
Abstract
This study presents the first annotated, haplotype-resolved, chromosome-scale genome of Lantana camara, a flowering shrub native to Central America and known for its dual role as an ornamental plant and an invasive species. Despite its widespread cultivation and ecological impact, the lack of a high-quality genome has hindered the investigation of traits of both ornamental and invasive. This research bridges the gap in genomic resources for L. camara, which is crucial for both ornamental breeding programs and invasive species management. Whole-genome and transcriptome sequencing were utilized to elucidate the genetic complexity of a diploid L. camara breeding line UF-T48. The genome was assembled de novo using HiFi and Hi-C reads, resulting in two phased genome assemblies with high Benchmarking Universal Single-Copy Orthologs (BUSCO) scores of 97.7%, indicating their quality. All 22 chromosomes were assembled with pseudochromosomes averaging 117 Mb. The assemblies revealed 29 telomeres and an extensive presence of repetitive sequences, primarily long terminal repeat transposable elements. The genome annotation identified 83,775 protein-coding genes, with 83% functionally annotated. In particular, the study mapped 42 anthocyanin and carotenoid candidate gene clusters and 12 herbicide target genes to the assembly, identifying 38 genes spread across the genome that are integral to flower color development and 53 genes for herbicide targeting in L. camara. This comprehensive genomic study not only enhances the understanding of L. camara’s genetic makeup but also sets a precedent for genomic research in the Verbenaceae family, offering a foundation for future studies in plant genetics, conservation, and breeding.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Lantana camara, commonly known as lantana, is a flowering shrub native to Central America. It has been introduced to various parts of the world, including India, Australia, Africa, and the United States, where it has become invasive in certain regions (Sharma et al. 2005; Bhagwat et al. 2012; Taylor et al. 2012; Shackleton et al. 2017). Despite its status as an invasive species, lantana remains a popular ornamental plant, contributing significantly to the flowering plant market in the United States. Its dual role as both an attractive ornamental and a problematic invasive species makes it a subject of interest for both ecological and economic reasons.
L. camara is a polyploid species with a base chromosome number of 11 (1x = 11). Ploidy levels in this species can range from diploid (2x) to hexaploid (6x), particularly in commercial varieties and breeding lines (Czarnecki et al. 2014; Parrish et al. 2021). It is believed that L. camara is an autopolyploid species, capable of increasing its ploidy levels due to the presence of unreduced female gametes (Czarnecki and Deng 2009). This polyploid nature potentially contributes to its adaptability and invasiveness, as well as its appeal as an ornamental plant.
Despite the rich genetic diversity inherent to L. camara, there is a conspicuous lack of comprehensive genomic resources to guide both breeding programs aimed at enhancing its ornamental traits and conservation efforts to manage its invasive characteristics. While a handful of transcriptome studies have been conducted focusing on aspects such as unreduced female gamete production genes and genes involved in phenylpropanoid biosynthesis (Peng et al. 2019; Shah et al. 2020), these offer only a partial view of the species’ genetic landscape. Moreover, a 2013 study that used chloroplast spacers and microsatellites to explore the population structure of lantana in India found high levels of genetic diversity at the examined loci (Ray and Quader 2014). This study suggested multiple introductions of the species into India, but it also underscored the need for more extensive genomic data. Given the high genetic diversity observed at just a few loci, there is a compelling case for a more comprehensive genomic exploration to unlock the full scope of lantana’s genetic makeup.
Whole-genome sequencing and de novo assembly have become indispensable tools for bioinformaticians and geneticists seeking to elucidate the traits inherent to plant species. The availability of such comprehensive genomic data empowers researchers to identify genes associated with key traits, develop molecular markers for breeding programs, and explore the phylogenetic relationships among plant species. For L. camara, the first step in this genomic exploration was the assembly and annotation of its chloroplast genome (Yaradua and Shah 2020). This study reported a chloroplast genome length of 154,388 bp and identified 90 protein-coding genes. Furthermore, a comparative analysis with other chloroplast genomes in the Verbenaceae family positioned L. camara as a sister taxon to Lippia origanoides. While this initial study laid important groundwork, it also highlighted the need for a more comprehensive genomic analysis to fully understand the genetic diversity and potential of this complex species.
Subsequent to the initial assembly of Lantana camara's chloroplast genome, two de novo genome assemblies have been published, both utilizing short-read sequencing data. The first, by Shah et al. (2022), was part of a broader study aimed at identifying gene targets for herbicide development across seven weed species. For L. camara, the study focused on a wild population in Queensland, Australia, and generated over 870 million 2 × 150 bp paired-end Illumina reads. These were assembled into 1,053,782 scaffolds with an N50 of 3 kb, resulting in a fragmented 1.57 Gb genome. This assembly had a Benchmarking Universal Single-Copy Orthologs (BUSCO) score of 79.8% and contained 18,369 protein-coding genes. Based on a k-mer estimated genome size of 6.36 Gb and other genome estimates, it can be inferred that the sequenced accession was tetraploid (Parrish et al. 2021). In the same year, Joshi et al. (2022) took a similar approach but used an accession with a 2.59 pg/2C DNA content. They generated over 500 million paired-end reads and assembled a 1.89 Gb genome with a notably higher BUSCO score of 99.3%. Although the total number of scaffolds was not reported, 26,057 were greater than 10 kb in size. While these two genomes provide valuable genomic data for the species, a chromosome-scale assembly is needed for more accurate and reliable genomics studies.
In the present study, a significant step forward is taken in the genomic exploration of L. camara. The first annotated, haplotype-resolved, chromosome-scale genome is presented, not only for this species but also for the Verbenaceae family as a whole. This comprehensive genomic resource aims to fill existing gaps in the understanding of lantana’s genetic diversity and complexity. By providing such a detailed genomic map, the study offers valuable insights that could be leveraged for both conservation efforts to control its invasive spread with new herbicides and breeding programs to enhance its ornamental traits. The work sets a new standard for genomic research in the Verbenaceae family and offers a robust foundation for future studies.
Materials and methods
Plant material and DNA extraction
Lantana breeding line UF-T48 plants were subjected to etiolation by enclosing them in dark cardboard boxes within a temperature-controlled greenhouse environment for a duration of three weeks. Subsequently, etiolated leaves were harvested, snap-frozen in liquid nitrogen, and preserved at -80°C. The frozen tissue samples were then shipped to CD Genomics (Shirley, New York, USA) for genomic DNA extraction and sequencing. The cetyl trimethylammonium bromide (CTAB) method was used to isolate high molecular weight DNA suitable for subsequent sequencing processes.
Library preparation and sequencing
The high molecular weight DNA was utilized to prepare SMRT-bell libraries following the protocol provided by Pacific Biosciences (Menlo Park, California, USA). Additionally, Arima-HiC libraries were prepared (Arima, Carlsbad, California, USA) for chromatin conformation capture sequencing. The PacBio libraries were sequenced using three 8 M SMRT cells on a PacBio Sequel II system. The Hi-C libraries underwent sequencing on an Illumina NovaSeq 6000 platform (Illumina, San Diego, California, USA). Validation of the Hi-C libraries was conducted using 12 Gb of Illumina paired-end reads, analyzed with qc3c v0.5 software (DeMaere and Darling 2021) in the absence of a reference genome.
RNA extraction and sequencing
For transcriptomic analysis, approximately 100 mg of tissue was collected from leaves, roots, green stems, and green fruits. The samples were immediately frozen in liquid nitrogen and stored at -80°C. Collection occurred at the University of Florida Institute of Food and Agricultural Sciences (UF/IFAS) Gulf Coast Research and Education Center in Wimauma, Florida, USA, between 8:00 and 9:00 AM in October 2022. RNA extraction was performed using the RNeasy Plant Mini Kit by Qiagen (Hilden, Germany). The extracted RNA was then sent to Novogene (Beijing, China) for library preparation and Illumina sequencing, targeting a yield of 6 Gb per sample.
Genome size estimation
The nuclear DNA content of the UF-T48 lantana breeding line was assessed following the protocol established by Doležel et al. (2007). Fresh leaf tissue was thoroughly rinsed with tap water. Approximately 30 mg of leaf tissue from both lantana and the internal standard, tomato (Solanum lycopersicum L. ‘Stupické polni rané’ (1.96 pg•2C−1)), were co-chopped in 1 mL of LB01 buffer. To this mixture, 50 µL of RNase (Sigma-Aldrich, St. Louis, Missouri, USA; 1 mg•mL−1) was added. The chopping was performed with a sharp razor blade to release the nuclei into the solution. The nuclei suspension was then filtered through a 50 µm pore nylon mesh filter to remove debris. Subsequently, 50 µL of the DNA fluorochrome propidium iodide (Sigma-Aldrich, St. Louis, Missouri, USA; 1 mg•mL−1) was added to stain the DNA. The stained nuclei were analyzed using a Cyflow® Ploidy Analyser (Sysmex Europe GmbH, Norderstedt, Germany) flow cytometer. Each leaf sample was subjected to three flow cytometric analyses, and three separate clonal plants were evaluated to ensure accuracy. The DNA content for each sample was calculated using the formula provided by Doležel et al. (2007), which is: nuclear DNA content of lantana = nuclear DNA content of internal standard × (mean fluorescence value of lantana sample ÷ mean fluorescence value of the internal standard). K-mer counting was performed on the raw DNA sequencing reads using KMC v3.2.1 (Kokot et al. 2017). The resulting K-mers were plotted in R v4.3.1 (R Core Team 2023) to estimate the genome size.
De novo assembly
For quality assessment, PacBio and Hi-C sequencing reads were analyzed using FastQC v0.11.7 (Andrews 2010). Hi-C reads underwent trimming at the GATC restriction enzyme site with HOMER v4.11 (Heinz et al. 2010). The genome assembly was performed de novo using hifiasm, integrating the PacBio data sets and trimmed Hi-C reads with default parameters on a 50-thread computational setup (Cheng et al. 2021). The processed Hi-C reads were mapped to the draft genome following the Arima-HiC mapping pipeline protocol (Arima Genomics 2019). BWA v0.7.17 (Li and Durbin 2009) was used for the mapping, and the mapped reads were filtered using SAMtools v1.15 (Li et al. 2009) and BEDtools v2.30.0 (Quinlan and Hall 2010). The yahs v1.1 tool (Zhou et al. 2023) utilized the mapped reads and draft assembly for scaffolding. To fill gaps in the chromosome assemblies, raw PacBio sequencing reads were applied using TGS GapCloser v1.2.1 (Xu et al. 2020), which is tailored for closing gaps in third-generation sequencing assemblies.
Assembly quality evaluation
The integrity and quality of both draft and final genome assemblies were evaluated using Quast v5.0.2 (Gurevich et al. 2013), which provided essential statistics such as contig number, N50, and total assembly length. To estimate the assembly quality value (QV), Merqury v1.3 (Rhie et al. 2020) was employed, offering a k-mer based quantification of accuracy. The completeness of the assemblies was gauged using the BUSCO database v5.3.0 (Simão et al. 2015). For the spatial organization of the genome, trimmed Hi-C reads were aligned to the phased assemblies with HiC-Pro v3.0.0 (Servant et al. 2015) and the resulting contact maps were visualized using Juicebox v1.11.08 (Durand et al. 2016), providing a chromosomal interaction overview. The two phased assemblies were aligned to each other and plotted to assess synteny using D-GENIES (Cabanettes and Klopp 2018). To further assess the assembly quality, the Long Terminal Repeat Assembly Index (LAI) (Ou et al. 2018) was calculated for each chromosome using LTR-retriever v2.5 (Ou and Jiang 2018). This index offers a measure of the completeness of long terminal repeat retrotransposons, which is indicative of the overall assembly quality, particularly in repeat-rich regions.
Repetitive sequence annotation
Transposable elements (TEs), which are crucial components of the genomic landscape, were annotated using EDTA v1.9.6 (Ou et al. 2019). This tool was employed with its default parameters to systematically identify and catalog the various classes of TEs within the assembly. Following the annotation, the identified TE regions were masked to mitigate their impact on subsequent analyses, utilizing RepeatMasker v4.1.1 (Tarailo-Graovac and Chen 2009). In parallel, the assembly was scanned for simple sequence repeats (SSRs) using PERF v0.4.6 (Avvaru et al. 2018), which extracted microsatellite sequences, a resource valuable for genetic mapping and marker development. Additionally, the search for telomeric sequences was conducted using tidk v0.2.31 (Brown et al. 2023), a specialized tool for identifying the repetitive DNA sequences that cap the ends of chromosomes, providing insights into chromosome structure and stability.
Gene annotation
For the prediction of protein-coding genes in the UF-T48 lantana genome, a comprehensive approach was employed utilizing RNA-seq data. This data encompassed a diverse range of tissues, including leaves, green stems, roots, and green fruits, ensuring a broad representation of the gene expression profile. Additionally, publicly available RNA-seq reads specific to UF-T48 flowers were incorporated, sourced from the NCBI project PRJNA956917 (Parrish et al. 2024). RNA-seq reads were trimmed using Trimmomatic v0.39 (Bolger et al. 2014) prior to input for gene prediction. The gene prediction was conducted using Braker v3.0.3 (Gabriel et al. 2023) a tool known for its accuracy in predicting gene structures in eukaryotic genomes, especially when guided by RNA-seq data. Following the prediction of protein-coding genes, functional annotation was carried out using eggNOG mapper v2.1.6 (Cantalapiedra et al. 2021). This tool is adept at categorizing genes into functional groups based on orthology and provides insights into potential gene functions by mapping them to known gene families and biological pathways.
Anthocyanin/Carotenoid pathway and herbicide target genes
Candidate genes with differential expression in anthocyanin and carotenoid pathways between white, yellow, and red flower colors were retrieved from NCBI project PRJNA956917 (Parrish et al. 2024). Herbicide target gene queries were obtained from the study published by Shah et al. (2022). To locate these candidate genes within the assembled UF-T48 genome, a DIAMOND search v2.1.8 (Buchfink et al. 2021) was employed.
Tissue specific RNA analysis
Trimmed RNA-seq reads were aligned to the assembled genome using HISAT2 v2.2.1 (Kim et al. 2019). Raw gene counts were obtained from the alignment files by employing HTSeq v2.0.3 (Anders et al. 2015).
Results
Genome and transcriptome sequencing
Ploidy analysis revealed that the somatic nuclei of the UF-T48 lantana breeding line contained approximately 3.02 ± 0.02 pg/2C of nuclear DNA which equates to approximately 2.95 Gb (3.02 pg/2C × 0.978) (Jaroslav Doležel et al. 2007). To achieve 60 × coverage, three 8 M single-molecule, real-time (SMRT) cells were utilized on a PacBio Sequel II sequencer (Table 1). This approach yielded 94.86 Gb of HiFi reads, generated from 5.6 million reads with an average read length of 16,816 bp. For Hi-C sequencing, an Illumina NovaSeq 6000 was employed, resulting in 30.92 Gb of data. This dataset comprised 103 million paired-end reads, each with an average length of 150 bp. Prior to scaling up the Hi-C sequencing to achieve 10 × coverage, the quality of Hi-C cross-linking was assessed using 12 Gb of Illumina paired-end reads. Analysis of the reads indicated that 80% of the reads were true products of proximity ligation, confirming the quality of the Hi-C data. To further assist with genome annotation, RNA-seq data were also generated for the UF-T48 breeding line. This resulted in 258 Gb of data, produced from 347 million paired-end reads, each 150 bp in length (Table 1).
Phased genome assembly
The genome of UF-T48 was assembled de novo, without the use of parental genomic data, by utilizing HiFi and Hi-C reads. K-mer analysis for genome size estimation aligned well with flow cytometry estimates, reporting an estimated genome size of 2.95 Gb. The k-mer frequency distribution (k = 23) exhibited a bimodal pattern, characteristic of a diploid organism with both homozygous and heterozygous genomic regions (Fig. 1). Numerical integration of the areas under the respective peaks of the distribution yielded an estimated heterozygosity of 72.31%. The assembly was phased into two separate datasets: the phased 1 assembly contained 1,295 contigs with an N50 of 104.99 Mb, while the phased 2 assembly had 426 contigs with an N50 of 85.09 Mb (Table 2). The largest contigs in the phased 1 and phased 2 assemblies measured 146.15 Mb and 170.47 Mb, respectively. Notably, 75% of the phased 2 assembly was composed of just 11 contigs, suggesting that the majority of each of the chromosomes are composed of just one contig. Both phased assemblies achieved a complete BUSCO score of 97.7%, indicating high-quality genome assemblies.

The k-mer (k = 23) distribution of T48 Lantana camara genome. The leftmost peak (~ 33 ×) represents the heterozygous region of the genome and the rightmost peak (~ 66 ×) represents the homozygous region of the genome
Hi-C reads were utilized to scaffold the phased genome assemblies. Of these, 99.65% of the first set of Hi-C reads (read 1) and 99.38% of the second set (read 2) were successfully mapped to the assembled genome. After filtering out unmapped reads, low-quality reads, and singletons, 37.20% of the uniquely mapped reads were retained for scaffolding. These filtered Hi-C read pairs were visualized using a Hi-C contact map, which revealed 11 chromosomes in both phased assemblies (Fig. 2). The density of the Hi-C pairs on the contact map suggests a low likelihood of mis-assemblies in the genome. Furthermore, a high degree of collinearity was observed between the two phased assemblies, with only a few small inversions and rearrangements evident (Fig. 3). All chromosomes were assembled gap-free with the exception of chromosome 5 that has one gap of unknown length at 26.17 Mb of chromosome 5A and 23.88 Mb of chromosome 5B (Fig. 4).

Hi-C contact map of phased 1 (a) and phased 2 (b) genome assemblies of UF-T48 Lantana camara. Each square corresponds to the chromosome listed along the horizontal axis. The color scale bar represents interaction frequencies. Higher values indicate more frequent interactions

Dotplot of aligned Lantana camara UF-T48 phased 1 and phased 2 genome assemblies

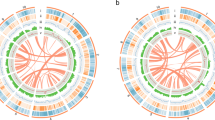
Circos plot displaying the characteristics of the UF-T48 Lantana camara genome assembly. Concentric circles from outside to inside show the following: 1) 22 assembled pseudomolecules (Mb); 2) heatmap of locations of predicted gene models with gene density increasing with darker shading; 3) heatmap of locations of predicted long terminal repeat (LTR) transposable elements (TEs) with LTR density increasing with darker shading; 4) locations of telomeric repeats; and 5) locations of gaps in the assembly
A total of 29 telomeres were identified using the telomeric motif (5'-AAACCCT-3') at the terminal ends of pseudo-chromosomes (Fig. 4). Telomere-to-telomere assembly was achieved for pseudo-chromosomes 1, 6, and 7 in both phased assemblies, as well as for pseudo-chromosomes 3B and 9B. Telomeres were identified at either the 5' or 3' end for all other pseudo-chromosomes, with the exception of chromosome 4A, which had no telomeric repeats detected. The Long Terminal Repeat (LTR) Assembly Index (LAI) for individual pseudo-chromosomes ranged from 18.51 to 23.1 (Fig. 5). The overall LAI scores were 19.61 for the phased 1 assembly and 19.12 for the phased 2 assembly, indicating high-quality genome assemblies.

The Long Terminal Repeat (LTR) Assembly Index (LAI) distribution in the UF-T48 Lantana camara genome assembly
Genome annotation
Repetitive sequences constitute 85.82% of the combined phased 1 and phased 2 lantana assemblies. Among these, long terminal repeat (LTR) transposable elements represent the majority, accounting for 70.23% of the repetitive sequences (Fig. 5; Supplementary Table 1). Simple sequence repeats (SSRs) comprise 2.12% of the genome, totaling 3,710,838 repeats (Supplementary Table 2). The genome contains 83,775 protein-coding genes, which give rise to 95,239 transcripts (Fig. 4; Supplementary Table 3). The average gene length is 2,415 bp, with a mean coding sequence length of 1,212 bp and an average of 4.5 exons per gene. Protein-coding genes span approximately 8.2% of the UF-T48 genome, equivalent to 202,281,879 bp. Out of the identified protein-coding genes, 83% were functionally annotated. A BUSCO analysis of these annotated genes revealed the presence of 2,176 complete core eudicot genes, accounting for 93.6%. Only 1.6% of these genes were fragmented, and 4.8% were missing.
Parrish et al. (2024) identified 40 anthocyanin and 2 carotenoid pathway genes that were differentially expressed in red and white flowers, respectively. Alignment of these clusters to the assembled UF-T48 genome revealed 38 genes located throughout the genome (Fig. 6). All of the gene clusters were representative of two alleles per locus. Chromosomes one, five, and seven contained the highest number of candidate genes with three candidates per chromosome.

Locations of differentially expressed anthocyanin and carotenoid genes in the UF-T48 Lantana camara genome assembly
Common herbicide gene targets
While eradicating invasive lantana genotypes from landscapes can be accomplished through herbicide applications, the hardy plant can take many applications for death to occur. This necessitates that more specialized herbicides be developed to control this invasive plant. To support this research, 12 common gene targets for herbicide development identified in the study by Shah et al. (2022) were extracted from the genome (Supplementary Table 4). All 12 gene targets were identified in full length within the genome including the two previously missing targets beta-isopropylmalate dehydrogenase (IMDH) and acetyl-CoA carboxylase 1 (accA) genes. These genes, integral to the branched-chain amino acid (BCAA) pathway and the acetyl-CoA carboxylase (ACCase) inhibitors, respectively, are crucial for the development of targeted herbicides.
Tissue specific RNA analysis
In the process of annotating the Lantana camara UF-T48 genome, RNA reads from various tissue types were aligned to the genome to quantify gene expression across different tissues. Out of the total 83,775 predicted genes in the genome, 41,729 genes (49.81%) were detected in the RNA-seq data derived from the six tissue types analyzed (Fig. 7). Notably, a significant number of genes, 22,344, were found to be expressed across all tissue types, indicating a broad spectrum of shared genetic activity. Among the different tissues, unopened flowers exhibited the highest number of expressed genes, with 40.45% of all predicted genes in the genome showing some level of expression in this tissue. In contrast, green fruit tissue had the fewest number of unique genes expressed, with only 465 genes uniquely expressed in this tissue type.

RNA gene expression counts from 6 tissue types that were used in the annotation of the UF-T48 Lantana camara genome assembly. This image was generated by ChatGPT-4 DALL•E 3, https://chat.openai.com
Discussion
The genome of the UF-T48 lantana breeding line, as revealed by this study, offers significant insights into the genetic composition of this ornamental plant. The findings align with prior research regarding genome size estimation techniques, with the ploidy analysis closely mirroring the K-mer analysis, a consistency observed in other plant genomes (Jaroslav Doležel et al. 2007).
A high level of genome heterozygosity as estimated by k-mers underscores the importance of having a haplotype phased assembly to capture the full genetic diversity present. The phased genome assembly, achieved without parental genomic data, underscores the advancements in sequencing technologies. The high N50 values of both phased assemblies, especially when compared to other plant genomes, indicate a high level of contiguity and completeness (Kersey 2019). The utilization of Hi-C reads for scaffolding further enhanced the quality of the assembly, as evidenced by the high mapping rates and the clear visualization of chromosome pairs on the Hi-C contact map. This approach, combined with the high BUSCO scores, suggests that the UF-T48 genome assembly is of superior quality and can serve as a reference for future lantana genomic studies.
The identification of telomeres in the UF-T48 genome is crucial for understanding chromosome stability and integrity. The presence of telomeres in most pseudo-chromosomes, and the achievement of telomere-to-telomere assembly in several, is indicative of a comprehensive and high-quality assembly. The LAI scores further corroborate the quality of the assembly, aligning with scores observed in other high-quality plant genome assemblies.
Repetitive sequences, particularly LTR transposable elements, dominate the UF-T48 genome. This high proportion of repetitive sequences is consistent with other complex plant genomes and underscores the challenges of assembling such genomes (Mehrotra and Goyal 2014; Macas et al. 2015).
Despite these challenges, the successful annotation of a significant number of protein-coding genes, with a high percentage being functionally annotated, is a testament to the robustness of the sequencing and annotation methodologies employed. While only half of the predicted protein-coding genes were supported by RNA-seq data, this likely reflects the limited depth of RNA-seq data coverage and the restricted range of tissue types analyzed. Nevertheless, the RNA-seq data proved adequate for training the ab initio model, enabling the prediction of the remaining genes in the genome. The BUSCO analysis results further emphasize the completeness of the UF-T48 genome assembly. The high percentage of complete core eudicot genes, coupled with a minimal number of fragmented or missing genes, places the UF-T48 genome among the top-tier of plant genome assemblies in terms of quality and completeness.
The alignment of anthocyanin and carotenoid biosynthetic pathway genes, previously identified in a de novo transcriptome study (Parrish et al. 2024), to the UF-T48 genome represents a significant step forward in connecting functional genomics with structural genomics in this species. The successful localization of genes such as anthocyanidin synthase (ANS), basic helix-loop-helix 42 (BHLH42), cinnamate-4-hydroxylase (C4H), and others not only emphasizes the UF-T48 assembly’s role as a robust scaffold for integrating transcriptomic and genomic data but also showcases its utility in diverse genomic explorations. This is further exemplified by identifying the 12 common gene targets for herbicide development, originally identified by Shah et al. (2022). The identification of these gene targets, including the previously missing IMDH and accA genes, within the UF-T48 genome signifies a parallel yet equally significant stride in understanding and combating herbicide development in lantana.
This dual achievement underscores the UF-T48 genome assembly’s versatility, serving both ornamental breeding programs and herbicide research. While the precise localization of biosynthetic pathway genes facilitates the manipulation of genes for vibrant coloration in lantana flowers, the mapping of herbicide-target genes offers a genetic blueprint for developing more effective herbicides. Thus, the UF-T48 genome emerges as a comprehensive tool, aiding in the creation of new floral varieties with desired characteristics and in controlling invasive genotypes, addressing both aesthetic and ecological concerns associated with lantana.
Conclusion
This study showcases the successful assembly of the UF-T48 lantana breeding line, a complex genome, using a combination of PacBio HiFi long-read sequencing and Hi-C data. This approach facilitated the creation of the first chromosome-scale, haplotype-phased assembly for Lantana camara. Remarkably, this high-quality assembly was achieved without the need for parental sequence data. The resulting genome provides a comprehensive genetic blueprint of this ornamental plant species. The availability of this UF-T48 genome assembly will undoubtedly pave the way for the identification of genes associated with key ornamental and invasive traits, furthering the development of advanced breeding tools and strategies for Lantana camara and the Verbenaceae family.
Availability of data and materials
The genome assembly files of the UF-T48 lantana genome are available from the NCBI Sequence Read Archive BioProject database with the accession numbers PRJNA1065478, PRJNA1069082, and PRJNA1069083.
References
Anders S, Pyl PT, Huber W. HTSeq – a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. https://doi.org/10.1093/BIOINFORMATICS/BTU638.
Andrews S. Babraham Bioinformatics - FastQC A quality control tool for high throughput sequence data. 2010. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 7 Mar 2023.
Arima Genomics. Arima-HiC mapping pipeline. 2019. https://github.com/ArimaGenomics/mapping_pipeline/tree/master. Accessed 7 Nov 2023.
Avvaru AK, Sowpati DT, Mishra RK. PERF: an exhaustive algorithm for ultra-fast and efficient identification of microsatellites from large DNA sequences. Bioinformatics. 2018;34:943–8. https://doi.org/10.1093/BIOINFORMATICS/BTX721.
Bhagwat SA, Breman E, Thekaekara T, Thornton TF, Willis KJ. A battle lost? Report on two centuries of invasion and management of Lantana camara L. in Australia, India and South Africa. PLoS One. 2012. https://doi.org/10.1371/journal.pone.0032407.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. https://doi.org/10.1093/BIOINFORMATICS/BTU170.
Brown M, De la GonzálezRosa PM, Mark B. A Telomer Identification toolkit. 2023. Zenodo. https://doi.org/10.5281/zenodo.10091385.
Buchfink B, Reuter K, Drost HG. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods. 2021;18:366–8. https://doi.org/10.1038/s41592-021-01101-x.
Cabanettes F, Klopp C. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ. 2018. https://doi.org/10.7717/PEERJ.4958.
Cantalapiedra CP, Hern̗andez-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. MOL BIOL EVOL. 2021;38:5825–9. https://doi.org/10.1093/MOLBEV/MSAB293.
Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18:170–5. https://doi.org/10.1038/s41592-020-01056-5.
Czarnecki DM, Deng Z. Occurrence of unreduced female gametes leads to sexual polyploidization in lantana. J Am Soc Hortic Sci. 2009;134:560–6. https://doi.org/10.21273/JASHS.134.5.560.
Czarnecki DM, Hershberger AJ, Robacker CD, Clark DG, Deng Z. Ploidy levels and pollen stainability of Lantana camara cultivars and breeding lines. HortScience. 2014;49:1271–6. https://doi.org/10.21273/HORTSCI.49.10.1271.
DeMaere MZ, Darling AE. qc3C: Reference-free quality control for Hi-C sequencing data. PLoS Comput Biol. 2021. https://doi.org/10.1371/JOURNAL.PCBI.1008839.
Doležel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc. 2007;2:2233–44. https://doi.org/10.1038/nprot.2007.310.
Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016;3:99–101. https://doi.org/10.1016/J.CELS.2015.07.012.
Gabriel L, Brůna T, Hoff KJ, Ebel M, Lomsadze A, Borodovsky M, et al. BRAKER3: fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS and TSEBRA. bioRxiv.:2023.06.10.544449 [Preprint]. 2023 [cited 2024 Mar 5]: [21 p.]. Available from: https://doi.org/10.1101/2023.06.10.544449.
Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013;29:1072–5. https://doi.org/10.1093/BIOINFORMATICS/BTT086.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. https://doi.org/10.1016/J.MOLCEL.2010.05.004.
Joshi AG, Praveen P, Ramakrishnan U, Sowdhamini R. Draft genome sequence of an invasive plant Lantana camara L. Bioinformation. 2022;18:739–41. https://doi.org/10.6026/97320630018739.
Kersey PJ. Plant genome sequences: past, present, future. Curr Opin Plant Biol. 2019;48:1–8. https://doi.org/10.1016/J.PBI.2018.11.001.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37:907–15. https://doi.org/10.1038/s41587-019-0201-4.
Kokot M, Dlugosz M, Deorowicz S. KMC 3: counting and manipulating k-mer statistics. Bioinformatics. 2017;33:2759–61. https://doi.org/10.1093/BIOINFORMATICS/BTX304.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. https://doi.org/10.1093/BIOINFORMATICS/BTP324.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. https://doi.org/10.1093/BIOINFORMATICS/BTP352.
Macas J, Novak P, Pellicer J, Cizkova J, Koblizkova A, Neumann P, et al. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE. 2015. https://doi.org/10.1371/JOURNAL.PONE.0143424.
Mehrotra S, Goyal V. Repetitive sequences in plant nuclear DNA: types, distribution, evolution and function. Genom Proteom Bioinform. 2014;12:164–71. https://doi.org/10.1016/J.GPB.2014.07.003.
Ou S, Jiang N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176:1410–22. https://doi.org/10.1104/PP.17.01310.
Ou S, Chen J, Jiang N. Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. 2018. https://doi.org/10.1093/NAR/GKY730.
Ou S, Su W, Liao Y, Chougule K, Agda JRA, Hellinga AJ, et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019;20:1–18. https://doi.org/10.1186/S13059-019-1905-Y.
Parrish SB, Qian R, Deng Z. Genome size and karyotype studies in five species of Lantana (Verbenaceae). HortScience. 2021;56:352–6. https://doi.org/10.21273/HORTSCI15603-20.
Parrish SB, Paudel D, Deng Z. Transcriptome analysis of Lantana camara flower petals reveals candidate anthocyanin biosynthesis genes mediating red flower color development. G3-Genes Genom Genet. 2024. https://doi.org/10.1093/G3JOURNAL/JKAD259.
Peng Z, Bhattarai K, Parajuli S, Cao Z, Deng Z. Transcriptome analysis of young ovaries reveals candidate genes involved in gamete formation in Lantana camara. Plants. 2019. https://doi.org/10.3390/PLANTS8080263.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. https://doi.org/10.1093/BIOINFORMATICS/BTQ033.
R Core Team. R: A Language and Environment for Statistical Computing. 2023. https://www.R-project.org/. Accessed 7 Nov 2023.
Ray A, Quader S. Genetic diversity and population structure of Lantana camara in India indicates multiple introductions and gene flow. Plant Biol. 2014;16:651–8. https://doi.org/10.1111/plb.12087.
Rhie A, Walenz BP, Koren S, Phillippy AM. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020;21:1–27. https://doi.org/10.1186/S13059-020-02134-9.
Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:1–11. https://doi.org/10.1186/S13059-015-0831-X.
Shackleton RT, Witt ABR, Aool W, Pratt CF. Distribution of the invasive alien weed, Lantana camara, and its ecological and livelihood impacts in eastern Africa. Afr J Range Forage Sci. 2017;34:1–11. https://doi.org/10.2989/10220119.2017.1301551.
Shah S, Lonhienne T, Murray CE, Chen Y, Dougan KE, Low YS, et al. Genome-guided analysis of seven weed species reveals conserved sequence and structural features of key gene targets for herbicide development. Front Plant Sci. 2022. https://doi.org/10.3389/FPLS.2022.909073.
Shah M, Alharby HF, Hakeem KR, Ali N, Rahman IU, Munawar M, et al. De novo transcriptome analysis of Lantana camara L. revealed candidate genes involved in phenylpropanoid biosynthesis pathway. Sci Rep. 2020. https://doi.org/10.1038/S41598-020-70635-5.
Sharma GP, Raghubanshi AS, Singh JS. Lantana invasion: an overview. Weed Biol Manag. 2005;5:157–65. https://doi.org/10.1111/J.1445-6664.2005.00178.X.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2. https://doi.org/10.1093/BIOINFORMATICS/BTV351.
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009. https://doi.org/10.1002/0471250953.BI0410S25.
Taylor S, Kumar L, Reid N. Impacts of climate change and land-use on the potential distribution of an invasive weed: a case study of Lantana camara in Australia. Weed Res. 2012;52:391–401. https://doi.org/10.1111/J.1365-3180.2012.00930.X.
Xu M, Guo L, Gu S, Wang O, Zhang R, Peters BA, et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience. 2020;9:1–11. https://doi.org/10.1093/GIGASCIENCE/GIAA094.
Yaradua SS, Shah M. The complete chloroplast genome of Lantana camara L. (Verbenaceae). Mitochondrial DNA Part B. 2020;5:918–9. https://doi.org/10.1080/23802359.2020.1719920.
Zhou C, McCarthy SA, Durbin R. YaHS: yet another Hi-C scaffolding tool. Bioinformatics. 2023. https://doi.org/10.1093/BIOINFORMATICS/BTAC808.
Acknowledgements
The authors would like to express their gratitude to their lab members for their valuable assistance and to the anonymous reviewers for their thorough and constructive feedback on the manuscript. Thank you to CD Genomics (Shirley, NY, USA) for PacBio and Hi-C sequencing services and Novogene (Beijing, China) for RNA sequencing services.
Funding
This work was supported in part by the U.S. Department of Agriculture Hatch projects (Projects No. FLA-GCC-005065 and No. FLA-GCC-005507).
Author information
Authors and Affiliations
Contributions
ZD designed and planned the project. SBP performed all RNA extractions, genome size estimations, and computational analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare that they have no competing interests. The corresponding author, ZD, is a member on this journal’s editorial team, and was not involved in the journal's review or decisions related to this mauscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Parrish, S.B., Deng, Z. The first high-quality genome assembly and annotation of Lantana camara, an important ornamental plant and a major invasive species. HORTIC. ADV. 2, 14 (2024). https://doi.org/10.1007/s44281-024-00043-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44281-024-00043-6