HIV prevalence, by making treatments more accessible and affordable. In the long term, nanocrystals may contribute to the pursuit of a functional cure for HIV, where the virus can be controlled without lifelong therapy. Overall, the future of nanocrystals in HIV treatment is an area of dynamic and impactful research that has the potential to greatly improve the lives of individuals living with HIV. These advancements represent a significant step forward in HIV treatment, offering more convenient dosing schedules and potentially improving patient adherence to therapy.
Data availability
No datasets were generated or analysed during the current study.
References
Zulfiqar HF, Javed A, Sumbal, Afroze B, Ali Q, Akbar K, et al. HIV diagnosis and treatment through advanced technologies. Front Public Health. 2017;5:32.
Sailaja I, Baghel MK, Shaker IA. Nanotechnology based drug delivery for HIV-AIDS treatment. In: AIDS updates-recent advances and new perspectives. IntechOpen; 2021.
Hanna G, Hirsch M. Antiretroviral therapy for human immunodeficiency virus infection. Principles Pract Infect eases. 2000;1:1655–78.
Cohen MS, Smith MK, Muessig KE, Hallett TB, Powers KA, Kashuba AD. Antiretroviral treatment of HIV-1 prevents transmission of HIV-1: where do we go from here? Lancet. 2013;382(9903):1515–24.
Hiv H. Human Immunodeficiency Virus (HIV). J Aust. 2000;172(6):266–9.
Yang L-L, Li Q, Zhou L-B, Chen S-Q. Meta-analysis and systematic review of the efficacy and resistance for human immunodeficiency virus type 1 integrase strand transfer inhibitors. Int J Antimicrob Agents. 2019;54(5):547–55.
Gomez C, Hope TJ. The ins and outs of HIV replication. Cell Microbiol. 2005;7(5):621–6.
Volberding PA, Deeks SG. Antiretroviral therapy and management of HIV infection. Lancet. 2010;376(9734):49–62.
de Béthune M-P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: a review of the last 20 years (1989–2009). Antiviral Res. 2010;85(1):75–90.
Patel PH, Zulfiqar H. Reverse transcriptase inhibitors. StatPearls (internet): StatPearls Publishing; 2023.
Marzinke MA. Therapeutic drug monitoring of antiretrovirals. In: Clinical challenges in therapeutic drug monitoring. Elsevier; 2016. p. 135–63.
Gupta K, Shrivastava P. Development and validation of UV spectrophotometric method for trimethoprim in pure and marketed formulation. Int J Health Sci. 2022. https://doi.org/10.53730/ijhs.v6nS2.7860.
Wang Y, Gu S-X, He Q, Fan R. Advances in the development of HIV integrase strand transfer inhibitors. Eur J Med Chem. 2021;225: 113787.
Arimide DA, Szojka ZI, Zealiyas K, Gebreegziabxier A, Adugna F, Sasinovich S, et al. Pre-treatment integrase inhibitor resistance and natural polymorphisms among HIV-1 subtype C infected patients in Ethiopia. Viruses. 2022;14(4):729.
Oliveira M, Ibanescu R-I, Anstett K, Mésplède T, Routy J-P, Robbins MA, Brenner BG. Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology. 2018;15:1–14.
Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV/AIDS Res Palliative Care. 2015;7:95–104.
Rao PKS. CCR5 inhibitors: emerging promising HIV therapeutic strategy. Indian J Sex Trans Dis AIDS. 2009;30(1):1–9.
Gadhe CG, Kothandan G, Madhavan T, Cho SJ. Molecular modeling study of HIV-1 gp120 attachment inhibitors. Med Chem Res. 2012;21:1892–904.
Kuritzkes DR. HIV-1 entry inhibitors: an overview. Curr Opin HIV AIDS. 2009;4(2):82–7.
Larson KB, Wang K, Delille C, Otofokun I, Acosta EP. Pharmacokinetic enhancers in HIV therapeutics. Clin Pharmacokinet. 2014;53:865–72.
Gibas KM, Kelly SG, Arribas JR, Cahn P, Orkin C, Daar ES, et al. Two-drug regimens for HIV treatment. The Lancet HIV. 2022;9(12):e868–83.
Pharma Specialists. https://www.pharmaspecialists.com.
FDA-Approved HIV medicines. https://hivinfo.nih.gov/home-page.
World health organization. https://www.who.int.
Saag MS, Gandhi RT, Hoy JF, Landovitz RJ, Thompson MA, Sax PE, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2020 recommendations of the International Antiviral Society–USA Panel. Jama. 2020;324(16):1651–69.
Zuo X, Huo Z, Kang D, Wu G, Zhou Z, Liu X, Zhan P. Current insights into anti-HIV drug discovery and development: a review of recent patent literature (2014–2017). Expert Opin Ther Pat. 2018;28(4):299–316.
Mamo T, Moseman EA, Kolishetti N, Salvador-Morales C, Shi J, Kuritzkes DR, et al. Emerging nanotechnology approaches for HIV/AIDS treatment and prevention. Nanomedicine. 2010;5(2):269–85.
Kumar L, Verma S, Prasad DN, Bhardwaj A, Vaidya B, Jain AK. Nanotechnology: a magic bullet for HIV AIDS treatment. Artif Cells Nanomed Biotechnol. 2015;43(2):71–86.
Dizaj SM, Vazifehasl Z, Salatin S, Adibkia K, Javadzadeh Y. Nanosizing of drugs: effect on dissolution rate. Res Pharm Sci. 2015;10(2):95–108.
Fan R, Wang H, Zhang L, Ma T, Tian Y, Li H. Nanocrystallized oleanolic acid better inhibits proliferation, migration and invasion in intracranial glioma via caspase-3 pathway. J Cancer. 2020;11(7):1949.
Zhou T, Su H, Dash P, Lin Z, Shetty BLD, Kocher T, et al. Creation of a nanoformulated cabotegravir prodrug with improved antiretroviral profiles. Biomaterials. 2018;151:53–65.
Lu L, Xu Q, Wang J, Wu S, Luo Z, Lu W. Drug nanocrystals for active tumor-targeted drug delivery. Pharmaceutics. 2022;14(4):797.
Tabasum S, Younas M, Zaeem MA, Majeed I, Majeed M, Noreen A, et al. A review on blending of corn starch with natural and synthetic polymers, and inorganic nanoparticles with mathematical modeling. Int J Biol Macromol. 2019;122:969–96.
Jarvis M, Krishnan V, Mitragotri S. Nanocrystals: a perspective on translational research and clinical studies. Bioeng Transl Med. 2019;4(1):5–16.
Gao Y, Wang J, Wang Y, Yin Q, Glennon B, Zhong J, et al. Crystallization methods for preparation of nanocrystals for drug delivery system. Current pharmaceutical design. 2015;21(22):3131–9.
Jahangir MA, Imam SS, Muheem A, Chettupalli A, Al-Abbasi FA, Nadeem MS, et al. Nanocrystals: characterization overview, applications in drug delivery, and their toxicity concerns. J Pharm Innov. 2020. https://doi.org/10.1007/s12247-020-09499-1.
Rodrigues M, Baptista B, Lopes JA, Sarraguça MC. Pharmaceutical cocrystallization techniques. Advances and challenges. Int J Pharm. 2018;547(1–2):404–20.
Zhang L, Kong D, Wang H, Jiao L, Zhao X, Song J, et al. Cocrystal of apixaban–quercetin: Improving solubility and bioavailability of drug combination of two poorly soluble drugs. Molecules. 2021;26(9):2677.
Singh M, Barua H, Jyothi VGS, Dhondale MR, Nambiar AG, Agrawal AK, et al. Cocrystals by design: a rational coformer selection approach for tackling the API problems. Pharmaceutics. 2023;15(4):1161.
Wienen-Schmidt B, Oebbeke M, Ngo K, Heine A, Klebe G. Two methods, one goal: Structural differences between cocrystallization and crystal soaking to discover ligand binding poses. ChemMedChem. 2021;16(1):292–300.
Haneef J, Amir M, Sheikh NA, Chadha R. Mitigating drug stability challenges through cocrystallization. AAPS PharmSciTech. 2023;24(2):62.
Junghanns JUA, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomed. 2008;3(3):295–310.
Fontana F, Figueiredo P, Zhang P, Hirvonen JT, Liu D, Santos HA. Production of pure drug nanocrystals and nano co-crystals by confinement methods. Adv Drug Deliv Rev. 2018;131:3–21.
Gujar K, Wairkar S. Nanocrystal technology for improving therapeutic efficacy of flavonoids. Phytomedicine. 2020;71: 153240.
Gao L, Zhang D, Chen M. Drug nanocrystals for the formulation of poorly soluble drugs and its application as a potential drug delivery system. J Nanopart Res. 2008;10:845–62.
Liu P. Nanocrystal formulation for poorly soluble drugs. Dissertationes bioscientiarum molecularium Universitatis Helsingiensis in Viikki. 2013:62.
Lu Y, Li Y, Wu W. Injected nanocrystals for targeted drug delivery. Acta Pharmaceutica Sinica B. 2016;6(2):106–13.
Castrillo P, Olmos D, Amador D, González-Benito J. Real dispersion of isolated fumed silica nanoparticles in highly filled PMMA prepared by high energy ball milling. J Colloid Interface Sci. 2007;308(2):318–24.
Krause K, Müller R. Production and characterisation of highly concentrated nanosuspensions by high pressure homogenisation. Int J Pharm. 2001;214(1–2):21–4.
Dong Y, Ng WK, Hu J, Shen S, Tan RB. Continuous production of redispersible and rapidly-dissolved fenofibrate nanoformulation by combination of microfluidics and spray drying. Powder Technol. 2014;268:424–8.
Schubert S, Delaney JT Jr, Schubert US. Nanoprecipitation and nanoformulation of polymers: from history to powerful possibilities beyond poly (lactic acid). Soft Matter. 2011;7(5):1581–8.
Kim M-S, Jin S-J, Kim J-S, Park HJ, Song H-S, Neubert RH, Hwang S-J. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2008;69(2):454–65.
Badens E, Masmoudi Y, Mouahid A, Crampon C. Current situation and perspectives in drug formulation by using supercritical fluid technology. J Supercritical Fluids. 2018;134:274–83.
Adibkia K, Jalali MB, Javadzadeh Y, Esfanjani HM. A review on the methods and applications of spray drying technology. Pharm Sci. 2013;18(2):119–32.
Peltonen L, Hirvonen J. Drug nanocrystals–versatile option for formulation of poorly soluble materials. Int J Pharm. 2018;537(1–2):73–83.
Hassan AS, Soliman GM. Rutin nanocrystals with enhanced anti-inflammatory activity: Preparation and ex vivo/in vivo evaluation in an inflammatory rat model. Pharmaceutics. 2022;14(12):2727.
Mashiach R, Weissman H, Avram L, Houben L, Diskin-Posner Y, Arunachalam V, et al. Cation-ligand complexation mediates the temporal evolution of colloidal fluoride nanocrystals through transient aggregation. Nano Lett. 2021;21(23):9916–21.
Yang H, Kim H, Jung S, Seo H, Nida SK, Yoo S-Y, Lee J. Pharmaceutical strategies for stabilizing drug nanocrystals. Curr Pharm Des. 2018;24(21):2362–74.
He Y, Ye Z, Liu X, Wei Z, Qiu F, Li H-F, et al. Can machine learning predict drug nanocrystals? J Control Release. 2020;322:274–85.
Tuomela A, Hirvonen J, Peltonen L. Stabilizing agents for drug nanocrystals: effect on bioavailability. Pharmaceutics. 2016;8(2):16.
Rossier B, Jordan O, Allémann E, Rodriguez-Nogales C. Nanocrystals and nanosuspensions: an exploration from classic formulations to advanced drug delivery systems. Drug Deliv Transl Res. 2024. https://doi.org/10.1007/s13346-024-01559-0.
Peltonen L, Hirvonen J. Pharmaceutical nanocrystals by nanomilling: critical process parameters, particle fracturing and stabilization methods. J Pharm Pharmacol. 2010;62(11):1569–79.
Li J, Wang Z, Zhang H, Gao J, Zheng A. Progress in the development of stabilization strategies for nanocrystal preparations. Drug Deliv. 2021;28(1):19–36.
Katari O, Jain S. Nanomedicines to improve oral delivery of antiretroviral drugs. In: Nanomedicines for the prevention and treatment of infectious diseases. Springer; 2023. p. 265–95.
Shete G, Jain H, Punj D, Prajapat H, Akotiya P, Bansal AK. Stabilizers used in nano-crystal based drug delivery systems. J Excipients Food Chem. 2016;5(4):184–209.
Lori MS, Ohadi M, Estabragh MA, Afsharipour S, Banat IM, Dehghannoudeh G. pH-sensitive polymer-based carriers as a useful approach for oral delivery of therapeutic protein: a review. Protein Pept Lett. 2021;28(11):1230–7.
Mohanraj V, Chen Y. Nanoparticles-a review. Trop J Pharm Res. 2006;5(1):561–73.
Marques BLM. Nanopartículas contendo óleo de quinoa (Chenopodium quinoa Willd.): síntese, caracterização físico química, avaliação in vitro da toxicidade e do potencial bioativo. Universidade Federal do Rio Grande do Norte; 2023.
Luo Y, Hong Y, Shen L, Wu F, Lin X. Multifunctional role of polyvinylpyrrolidone in pharmaceutical formulations. AAPS PharmSciTech. 2021;22:1–16.
Praveen N, Preetha RST, Pagare V, Devasia J, Nizam A, Mukherjee E, et al. Plant-based metabolites as source of antimicrobial therapeutics: prospects and challenges. In: Antimicrobials in pharmaceutical and medicinal research. CRC Press; 2023. p. 165–201.
Elzoghby AO, Samy WM, Elgindy NA. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157(2):168–82.
Pas T, Verbert S, Appeltans B, Van den Mooter G. The influence of crushing amorphous solid dispersion dosage forms on the in-vitro dissolution kinetics. Int J Pharm. 2020;573: 118884.
Uttaro E, Pudipeddi M, Schweighardt A, Zhao F. To crush or not to crush: a brief review of novel tablets and capsules prepared from nanocrystal and amorphous solid dispersion technologies. Am J Health Syst Pharm. 2021;78(5):389–94.
Singh D, McMillan J, Hilaire J, Gautam N, Palandri D, Alnouti Y, et al. Development and characterization of a long-acting nanoformulated abacavir prodrug. Nanomedicine. 2016;11(15):1913–27.
Malviya V, Burange P, Thakur Y, Tawar M. Enhancement of solubility and dissolution rate of atazanavir sulfate by nanocrystallization. Indian J Pharm Educ Res. 2021;55(3):S672–80.
Nguyen DN, Clasen C, Van den Mooter G. Encapsulating darunavir nanocrystals within Eudragit L100 using coaxial electrospraying. Eur J Pharm Biopharm. 2017;113:50–9.
Banoub MG, Bade AN, Lin Z, Cobb D, Gautam N, Dyavar Shetty BL, et al. Synthesis and characterization of long-acting darunavir prodrugs. Mol Pharm. 2019;17(1):155–66.
Bhairam M, Pandey RK, Shukla SS, Gidwani B. Preparation, optimization, and evaluation of dolutegravir nanosuspension: in vitro and in vivo characterization. J Pharm Innov. 2023;18(4):1798–811.
Sartori GJ, Prado LD, Rocha HVA. Efavirenz dissolution enhancement V-A combined top down/bottom up approach on nanocrystals formulation. Braz J Pharm Sci. 2022;58: e18800.
Jakune V, Ghurghure S, Dyawarkonda M, Khadtare J, Yanjane S. Design and characterization of efavirenz nanocrystals for solubility enhancement. J Global Trends Pharm Sci. 2021;12(2):9168–9178.
Sartori GJ, Prado LD, Rocha HVA. Efavirenz dissolution enhancement IV—antisolvent nanocrystallization by sonication, physical stability, and dissolution. AAPS PharmSciTech. 2017;18:3011–20.
Ibrahim IM, Bade AN, Lin Z, Soni D, Wojtkiewicz M, Dyavar Shetty BL, et al. Synthesis and characterization of a long-acting emtricitabine prodrug nanoformulation. Int J Nanomed. 2019;14:6231–47.
Smith N, Bade AN, Soni D, Gautam N, Alnouti Y, Herskovitz J, et al. A long acting nanoformulated lamivudine ProTide. Biomaterials. 2019;223: 119476.
Shegokar R, Jansch M, Singh KK, Müller RH. In vitro protein adsorption studies on nevirapine nanosuspensions for HIV/AIDS chemotherapy. Nanomed Nanotechnol Biol Med. 2011;7(3):333–40.
Mukadam IZ, Machhi J, Herskovitz J, Hasan M, Oleynikov MD, Blomberg WR, et al. Rilpivirine-associated aggregation-induced emission enables cell-based nanoparticle tracking. Biomaterials. 2020;231: 119669.
Baert L, Klooster G, Dries W, François M, Wouters A, Basstanie E, et al. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. Eur J Pharm Biopharm. 2009;72(3):502–8.
Cobb DA, Smith N, Deodhar S, Bade AN, Gautam N, Shetty BLD, et al. Transformation of tenofovir into stable ProTide nanocrystals with long-acting pharmacokinetic profiles. Nat Commun. 2021;12(1):5458.
Adetokunboh OO, Schoonees A, Balogun TA, Wiysonge CS. Efficacy and safety of abacavir-containing combination antiretroviral therapy as first-line treatment of HIV infected children and adolescents: a systematic review and meta-analysis. BMC Infect Dis. 2015;15:1–13.
Cruciani M, Mengoli C, Malena M, Serpelloni G, Parisi SG, Moyle G, Bosco O. Virological efficacy of abacavir: systematic review and meta-analysis. J Antimicrob Chemother. 2014;69(12):3169–80.
Gurumukhi VC, Bari SB. Quality by design (QbD)–based fabrication of atazanavir-loaded nanostructured lipid carriers for lymph targeting: bioavailability enhancement using chylomicron flow block model and toxicity studies. Drug Deliv Transl Res. 2022;12(5):1230–52.
Zolotov S, Demina N, Zolotova A. Influence of water-soluble pharmaceutically acceptable polymers on the solubility of darunavir and darunavir ethanolate. Pharm Chem J. 2021;54:1274–7.
Sillman B, Bade AN, Dash PK, Bhargavan B, Kocher T, Mathews S, et al. Creation of a long-acting nanoformulated dolutegravir. Nat Commun. 2018;9(1):443.
Rao MR, Babrekar LS. Liposomal drug delivery for solubility and bioavailability enhancement of efavirenz. Indian J Pharm Sci. 2018;80(6):1115–24.
Singh L, Kruger HG, Maguire GE, Govender T, Parboosing R. The role of nanotechnology in the treatment of viral infections. Ther Adv Infect Dis. 2017;4(4):105–31.
Anderson PL, Rower JE. Zidovudine and lamivudine for HIV infection. Clin Med Rev Ther. 2010;2: a2004.
Ranjita S. Nanosuspensions: a new approach for organ and cellular targeting in infectious diseases. J Pharm Investig. 2013;43(1):1–26.
Singh K, Sarafianos SG, Sönnerborg A. Long-acting anti-HIV drugs targeting HIV-1 reverse transcriptase and integrase. Pharmaceuticals. 2019;12(2):62.
Serpi M, Pertusati F. An overview of ProTide technology and its implications to drug discovery. Expert Opin Drug Discov. 2021;16(10):1149–61.
De Clercq E. Tenofovir alafenamide (TAF) as the successor of tenofovir disoproxil fumarate (TDF). Biochem Pharmacol. 2016;119:1–7.
Acknowledgements
Gratitude to Copilot for its valuable help in constructing some of the tables.
Author information
Authors and Affiliations
Contributions
J.B and R.B wrote the main manuscript text, A.SH and A.A prepared figures and tables F.H wrote introduction and reviewed manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Babaei, J., Hosseini, F., Shadab, A. et al. Nanocrystallization strategies for enhanced HIV drug performance from solubility to sustained action. Discov Med 1, 43 (2024). https://doi.org/10.1007/s44337-024-00049-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44337-024-00049-z