
Abstract
Sequencing strategies have continually advanced, with high-throughput sequencing (HTS) technologies emerging as pivotal tools in plant phylogenomics. As a standard form of target capture sequencing, hybridization target enrichment sequencing (Hyb-seq) is innovative and flexible, and then HTS strategy is widely adopted in phylogenomics. The universal Angiosperms353 probe sets (Angiosperms353) are instrumental in Hyb-seq and have been witnessed increased application in recent years. This review integrates numerous publications and empirical datasets to comprehensively assess the potential applications of Angiosperms353. Through evaluations using 18-taxa and 343-taxa genomic datasets, this review explores potential factors influencing Angiosperms353 gene capture. The RNA-seq strategy yielded the highest number of Angiosperms353 genes, followed by whole-genome sequencing and genome skimming. Increased sequencing depth enhanced gene capture yields, and no evident bias was observed across all tested angiosperm groups. In the genomics era with extensive HTS data, this review provides comprehensive guidelines for the practical application of Angiosperms353, promoting resource recycling and maximizing genomic data sharing and utilization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A taxonomy-based phylogenetic tree serves as the foundation for biodiversity conservation and offers guidelines for mining and utilizing germplasm resources in horticulture and agriculture. Previous plant phylogenetic studies primarily relied on Sanger sequencing, utilizing specific region markers, such as ITS, ETS, rbcL, atpB, matK, and trnL-trnF (Shi et al. 2013; Zhao et al. 2016; Liao et al. 2023; Phang et al. 2023). However, these markers have inherent limitations, including short lengths (usually <1000 base pairs (bp)), high homogeneity (e.g., plastid loci), or gene paralogs (e.g., ITS). Individual molecular markers may introduce biases, resulting in unresolved phylogenetic relationships with limited evolutionary history (Coissac et al. 2016; Wilkinson et al. 2017).
Because of these limitations, there has been a noticeable decline in the generation of Sanger sequencing data (for individual genes/loci or molecular markers) in recent years (Supplementary Fig. S1A). However, high-throughput sequencing (HTS) technology, including whole-genome sequencing (WGS) (Ng and Kirkness 2010), restriction site-associated DNA sequencing (RAD-seq) (Shafer et al. 2016), transcriptome sequencing (RNA-seq) (Kumar et al. 2012), genome skimming (Dodsworth 2015) and hybridization target enrichment sequencing (Hyb-seq) (Weitemier et al. 2014), has played a pivotal role in advancing biological discoveries (summarized in Table 1). HTS not only enhances the convenience of genome sequencing for various plant groups (Dodsworth et al. 2019), but also overcomes challenges related to DNA degradation (Bakker et al. 2016; Robillard et al. 2020; Folk et al. 2021; Zhao et al. 2023). Additionally, HTS yields additional DNA data with genetic information, significantly increasing genome-level sequencing data (Supplementary Fig. S1B). Initially, researchers preferred using plastid genomes due to their relatively small size, conserved structure, low recombination rates, ease of assembly, high sequence homogeneity, and high stability in specific regions. Plastid genomes have been widely used for reconstructing phylogenetic relationships in various plants (Gitzendanner et al. 2018; Forrest et al. 2019; Li et al. 2019; Guo et al. 2022). Nevertheless, plastid data may lack informative sites for addressing complex evolutionary radiations (Turner et al. 2016; Huang et al. 2023). Furthermore, since only maternally inherited information is provided, plastid genomes are represented as a single nonrecombinant linkage group. Consequently, all genes within this group are considered to represent the same evolutionary history. Moreover, complex evolutionary processes such as hybridization, introgression, horizontal gene transfer, polyploidy, and incomplete line sorting can hinder the recovery of the true species history (Wolf et al. 2018; Pillon et al. 2021; Thureborn et al. 2022).
In recent years, nuclear genomes have become increasingly accessible, and various international genome-related collaborations have promoted the generation and analysis of global biological genomes. Notably, the Earth BioGenome Project aims to sequence and annotate the genomes of approximately 1.5 million eukaryotic species within a decade (Lewin et al. 2018). Similarly, projects such as OneKP (Leebens-Mack et al. 2019), GoFlag (Breinholt et al. 2021), and PAFTOL (Baker et al. 2022) have emerged. These endeavors will establish the genomic foundation to address crucial biological issues and enhance our understanding of global biodiversity and resource management.
Despite remarkable advances in sequencing technology and the generation of extensive genome-level sequencing data, these invaluable and crucial genomic resources yet to be explored. Therefore, target capture sequencing has rapidly evolved, by enriching and sequencing specific genomic regions of interest using capture reagent kits (Mamanova et al. 2010). The process begins with capturing on-target sequences or genomic regions and identifying target genes (e.g., genes, exons, or ultraconserved elements) through various genomic data resources (WGS, RNA-seq, and genome skimming), and it focuses on low- or single-copy genes (McKain et al. 2018). Target capture sequencing enables cost-effective and reliable generation of sequences for hundreds or even thousands of target gene loci (Cronn et al. 2012; Bragg et al. 2015; Leebens-Mack et al. 2019).
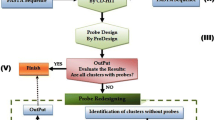
Hyb-seq, a method that integrates targeted sequencing with genome skimming, has revolutionized phylogenetic reconstruction by providing abundant molecular data and numerous low- and multicopy nuclear genes for various phylogenetic studies (Weitemier et al. 2014; Dodsworth et al. 2019). The process begins by screening conserved regions for enrichment through reference genome sequencing alignment analysis. Subsequently, single-stranded DNA or RNA probes with a length of 80–120 bp and a streptavidin coat based on sequence conservation are synthesized. These probes are then hybridized and paired with the genomic library of the samples. The on-target sequences are enriched and fixed through streptavidin-coated magnetic beads that bind biotinylated baits (and bait-bound DNA). Finally, the library fragments that have not been hybridized are eluted, and the target library fragments are retrieved for HTS (Dodsworth et al. 2019) (reviewed in Fig. 1). This technique has become a standard approach in phylogenetics, outperforming Sanger sequencing, particularly in resolving plant phylogenetic relationships (Wang et al. 2021; Sun et al. 2022; Sundararaman et al. 2023).

The technical workflow of Hyb-seq, modified from the past research (Johnson et al. 2019)
Compared with other HTS methods, Hyb-seq reduces workload and computational complexity, and offers flexibility in designing probes to effectively address phylogenetic relationships at various taxonomic levels (McKain et al. 2018; Ogutcen et al. 2021). Among widely used probe sets, Angiosperms353, derived from 600 angiosperm species, is highly favored. This specialized capture probe, consisting of 120 bp, was meticulously designed using the k-medoid clustering method. Subsequently, target enrichment sequencing is conducted for these 353 single-copy nuclear genes (Johnson et al. 2019). Angiosperms353 has been extensively applied to elucidate various levels of phylogenetic relationships, providing a more extensive molecular dataset for reconstructing evolutionary relationships (Baker et al. 2021; Joyce et al. 2023; Larson et al. 2023; Masters et al. 2023). In addition, the development of tools for retrieving Angiosperms353 data has further facilitated its broader applications.
As Angiosperms353 gains popularity among researchers, publications using this approach have gradually increased since its discovery (Supplementary Fig. S1a). Nevertheless, the lack of essential review studies comparing the capture efficiency of Angiosperms353 among various software/tools using empirical data and providing practical guidance for handling massive high-throughput data complicates achieving cost-effectiveness in this context. Hence, retrieving Angiosperms353 from diverse HTS datasets obtained from various platforms and methods still poses a challenge.
In this article, we comprehensively evaluated the potential application of this approach in Angiosperms353. This assessment is based on the literature review and testing with extensive empirical data. We began by presenting an overview of the research progress on Angiosperms353’s application. Subsequently, we assembled an 18-taxa genomic dataset to evaluate the performance of two commonly used tools for capturing Angiosperms353. Furthermore, we included a 343-taxa dataset to comprehensively assess various potential factors impacting Angiosperms353 yields (Fig. 2). The primary goal of this review is to provide an optimal and cost-effective solution for applying Angiosperms353 in phylogenomics across multiple biology disciplines, ultimately facilitating the integration of extensive HTS resources and maximizing genomic data sharing and utilization.

Workflow of the evaluation on Angiosperms353 using empirical data from this study. ANA grade, Amborellales, Nymphaeales, and Austrobaileyales orders recognized by APG IV (2016); WGS, whole-genome sequencing; RAD-seq, restriction site-associated DNA sequencing; RNA-seq, transcriptome sequencing
Utilizing Angiosperms353 and comparing it with lineage-specific probe sets
Angiosperms353, widely adopted for its high universality, offers the advantage of off-the-shelf use without requiring costly start-up investments. Moreover, it diminishes the requirement for specialized bioinformatics expertise in probe design (Johnson et al. 2019). Therefore, it is applied across all levels of angiosperms in phylogenomics, spanning the order level (Antonelli et al. 2021; Zuntini et al. 2021), family level (Hendriks et al. 2021; Yardeni et al. 2021; Haigh et al. 2023; Joyce et al. 2023; Larson et al. 2023), genus level (Frost et al. 2021; Howard et al. 2022; Simões et al. 2022), and even species and population levels (Crowl et al. 2022; Masters et al. 2023).
Utilizing Angiosperms353 at various taxonomic levels
There is substantial evidence supporting the efficacy of Angiosperms353 in elucidating relationships among families across various orders. Notable examples include its application in Cornales (Thomas et al. 2021), Myrtales (Maurin et al. 2021), Gentianales (Antonelli et al. 2021), Commelinales (Zuntini et al. 2021), Oxalidales (Pillon et al. 2021), Orchids (Eserman et al. 2021), and Sapindales (Joyce et al. 2023). Angiosperms353 demonstrated remarkable effectiveness in Gentianales, offering valuable insights into its evolution. Antonelli et al. (2021) used Angiosperms353 to study approximately 150 Gentianales species, revealing well-resolved relationships within this order. Remarkably, over 80% of the nodes in the phylogenetic tree received robust support, and the research strongly supported the monophyly of each of the five families within Gentianales. Furthermore, Angiosperms353 offered reliable evidence for resolving phylogenetic relationships among families within Commelinales. Zuntini et al. (2021) conducted comprehensive phylogenomics on 290 species in Commelinales, capturing 352 genes with Angiosperms353. This effort supported the monophyly of Commelinales and its five families, effectively clarifying relationships among and within these families.
Similarly, the abundance of Angiosperms353 proves sufficient for inferring high-resolution phylogenetic relationships at the genus level. Multiple instances highlight its effectiveness in this context, such as in Apiaceae (Clarkson et al. 2021), Araceae (Haigh et al. 2023), Cyperaceae (Larridon et al. 2020), Cactaceae (Acha and Majure 2022), Convolvulaceae (Simões et al. 2022), Rubioideae (Thureborn et al. 2022), and Primulaceae (Larson et al. 2023). Angiosperms353 played a crucial role in establishing a new classification system for Cyperaceae. This research sequenced 311 samples using Angiosperms353, providing robust support for the monophyly of Cyperaceae. The phylogenomic framework of Cyperaceae received significant support for two subfamilies, 24 tribes, 10 subtribes, and most previously recognized genera (Larridon et al. 2021).
Increasing evidence suggests that the sequence data obtained through Angiosperms353 exhibit sufficient variability for reconstructing relationships at the species level. Several studies have explored the applicability of Angiosperms353 in species-level phylogenetic studies, including those involving Otoba (Frost et al. 2021), Solanum (Gagnon et al. 2022), Palaquium (Phang et al. 2023), Corydalis (Chen et al. 2023), Vaccinium section Cyanococcus (Crowl et al. 2022), and Urochloa sensu lato (Masters et al. 2023). For instance, Frost et al. (2021) analyzed the phylogenetic relationships of 20 Otoba samples, strongly supporting its monophyly. The authors revealed three clades within the genus and resolved the first phylogeny of Otoba using targeted enrichment sequencing. Moreover, Crowl et al. (2022) utilized Angiosperms353 for a phylogenetic analysis of Vaccinium section Cyanococcus. This research successfully captured 323 genes, revealing that the northern lineages of V. boreale and V. myrtilloides were sisters, V. boreale was nonmonophyletic, and V. caesariense was nested in the V. fuscatum clade.
In summary, Angiosperms353 is widely used across different taxonomic levels and plays a vital role in advancing the establishment of a complete and unified tree of life.
Comparison between Angiosperms353 and lineage-specific probe sets
While Angiosperms353 is an attractive choice for phylogenetic studies, it may face challenges in fully exploring sufficient information sites to resolve phylogenetic relationships for all taxa, particularly at lower taxonomic levels, such as species or populations. This limitation arises from its universal design intended for all angiosperms. Factors such as rapid species radiation, low sequence differentiation, and gene and genome duplications can diminish the efficiency of Angiosperms353 application (Eserman et al. 2021). Consequently, there is a growing inclination toward designing lineage-specific probe sets for specific taxonomic groups. These probes integrate local information from single-copy genes, ensuring higher fidelity between the probe and the target. This approach successfully selects and restores a larger portion of orthologs, providing more phylogenetic information and sufficient variable sites to improve the phylogenetic resolution for target groups. It also allows for maximizing the acquisition of phylogenetic signals for each sequencing region (Folk et al. 2015). Hence, lineage-specific probe sets prove more suitable for addressing challenges with recalcitrant nodes in a phylogeny, particularly for taxa at shallow levels (Gomez et al. 2019; Eserman et al. 2021; Hendriks et al. 2021; Mandel 2021; McDonnell et al. 2021; Yardeni et al. 2021; Acha and Majure 2022). Compared to Angiosperms353, lineage-specific probe sets may be more effective in elucidating phylogenetic relationships within rapidly radiating clades (Lamesch et al. 2012; Romeiro-Brito et al. 2022).
However, the prevalence of genome duplications in angiosperms poses challenges in distinguishing paralogs from orthologs when designing lineage-specific probe sets, leading to potential false-positive relationships in phylogenetic inference (Cheng et al. 2017; Romeiro-Brito et al. 2022). Additionally, lineage-specific probe sets typically exhibit minimal gene overlap and are frequently incompatible with data generated by other probe sets or sequencing strategies. This limitation significantly restricts opportunities to share and reuse data across diverse studies and projects. In contrast, Angiosperms353 offers the advantage of eliminating the need to develop specific probes, saving both time and cost. The data generated through Angiosperms353 can be integrated with genomic sequence data from other taxa produced by unrelated projects, enabling a larger scale of phylogenomics (Hendriks et al. 2021; Yardeni et al. 2021; Simões et al. 2022). For example, Yardeni et al. (2021) compared the relative efficacy of the lineage-specific probe Bromelia1776 with Angiosperms353 in terms of gene capture success, considering bait design, data processing, and other factors. Although lineage-specific probe sets exhibited a higher target capture rate and phylogenetic resolution, their development proved time-consuming and required extensive bioinformatics expertise.
Compared with lineage-specific probe sets, Angiosperms353 provides a cost-effective and time-efficient alternative, proving undoubtedly effective for groups lacking genomic resources, albeit potentially providing relatively fewer informative sites. It has been reported that, Angiosperms353 is as effective as lineage-specific probe sets and highly consistent with inferred phylogenetic relationships (Chau et al. 2018; Larridon et al. 2021; Siniscalchi et al. 2021; Ufimov et al. 2021; Yardeni et al. 2021; Simões et al. 2022; Thureborn et al. 2022). Thureborn et al. (2022) argued while lineage-specific probe sets typically enhance capture efficiency in target regions due to their specificity, the proportion of phylogenetic information sites of Angisperms353 slightly exceeded that of lineage-specific probe sets. Angiosperms353 and lineage-specific probe sets produced similar results at the genus and subgenus levels. Eserman et al. (2021) compared the phylogenetic outcomes of Angiosperms353 and Orchidaceae-specific probes Orchidaceae963 in three major Orchid subfamilies—Orchidaceae, Epidendroideae, and Orchidaceae. Although Orchidaceae963 strongly supported the reconstructed phylogenetic relationships and clarified relationships within Orchidaceae, the topological structure of the trees generated by Orchidaceae963 was generally consistent with the topology recovered by Angiosperms353. Therefore, Angiosperms353 holds substantial potential for conducting phylogenomic research at all taxonomic levels.
Comprehensive evaluation of factors impacting Angiosperms353 application
Given the substantial potential of Angiosperms353, the potential factors affecting the capture of Angiosperms353 genes were comprehensively evaluated in terms of software/tools, sequencing strategies, sequencing depths, and entire representative angiosperm groups, following the workflow illustrated in Fig. 1. This assessment was conducted in two steps. In the first step, an 18-taxa genomic dataset was assembled to assess the performance of two commonly used tools, HybPiper and Easy353. This step aimed to investigate how different software programs impact the yields of Angiosperms353 captured. In the second step, a more extensive 343-taxa genomic dataset was assembled for in-depth testing using the ascendant Easy353. This step aimed to assess how different sequencing strategies, sequencing depths, and angiosperm groups might influence the yields of Angiosperms353 captured.
Assessing the performance of tools designed for capturing Angiosperms353 genes is crucial to ensure the effectiveness and practical application of Angiosperms353 data. Notable tools for Angiosperms353 capture include MarkerMiner (Chamala et al. 2015), HybPiper (Johnson et al. 2016), Phyluce (Faircloth 2016), HybPhaser (Nauheimer et al. 2021), Easy353 (Zhang et al. 2022), PhyloHerb (Cai et al. 2022), and GeneMiner (Xie et al. 2023). Phyluce is a software package originally developed for analyzing data from ultraconserved elements in organismal genomes. PhyloHerb extracts low-copy nuclear genes (e.g., Angiosperms353) from genome skimming data using reference sequences and raw reads. HybPiper, designed for targeted sequence capture, enriches DNA sequencing libraries for gene regions of interest, particularly capturing exons and flanking introns from the Hyb-seq platform. The assembled sequence data can be utilized for phylogenetic analysis at different levels. A companion pipeline, putative paralog detection (PPD; Zhou et al. 2022), is an extension to the HybPiper pipeline, and identifies putative paralogs based on sequence similarity and the presence of heterozygous sites at each locus (Zhou et al. 2022). Furthermore, Easy353 efficiently retrieves Angiosperms353 genes, and an enhanced version called GeneMiner has recently been developed (Xie et al. 2023). GeneMiner is flexible with input formats and accommodating options such as the GenBank file format and Fasta format. Although these tools possess distinct features, researchers commonly aim to use Angiosperms353 genes retrieved by these tools to reconstruct phylogenies. This can be achieved by concatenating all gene alignments (McVay and Carstens 2013) or obtaining a species tree based on all individual gene trees in a coalescence model (Liu et al. 2015; Zhang et al. 2018).
HybPiper and Easy353 are two widely acclaimed software programs with outstanding features in the community. HybPiper, a set of Python scripts, effectively packages bioinformatic tools for extracting target sequences from high-throughput DNA sequencing reads. It specializes in retrieving on-target sequences of nuclear genes and flanking off-target regions. The method involves read mapping using BWA or Bowtie2 (Li and Durbin 2009), assembling reads into contigs, and extracting the target sequence. The primary output comprises nucleotide and translated amino acid sequences of each gene, assembled from sequencing reads. HybPiper further provides postprocessing scripts for retrieving sequences from multiple samples, including visually summarizing statistics like capture efficiency and coverage depth and extracting flanking intron sequences. Competitively, Easy353 is an exclusive Angiosperms353 mining tool employing a reference-guided strategy. It implements an optimized filtering approach based on k-mers and an assembly algorithm integrated with the weighted de Bruijn graph (DBG) (Compeau et al. 2011). Conservative regions from reference sequences enhance assembly, ensuring high accuracy and sensitivity. The output from Easy353 can be refined using the PPD script to identify putative paralogs (Zhou et al. 2022). The tool involves three main steps: “reference database building”, “read filtering”, and “read assembly” (see “Data retrieval, processing, and analysis” below).
To evaluate the capture efficiency of different software programs for Angiosperms353, we initially compiled a genomic dataset comprising 18 taxa from three sequencing strategies, including genome skimming, WGS, and RNA-seq (Supplementary Table S2). Using the same set of reference sequences, we assessed the performance and efficiency of the two most popular tools, HybPiper V2.1.6 (Johnson et al. 2016) and Easy353 V1.5.0 (Zhang et al. 2022). Our evaluation considered the number of captured Angiosperms353 genes, the sequence length of the retrieved genes, and the runtime. Statistically, no significant difference was detected between HybPiper and Easy353 in terms of the capture rate (average gene count: 218 vs. 202). However, HybPiper requires a shorter runtime (13 min vs. 53 min). In terms of the sequence length of the captured Angiosperms353 genes, Easy353 showed a longer length (HybPiper: 644 bp vs. Easy353: 755 bp; Supplementary Table S2). Furthermore, the average count of captured Angiosperms353 genes varied across all three sequencing strategies (genome skimming, WGS, and RNA-seq). In genome skimming and WGS, HybPiper captured more Angiosperms353 genes than Easy353 (genome skimming: WGS, 97:266 vs. 88:220; Fig. 3a, b). However, in RNA-seq data, Easy353 captured more genes than HybPiper (299 vs. 292; Fig. 3c). Similarly, Easy353 consistently yielded longer sequences on average in genome skimming and RNA-seq but not in WGS. In genome skimming, the average sequence length retrieved by Easy353 was notably longer than that retrieved from HybPiper (450 bp vs. 385 bp). In particular, in RNA-seq, the average sequence length retrieved by Easy353 was significantly longer (Fig. 3d, f). In contrast, the average sequence length retrieved by Easy353 in WGS data was slightly shorter than that retrieved by HybPiper (570 bp vs. 614 bp; Fig. 3e). Additionally, across all three sequencing strategies, HybPiper had a shorter average runtime than Easy353 (genome skimming:WGS:RNA-seq, 8:20:9 min vs. 36:108:26 min; Fig. 3g, h, i), and the dataset generated from WGS took more time for both Angiosperms353 mining tools.

Comparison of the performances of Easy353 and HybPiper across the three sequencing strategies. (a-c) Comparison of the numbers of genes identified in the Angiosperms353 gene sets. (d–f) Comparison of the average sequence length for the same captured Angiosperms353 gene. (g–i) Comparison of runtimes. (a, d, g) panels for Genome skimming; (b, e, h) for WGS, and (c, f, i) for RNA-seq. Abbreviations for species used in this test are as follows: Gd, Guaduella densiflora; Ph, Prunus hypoxantha; Pj, Prunus jenkinsii; Pl, Prunus laurocerasus; Pi, Prunus incisoserrata; Oe, Osmanthus enervius; Dh, Dendrobium heterocarpum; Sb, Sorghum bicolor; Nn, Nelumbo nucifera; Vr, Vitis romanetii; Vh, Vitis hancockii; Bm, Begonia microsperma; Pe, Phalaenopsis equestris; Ca, Catabrosa aquatica; Cy, Camellia yunnanensis; Ce, Camellia euphlebia; Cd, Camellia danzaiensis
Overall, HybPiper and Easy353 exhibited distinct advantages across all three sequencing strategies. HybPiper outperformed Easy353 in capturing more Angiosperms353 genes in genome skimming and RNA-seq datasets while requiring a shorter runtime. However, Easy353 consistently yielded longer sequence lengths than HybPiper. Regarding accessibility, HybPiper is exclusively accessible through a command-line interface. Moreover, identifying the final set of Angiosperms353 genes in the later stages of the HybPiper pipeline can be challenging due to their burial with numerous intermediate sequence files with limited visualization. In contrast, Easy353 provides a well-structured and user-friendly result indexing and display, accessible through both graphical and command-line interfaces. Additionally, Easy353 is compatible with multiple operating systems and building reference databases.
In addition to assessing the impact of different tools on Angiosperms353 yields, we further assembled 343-taxa genomic datasets. Through comprehensive testing with the ascendant Easy353, we examined the capture efficiency of Angiosperms353 across various sequencing strategies, depths, and representative angiosperm groups (Figs. 4, 5 and 6). By performing the aforementioned tests, this review also provides practical guidelines for the optimal and cost-effective utilization of Angiosperms353 in relevant research fields. To investigate the influence of various sequencing strategies on Angiosperms353 gene yields, we expanded the initial three sequencing strategies to four—RAD-seq, genome skimming, WGS, and RNA-seq. This expansion comprised 89 RAD-seq, 78 genome skimming, 89 WGS, and 87 RNA-seq datasets (Supplementary Table S3). These results indicate that RNA-seq is the most effective strategy for identifying Angiosperms353 genes (Figs. 4a and 5a), exhibiting the highest capture rate (> 300 genes in most cases) compared to other sequencing strategies, i.e., WGS (average of 144 genes) and genome skimming (average of 57 genes) (Figs. 4a and 5a). The lowest yield was observed in the RAD-seq dataset, with only nine genes on average per dataset (Figs. 4a and 5a). The percentage of Angiosperms353 individuals captured from the RNA-seq datasets was approximately 35 times greater than that from the RAD-seq datasets.

Capture efficiency of Angiosperms353 genes under different sequencing strategies and sequencing depths. (a) Four sequencing strategies (RAD-Seq, Genome skimming, WGS, and RNA-Seq). (b) Three sequencing depths (0–10×, 10–30×, and > 30×). The black dot in (b) indicates poor Angiosperms353 capture yield in the RAD-seq data (with the majority of data concentrated around this point). Note: Sequencing depth, defined as the ratio of the total number of sequenced base pairs (bp) to the genome size, serves as a metric to evaluate sequencing quantity. WGS, whole-genome sequencing; RAD-seq, restriction site-associated DNA sequencing; RNA-seq, transcriptome sequencing

Average counts of Angiosperms353 genes identified by Easy353 across various datasets. (a) Counts of Angiosperms353 genes from datasets employing four sequencing strategies (RAD-Seq, Genome skimming, WGS, and RNA-Seq). (b) Counts of Angiosperms353 genes from datasets with three sequencing depths (0–10×, 10–30×, and >30×). (c) Counts of Angiosperms353 genes in representative angiosperm groups: ANA grade (represented by Magnoliaceae), monocot (Orchidaceae and Poaceae), eudicot (Nelumbonaceae), suprosid (Vitaceae and Rosaceae), and superasterid (Oleaceae and Theaceae) families based on APG IV (2016). Average counts are presented as mean ± SE; the letters “a” to “d” indicate significant differences (p <0.05) based on the Least Significant Difference (LSD) method after one-way analysis of variance (ANOVA); same letters indicate no significant difference (p <0.05). ANA grade refers to the Amborellales, Nymphaeales, and Austrobaileyales orders recognized by APG IV (2016); WGS, whole genome sequencing; RAD-seq, restriction site-associated DNA sequencing; RNA-seq, transcriptome sequencing

The overall yield of Angiosperms353 showed no bias across all Angiosperm groups. Each group is represented by an iconic image of a well-known species in the family. Angiosperms353 average counts are presented as the mean ± SE; letter “a” indicates significant differences (p <0.05) based on the Least Significant Difference (LSD) method after one-way analysis of variance (ANOVA); same letters indicate no significant difference (p <0.05). ANA grade refers to the Amborellales, Nymphaeales, and Austrobaileyales orders recognized by APG IV (2016)
To investigate the impact of sequencing depth on Angiosperms353 gene capture, the datasets were categorized into three sequencing depth ranges: 0–10 × (285-taxa dataset), 10–30 × (38-taxa dataset), and >30 × (20-taxa dataset) (Supplementary Table S3). Among these depths, 30 × was the most effective, yielding approximately 300 genes in most cases (Figs. 4b and 5b). The percentage of captured Angiosperms353 was significantly greater (p <0.05) at sequencing depths >30 × , while it significantly decreased at 0–10 × or 10–30 × depths (Fig. 5b). At 0–10 × , an average of 109 Angiosperms353 genes were captured per dataset, increasing to 222 at 10–30 × and reaching 312 at >30 × (Fig. 5b). Therefore, a positive trend in the capture rate was observed with increasing sequencing depth.
In the RNA-seq datasets, the number of genes captured by Easy353 remained relatively constant despite increased sequence depth (Fig. 4b). This could be due to the stringent tissue sampling requirements (i.e., young, fresh tissues preserved in liquid nitrogen or dry ice, followed by –80℃ storage), enhancing library construction and sequencing efficiency. Although this method easily yields numerous protein-coding genes, gene expression variations among different biological tissues and the inclusion of non-phylogenetically informative transcripts may limit its effectiveness (Johnson et al. 2019). With increasing sequencing depth, WGS and genome skimming may yield additional genetically informative DNA, particularly for herbarium specimens, resulting in a gradual increase in the number of captured Angiosperms353 genes. WGS requires reference genome data from closely related species. By mapping the obtained reads to the reference genome for sequence assembly and construction, they can facilitate population evolution analysis and functional gene discovery. Although its sequencing cost is relatively low, the absence of a closely related reference genome can pose obstacles for non-model species research (Hollingsworth et al. 2016; Supple and Shapiro 2018). Genome skimming is a cost-effective method that efficiently reveals repetitive elements, such as satellite DNA and transposable elements. However, these replicates possess minimal variation, and the utility of this technology in capturing direct homologous regions of nuclear genes used for sequence alignment is limited due to a lack of coding region information (Dodsworth 2015). RAD-seq serves as a substitute for WGS, being independent of the reference genome. This method uses HTS of DNA related to restriction endonuclease recognition sites to identify high-density single nucleotide polymorphism (SNP) sites, reducing genome complexity and library construction and sequencing costs (Shafer et al. 2016). However, RAD-seq markers are lineage-specific, introducing bias when screening homologous sequences across distantly related lineages. This may result in datasets falling short of comprehensive representation (Andrews et al. 2016; Heckenhauer et al. 2018). Moreover, using short and inconsistently represented loci in phylogenetic sampling may lead to reduced phylogenetic signals and challenges in assessing phylogenetic relationships (Jones and Good 2015; McKain et al. 2018).
Furthermore, to explore potential bias in capturing Angiosperms353 genes across different angiosperm groups, datasets from diverse angiosperm clades were selected, including ANA grade (represented by Magnoliaceae, 41-taxa dataset), monocots (Orchidaceae and Poaceae, 48-taxa and 47-taxa dataset), eudicots (Nelumbonaceae, 31-taxa dataset), superrosids (Vitaceae and Rosaceae, 40-taxa and 53-taxa dataset), and superasterids (Oleaceae and Theaceae, 42-taxa and 41-taxa dataset) (Supplementary Table S3). Notably, Rosaceae exhibited the highest average capture of Angiosperms353 genes (211 genes captured), significantly differing from the remaining groups (p < 0.05; Fig. 5c). While Orchidaceae captured 140 Angiosperms353 genes, and Magnoliaceae captured the fewest (91 genes), these two groups did not significantly differ (p < 0.05; Fig. 5c).
To mitigate the potential influence from sequencing depth and strategies, a 155-taxa dataset was generated from the original 343-taxa dataset, maintaining similar sequencing depths (except for Nelumbonaceae, which used a 15-taxa dataset due to the unavailability of genome skimming data; Supplementary Table S4). The results obtained from the 155-taxa dataset (Fig. 6) differed from those obtained from the original 343-taxa dataset (Fig. 5c). Orchidaceae exhibited the highest yield (166 genes captured), whereas Magnoliaceae had the lowest yield (92 genes). The remaining groups (Theaceae, Oleaceae, Rosaceae, Vitaceae, Nelumbonaceae, and Poaceae) captured over 100 genes, but less than the Orchidaceae, which may be due to higher yields in RAD-seq only.
Therefore, to further investigate potential bias among representative groups in the RAD-seq data, another 30-taxa dataset was added, encompassing five angiosperm groups (Lauraceae, Cyperaceae, Betulaceae, Rutaceae, and Asteraceae; Supplementary Table S5). Despite non-significant differences, the gene yields were lower than Orchidaceae yields in all cases, indicating no lineage-based capture bias in RAD-seq data, except for Orchidaceae (Supplementary Tables S4 and S5). Notably, the greater number of genes captured for Orchidaceae in the RAD-seq dataset did not translate to a similar pattern in Poaceae (another monocot representative) or any other sampled group (Supplementary Table S5).
Beside Orchidaceae, most genes captured from Superrosid clade, whereas the lowest yields were found in the ANA grade. Overall, significant differences in the number of genes captured among different angiosperm groups were not observed (Fig. 6). Therefore, there was no discernible bias among the different angiosperm groups across all the sequencing strategies.
Discussion and conclusions
This review summarizes the extensive literature on plant phylogenomics, leveraging high-throughput sequencing technologies and diverse empirical genomic datasets. It emphasizes the utility of Angiosperms353 by integrating various genomic resources. Additionally, the RNA-seq strategy demonstrates the highest Angiosperms353 yields among the four major sequencing strategies. Higher sequencing depth correlates with increased gene capture, and no obvious yield bias was observed across different angiosperm groups.
In the age of phylogenomics, the universal probe set Angiosperms353 has standardized the use of genomic data for inferring angiosperm phylogenetic relationships, facilitating phylogenomic analyses at any scale. It enables the integration of different genomic datasets and taxonomic groups (Johnson et al. 2019; Baker et al. 2021). Moreover, its efficacy with degraded DNA, often found in ‘genomic treasure troves’, extends its applicability to sequencing ancient specimens from herbariums and museums, revitalizing this field (Brewer et al. 2019; Slimp et al. 2021). The enriched Angiosperms353-related phylogenomics (Forrest et al. 2019; Clarkson et al. 2021; Frost et al. 2021; Pillon et al. 2021) opens avenues for a comprehensive exploration of the evolutionary history of plant species, ranging from historical and archaeological records to freshly collected specimens in the field. This approach is capable of providing evolutionary insights into how species have evolved and adapted over time.
Despite Angiosperms353 designed for single-copy genes, challenges persist due to whole-genome duplication events and angiosperm polyploidy, causing inconsistencies in gene trees and introducing phylogenetic noise (McKain et al. 2018; Gomez et al. 2019). Some taxa exhibit low capture efficiency, yielding insufficient resolution for specific lineages. Observations from test datasets suggest that RNA-seq is the most suitable sequencing strategy, capturing the highest number of Angiosperms353 with less impact from sequencing depth or lineage constraints. When sampling conditions are constrained, and RNA-seq is not possible, WGS and genome skimming offer viable alternatives, with sequencing depth significantly influencing gene capture efficiency in genome skimming. Across these sequencing strategies, increased sequencing depth generally leads to higher Angiosperms353 yields.
This review also underscores the potential of integrating single-copy gene capture tools. For example, HybPiper, when employed after establishing the Easy353 reference data, yields a comparable number of Angiosperms353 genes as Easy353 but with a more efficient runtime. This combination of HybPiper and Easy353 enhances gene capture, contributing to the development of accessible tools for data storage and distribution and fostering further advancements. The combination of HybPiper and PPD proves valuable by eliminating collateral homologous genes identified by both pipelines. This synergy allows the construction of a more robust orthologous gene dataset. Zhou et al. (2022) captured Angiosperms353 data from Castanea (Fagaceae) and Hamamelis (Hamamelidaceae), revealing the ability of PPD to identify more putative paralogs than HybPiper alone (e.g., 31 genes via PPD vs. four genes with HybPiper), resulting in a more robust phylogeny. The integration of HybPiper and PPD aids in removing paralogous genes identified by both methods, facilitating the construction of a more robust homologous gene dataset for phylogenomic and divergence time analyses.
Researchers employing target enrichment methods often face the dilemma between adopting universal or lineage-specific probe sets. To address this dilemma, exploring the simultaneous enrichment of combining multiple probe sets, such as lineage-specific integrated universal or multiple lineage-specific sets, is crucial. This approach leverages the increasing availability of probes and decreasing sequencing costs. In evolutionary biology, Angiosperms353 maximizes the utility of genomics across different scales. It introduces new possibilities for resolving phylogenetic relationships at various taxonomic scales (Hendriks et al. 2021, 2023; Pillon et al. 2021; Yardeni et al. 2021; Thureborn et al. 2022; Phang et al. 2023). For example, Hendriks et al. (2023) effectively combined Angiosperms353 with the Brassicaceae-specific probe set (Nikolov1827) in a single hybridization reaction, achieving comprehensive outcomes with minimal additional cost and effort. Similar studies have demonstrated the effectiveness of combining Angiosperms353 with lineage-specific probe sets to enhance phylogenetic resolution, shedding light on species delimitation (Phang et al. 2023). While lineage-specific probe sets boast higher success rate in capturing specific groups, offering advantages for detailed phylogenetics and population genetic analyses, such as exploring gene tree consistency, nucleotide diversity, or population structure, they are susceptible to indistinguishable paralogs (Dornburg et al. 2019; Yardeni et al. 2021). Conversely, Angiosperms353 excels in integrating genomic resources from various HTS platforms, laying the foundation for large-scale phylogenomic research, particularly when constructing tree of life using complete genomes is impractical or impossible.
In summary, the future of phylogenomics using Angiosperms353 appears promising, fueled by ongoing improvements in probe design and broader taxonomic coverage. Consequently, improving the efficiency of bioinformatic tools, collaboration, data sharing, and technological advancements will play pivotal roles in advancing our understanding of the evolutionary relationships across the entirety of life using the available data.
Data retrieval, processing, and analysis
The NCBI website (https://www.ncbi.nlm.nih.gov/) was used to query and download nucleotide sequence data. Raw reads from 375 accessions, encompassing RAD-seq, genome skimming, WGS, and RNA-seq projects, were downloaded from the NCBI SRA (https://www.ncbi.nlm.nih.gov/sra/) and described in Supplementary Table S1.
Raw sequencing data from the SRA database were downloaded using the NCBI SRA Toolkit (Edwards 2022), with the “fastq-dump” command converting SRA format to fastq format. Trimmomatic V0.38 (Bolger et al. 2014) was then applied to trim adaptor sequences and low-quality bases from the ends of sequencing files, employing a Phred33 score and setting the minimum read length to 36 bp. Base pairs falling below the quality threshold were removed. Trimmed reads were used for locus assembly via HybPiper V2.1.6 (Johnson et al. 2016) and Easy353 V1.5.0 (Zhang et al. 2022).
To enable efficient locus recovery, a variable reference sequence file was constructed from sequences from multiple species across the Easy353 reference database. Trimmed reads were mapped to HybPiper reference sequences using BWA (Li and Durbin 2009) and assembled into contigs using SPADES (Bankevich et al. 2012). Easy353, with its three primary modules for recovering Angiosperms353 from sequencing data (“reference database building”, “read filtering”, and “read assembly”), used k-mers and a hash table (Schbath et al. 2012) for read filtering and a de Bruijn graph (DBG) (Compeau et al. 2011) for de novo assembly.
All statistical analyses and plots were conducted in R (R Core Team 2023) using the packages “agricolae” (Mendiburu 2023), “ggplot2” (Wickham 2016), “tidyverse” (Wickham et al. 2019), and “rentrez” (Winter 2017).
All necessary data to evaluate the conclusions in this manuscript are presented in the main text and supplementary materials (Fig. S1, Tables S1–S5). Raw reads were obtained from NCBI databases (https://www.ncbi.nlm.nih.gov/), and the complete list of NCBI accession numbers is available in Tables S1–S5.
Availability of data and materials
Data were obtained from the NCBI and will be shared upon request by the readers.
References
Acha S, Majure LC. A new approach using targeted sequence capture for phylogenomic studies across Cactaceae. Genes. 2022;13:350. https://doi.org/10.3390/genes13020350.
Andrews KR, Good JM, Miller MR, Luikart G, Hohenlohe PA. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat Rev Genet. 2016;17:81–92. https://doi.org/10.1038/nrg.2015.28.
Antonelli A, Clarkson JJ, Kainulainen K, Maurin O, Brewer GE, Davis AP, et al. Settling a family feud: a high-level phylogenomic framework for the Gentianales based on 353 nuclear genes and partial plastomes. Am J Bot. 2021;108:1143–65. https://doi.org/10.1002/ajb2.1697.
APG IV. An updated of the angiosperm phylogeny group classifications for orders and families of flowering plants: APG IV. Bot J Linnean Soc. 2016;181:1–20. https://doi.org/10.1111/boj.12385.
Baker WJ, Dodsworth S, Forest F, Graham SW, Johnson MG, McDonnell A, et al. Exploring Angiosperms353: an open, community toolkit for collaborative phylogenomic research on flowering plants. Am J Bot. 2021;108:1059–65. https://doi.org/10.1002/ajb2.1703.
Baker WJ, Bailey P, Barber V, Barker A, Bellot S, Bishop D, et al. A comprehensive phylogenomic platform for exploring the angiosperm tree of life. Syst Biol. 2022;71:301–19. https://doi.org/10.1093/sysbio/syab035.
Bakker FT, Lei D, Yu J, Mohammadin S, Wei Z, van de Kerke S, et al. Herbarium genomics: plastome sequence assembly from a range of herbarium specimens using an iterative organelle genome assembly pipeline. Biol J Linnean Soc. 2016;117:33–43. https://doi.org/10.1111/bij.12642.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. https://doi.org/10.1089/cmb.2012.0021.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Bragg JG, Potter S, Bi K, Moritz C. Exon capture phylogenomics: efficacy across scales of divergence. Mol Ecol Resour. 2015;16:1059–68. https://doi.org/10.1111/1755-0998.12449.
Breinholt JW, Carey SB, Tiley GP, Davis EC, Endara L, McDaniel SF, et al. A target enrichment probe set for resolving the flagellate land plant tree of life. Appl Plant Sci. 2021;9:e11406. https://doi.org/10.1002/aps3.11406.
Brewer GE, Clarkson JJ, Maurin O, Zuntini AR, Barber V, Bellot S, et al. Factors affecting targeted sequencing of 353 nuclear genes from herbarium specimens spanning the diversity of angiosperms. Front Plant Sci. 2019;10:1102. https://doi.org/10.3389/fpls.2019.01102.
Cai L, Zhang H, Davis CC. PhyloHerb: a high-throughput phylogenomic pipeline for processing genome skimming data. Appl Plant Sci. 2022;10:e11475. https://doi.org/10.1002/aps3.11475.
Chamala S, García N, Godden GT, Krishnakumar V, Jordon-Thaden IE, De Smet R, et al. MarkerMiner 1.0: a new application for phylogenetic marker development using angiosperm transcriptomes. Appl Plant Sci. 2015;3:apps.1400115. https://doi.org/10.3732/apps.1400115.
Chau J, Rahfeldt WA, Olmstead RG. Comparison of taxon-specific versus general locus sets for targeted sequence capture in plant phylogenomics. Appl Plant Sci. 2018;6:e1032. https://doi.org/10.1002/aps3.1032.
Chen JT, Lidén M, Huang XH, Zhang L, Zhang XJ, Kuang TH, et al. An updated classification for the hyper-diverse genus Corydalis (Papaveraceae: Fumarioideae) based on phylogenomic and morphological evidence. J Integr Plant Biol. 2023;65:2138–56. https://doi.org/10.1111/jipb.13499.
Cheng CY, Krishnakumar V, Chan AP, Thibaud-Nissen F, Schobel S, Town CD. Araport11: a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017;89:789–804. https://doi.org/10.1111/tpj.13415.
Clarkson JJ, Zuntini AR, Maurin O, Downie SR, Plunkett GM, Nicolas AN, et al. A higher-level nuclear phylogenomic study of the carrot family (Apiaceae). Am J Bot. 2021;108:1252–69. https://doi.org/10.1002/ajb2.1701.
Coissac E, Hollingsworth PM, Lavergne S, Taberlet P. From barcodes to genomes: extending the concept of DNA barcoding. Mol Ecol. 2016;25:1423–8. https://doi.org/10.1111/mec.13549.
Compeau PEC, Pevzner PA, Tesler G. How to apply de Bruijn graphs to genome assembly. Nat Biotechnol. 2011;29:987–91. https://doi.org/10.1038/nbt.2023.
Cronn R, Knaus BJ, Liston A, Maughan PJ, Parks M, Syring JV, et al. Targeted enrichment strategies for next-generation plant biology. Am J Bot. 2012;99:291–311. https://doi.org/10.3732/ajb.1100356.
Crowl AA, Fritsch PW, Tiley GP, Lynch NP, Ranney TG, Ashrafi H, et al. A first complete phylogenomic hypothesis for diploid blueberries (Vaccinium section Cyanococcus). Am J Bot. 2022;109:1596–606. https://doi.org/10.1002/ajb2.16065.
Dodsworth S. Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 2015;20:525–7. https://doi.org/10.1016/j.tplants.2015.06.012.
Dodsworth S, Pokorny L, Johnson MG, Kim JT, Maurin O, Wickett NJ, et al. Hyb-seq for flowering plant systematics. Trends Plant Sci. 2019;24:887–91. https://doi.org/10.1016/j.tplants.2019.07.011.
Dornburg A, Su Z, Townsend JP, Mueller R. Optimal rates for phylogenetic inference and experimental design in the era of genome-scale data sets. Syst Biol. 2019;68:145–56. https://doi.org/10.1093/sysbio/syy047.
Edwards D. Methods in molecular biology. In: Edwards D, editor. Plant bioinformatics: methods and protocols. New York: Humana; 2022. p. 1374.
Eserman LA, Thomas SK, Coffey EED, Leebens-Mack JH. Target sequence capture in orchids: developing a kit to sequence hundreds of single-copy loci. Appl Plant Sci. 2021;9:e11416. https://doi.org/10.1002/aps3.11416.
Faircloth BC. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics. 2016;32:786–8. https://doi.org/10.1093/bioinformatics/btv646.
Folk RA, Mandel JR, Freudenstein JV. A protocol for targeted enrichment of intron-containing sequence markers for recent radiations: a phylogenomic example from Heuchera (Saxifragaceae). Appl Plant Sci. 2015;3:apps.1500039. https://doi.org/10.3732/apps.1500039.
Folk RA, Kates HR, LaFrance R, Soltis DE, Soltis PS, Guralnick RP. High-throughput methods for efficiently building massive phylogenies from natural history collections. Appl Plant Sci. 2021;9:e11410. https://doi.org/10.1002/aps3.11410.
Forrest LL, Hart ML, Hughes M, Wilson HP, Chung KF, Tseng YH, et al. The limits of Hyb-seq for herbarium specimens: impact of preservation techniques. Front Ecol Evol. 2019;7:439. https://doi.org/10.3389/fevo.2019.00439.
Frost L, Santamaría-Aguilar DA, Singletary D, Lagomarsino LP. Niche evolution of the neotropical tree genus Otoba in the context of global biogeography of the nutmeg family, Myristicaceae. bioRxiv. 2021;10. https://doi.org/10.1101/2020.10.02.324368.
Gagnon E, Hilgenhof R, Orejuela A, McDonnell A, Sablok G, Aubriot X, et al. Phylogenomic discordance suggests polytomies along the backbone of the large genus Solanum. Am J Bot. 2022;109:580–601. https://doi.org/10.1002/ajb2.1827.
Gitzendanner MA, Soltis PS, Wong GKS, Ruhfel BR, Soltis DE. Plastid phylogenomic analysis of green plants: a billion years of evolutionary history. Am J Bot. 2018;105:291–301. https://doi.org/10.1002/ajb2.1048.
Gomez SM, Pokorny L, Kantar MB, Forest F, Leitch IJ, Gravendeel B, et al. A customized nuclear target enrichment approach for developing a phylogenomic baseline for Dioscorea yams (Dioscoreaceae). Appl Plant Sci. 2019;7:e11254. https://doi.org/10.1002/aps3.11254.
Guo C, Luo Y, Gao LM, Yi TS, Li HT, Yang JB, et al. Phylogenomics and the flowering plant tree of life. J Integr Plant Biol. 2022;65:299–323. https://doi.org/10.1111/jipb.13415.
Haigh AL, Gibernau M, Maurin O, Bailey P, Carlsen MM, Hay A, et al. Target sequence data shed new light on the infrafamilial classification of Araceae. Am J Bot. 2023;110:e16117. https://doi.org/10.1002/ajb2.16117.
Heckenhauer J, Samuel R, Ashton PS, Abu Salim K, Paun O. Phylogenomics resolves evolutionary relationships and provides insights into floral evolution in the tribe Shoreeae (Dipterocarpaceae). Mol Phylogenet Evol. 2018;127:1–13. https://doi.org/10.1016/j.ympev.2018.05.010.
Hendriks KP, Mandáková T, Hay NM, Ly E, Hooft van Huysduynen A, Tamrakar R, et al. The best of both worlds: combining lineage-specific and universal bait sets in target-enrichment hybridization reactions. Appl Plant Sci. 2021;9:aps3.11438. https://doi.org/10.1002/aps3.11438.
Hendriks KP, Kiefer C, Al-Shehbaz IA, Bailey CD, Hooft van Huysduynen A, Nikolov LA, et al. Global Brassicaceae phylogeny based on filtering of 1,000-gene dataset. Curr Biol. 2023;33:4052-4068.e6. https://doi.org/10.1016/j.cub.2023.08.026.
Hollingsworth PM, Li DZ, van der Bank M, Twyford AD. Telling plant species apart with DNA: from barcodes to genomes. Philos Trans R Soc Lond B Biol Sci. 2016;371:20150338. https://doi.org/10.1098/rstb.2015.0338.
Howard CC, Crowl AA, Harvey TS, Cellinese N. Peeling back the layers: first phylogenomic insights into the Ledebouriinae (Scilloideae, Asparagaceae). Mol Phylogenet Evol. 2022;169:107430. https://doi.org/10.1016/j.ympev.2022.107430.
Huang Y, He J, Xu Y, Zheng W, Wang S, Chen P, et al. Pangenome analysis provides insight into the evolution of the orange subfamily and a key gene for citric acid accumulation in citrus fruits. Nat Genet. 2023;55(11):1964–75. https://doi.org/10.1038/s41588-023-01516-6.
Johnson MG, Gardner EM, Liu Y, Medina R, Goffinet B, Shaw AJ, et al. HybPiper: extracting coding sequence and introns for phylogenetics from high-throughput sequencing reads using target enrichment. Appl Plant Sci. 2016;4:1600016. https://doi.org/10.3732/apps.1600016.
Johnson MG, Pokorny L, Dodsworth S, Botigue LR, Cowan RS, Devault A, et al. A universal probe set for targeted sequencing of 353 nuclear genes from any flowering plant designed using k-medoids clustering. Syst Biol. 2019;68:594–606. https://doi.org/10.1101/361618.
Jones MR, Good JM. Targeted capture in evolutionary and ecological genomics. Mol Ecol. 2015;25:185–202. https://doi.org/10.1111/mec.13304.
Joyce EM, Appelhans MS, Buerki S, Cheek M, de Vos JM, Pirani JR, et al. Phylogenomic analyses of Sapindales support new family relationships, rapid mid-Cretaceous hothouse diversification, and heterogeneous histories of gene duplication. Front Plant Sci. 2023;14:1063174. https://doi.org/10.3389/fpls.2023.1063174.
Kumar R, Ichihashi Y, Kimura S, Chitwood DH, Headland LR, Peng J, et al. A high-throughput method for Illumina RNA-seq library preparation. Front Plant Sci. 2012;202:3. https://doi.org/10.3389/fpls.2012.00202.
Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012;40:D1202–10. https://doi.org/10.1093/nar/gkr1090.
Larridon I, Villaverde T, Zuntini AR, Pokorny L, Brewer GE, Epitawalage N, et al. Tackling rapid radiations with targeted sequencing. Front Plant Sci. 2020;10:1655. https://doi.org/10.3389/fpls.2019.01655.
Larridon I, Zuntini AR, Léveillé-Bourret É, Barrett RL, Starr JR, Muasya AM, et al. A new classification of Cyperaceae (Poales) supported by phylogenomic data. J Syst Evol. 2021;59:852–95. https://doi.org/10.1111/jse.12757.
Larson DA, Chanderbali AS, Maurin O, Gonçalves DJP, Dick CW, Soltis DE, et al. The phylogeny and global biogeography of Primulaceae based on high-throughput DNA sequence data. Mol Phylogenet Evol. 2023;182:107702. https://doi.org/10.1016/j.ympev.2023.107702.
Leebens-Mack JH, Barker MS, Carpenter EJ, Deyholos MK, Gitzendanner MA, Graham SW, et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature. 2019;574:679–85. https://doi.org/10.1038/s41586-019-1693-2.
Lewin HA, Robinson GE, Kress WJ, Baker WJ, Coddington J, Crandall KA, et al. Earth BioGenome Project: sequencing life for the future of life. Proc Natl Acad Sci USA. 2018;115:4325–33. https://doi.org/10.1073/pnas.1720115115.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. https://doi.org/10.1093/bioinformatics/btp324.
Li HT, Yi TS, Gao LM, Ma PF, Zhang T, Yang JB, et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat Plants. 2019;5:461–70. https://doi.org/10.1038/s41477-019-0421-0.
Liao M, Shepherd LD, Zhang JY, Feng Y, Mattapha S, Zhang LB, et al. Phylogeny, biogeography, and character evolution of the genus Sophora s.l. (Fabaceae, Papilionoideae). Mol Phylogenet Evol. 2023;181:107713. https://doi.org/10.1016/j.ympev.2023.107713.
Liu L, Wu S, Yu L. Coalescent methods for estimating species trees from phylogenomic data. J Syst Evol. 2015;53:380–90. https://doi.org/10.1111/jse.12160.
Mamanova L, Coffey AJ, Scott CE, Kozarewa I, Turner EH, Kumar A, et al. Target-enrichment strategies for next-generation sequencing. Nat Methods. 2010;7:111–8. https://doi.org/10.1038/nmeth.1419.
Mandel JR. What’s all the hype about HybSeq? A brief history and introduction to target enrichment in Compositae. Capitulum. 2021;1:44–7. https://doi.org/10.53875/capitulum.01.1.04.
Masters LE, Tomaszewska P, Schwarzacher T, Zuntini AR, Heslop-Harrison P, Vorontsova MS. Phylogenomic analysis reveals the evolutionary origins of five independent clades of forage grasses within the African genus Urochloa. bioRxiv. 2023. https://doi.org/10.1101/2023.07.03.547487.
Maurin O, Anest A, Bellot S, Biffin E, Brewer G, Charles-Dominique T, et al. A nuclear phylogenomic study of the angiosperm order Myrtales, exploring the potential and limitations of the universal Angiosperms353 probe set. Am J Bot. 2021;108:1087–111. https://doi.org/10.1002/ajb2.1699.
McDonnell AJ, Baker WJ, Dodsworth S, Forest F, Graham SW, Johnson MG, et al. Exploring Angiosperms353: developing and applying a universal toolkit for flowering plant phylogenomics. Appl Plant Sci. 2021;9:aps3.11443. https://doi.org/10.1002/aps3.11443.
McKain MR, Johnson MG, Uribe-Convers S, Eaton D, Yang Y. Practical considerations for plant phylogenomics. Appl Plant Sci. 2018;6:e1038. https://doi.org/10.1002/aps3.1038.
McVay J, Carstens B. Phylogenetic model choice: justifying a species tree or concatenation analysis. J Phylogen Evolution Biol. 2013;1:1000114. https://doi.org/10.4172/jpgeb.1000114.
Mendiburu Fd. Agricolae: statistical procedures for agricultural research. 2023. https://CRAN.R-project.org/package=agricolae.
Nauheimer L, Weigner N, Joyce E, Crayn D, Clarke C, Nargar K. HybPhaser: a workflow for the detection and phasing of hybrids in target capture data sets. Appl Plant Sci. 2021;9:aps3.11441. https://doi.org/10.1002/aps3.11441.
Ng P, Kirkness E. Whole genome sequencing. In: Barnes M, Breen G, editors. Genetic variation: methods and protocols. Totowa: Humana Press; 2010. p. 215–26.
Ogutcen E, Christe C, Nishii K, Salamin N, Möller M, Perret M. Phylogenomics of Gesneriaceae using targeted capture of nuclear genes. Mol Phylogenet Evol. 2021;157:107068. https://doi.org/10.1016/j.ympev.2021.107068.
Phang A, Pezzini FF, Burslem DFRP, Khew GS, Middleton DJ, Ruhsam M, et al. Target capture sequencing for phylogenomic and population studies in the southeast Asian genus Palaquium (Sapotaceae). Bot J Linnean Soc. 2023;203:134–47. https://doi.org/10.1093/botlinnean/boad022.
Pillon Y, Hopkins HCF, Maurin O, Epitawalage N, Bradford J, Rogers ZS, et al. Phylogenomics and biogeography of Cunoniaceae (Oxalidales) with complete generic sampling and taxonomic realignments. Am J Bot. 2021;108:1181–200. https://doi.org/10.1002/ajb2.1688.
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2023. https://www.R-project.org.
Robillard T, Strijk JS, Binh HT, Ngoc NV, Pereira JT, Slik JWF, et al. Museomics for reconstructing historical floristic exchanges: divergence of stone oaks across Wallacea. PLoS One. 2020;15:e0232936. https://doi.org/10.1371/journal.pone.0232936.
Romeiro-Brito M, Telhe MC, Amaral DT, Franco FF, Moraes EM. A target capture probe set useful for deep- and shallow-level phylogenetic studies in Cactaceae. Genes. 2022;13:707. https://doi.org/10.3390/genes13040707.
Schbath S, Martin V, Zytnicki M, Fayolle J, Loux V, Gibrat JF. Mapping reads on a genomic sequence: an algorithmic overview and a practical comparative analysis. J Comput Biol. 2012;19:796–813. https://doi.org/10.1089/cmb.2012.0022.
Shafer ABA, Peart CR, Tusso S, Maayan I, Brelsford A, Wheat CW, et al. Bioinformatic processing of RAD-seq data dramatically impacts downstream population genetic inference. Methods Ecol Evol. 2016;8:907–17. https://doi.org/10.1111/2041-210x.12700.
Shi S, Li J, Sun J, Yu J, Zhou S. Phylogeny and Classification of Prunus sensu lato (Rosaceae). J Integr Plant Biol. 2013;55:1069–79. https://doi.org/10.1111/jipb.12095.
Simões ARG, Eserman LA, Zuntini AR, Chatrou LW, Utteridge TMA, Maurin O, et al. A bird’s eye view of the systematics of Convolvulaceae: novel insights from nuclear genomic data. Front Plant Sci. 2022;13:889988. https://doi.org/10.3389/fpls.2022.889988.
Siniscalchi CM, Hidalgo O, Palazzesi L, Pellicer J, Pokorny L, Maurin O, et al. Lineage-specific vs. universal: a comparison of the Compositae1061 and Angiosperms353 enrichment panels in the sunflower family. Appl Plant Sci. 2021;9:aps3.11422. https://doi.org/10.1002/aps3.11422.
Slimp M, Williams LD, Hale H, Johnson MG. On the potential of Angiosperms353 for population genomic studies. Appl Plant Sci. 2021;9:aps3.11419. https://doi.org/10.1002/aps3.11419.
Sun QH, Morales-Briones DF, Wang HX, Landis JB, Wen J, Wang HF. Phylogenomic analyses of the East Asian endemic Abelia (Caprifoliaceae) shed insights into the temporal and spatial diversification history with widespread hybridization. Ann Bot. 2022;129:201–16. https://doi.org/10.1093/aob/mcab139.
Sundararaman B, Sylvester MD, Kozyreva VK, Berrada ZL, Corbett-Detig RB, Green RE. A hybridization target enrichment approach for pathogen genomics. Mbio. 2023:e01889–01823. https://doi.org/10.1128/mbio.01889-23.
Supple MA, Shapiro B. Conservation of biodiversity in the genomics era. Genome Biol. 2018;19:131. https://doi.org/10.1186/s13059-018-1520-3.
Thomas SK, Liu X, Du ZY, Dong Y, Cummings A, Pokorny L, et al. Comprehending Cornales: phylogenetic reconstruction of the order using the Angiosperms353 probe set. Am J Bot. 2021;108:1112–21. https://doi.org/10.1002/ajb2.1696.
Thureborn O, Razafimandimbison SG, Wikström N, Rydin C. Target capture data resolve recalcitrant relationships in the coffee family (Rubioideae, Rubiaceae). Front Plant Sci. 2022;13:967456. https://doi.org/10.3389/fpls.2022.967456.
Turner B, Paun O, Munzinger J, Chase MW, Samuel R. Sequencing of whole plastid genomes and nuclear ribosomal DNA of Diospyros species (Ebenaceae) endemic to New Caledonia: many species, little divergence. Ann Bot. 2016;117:1175–85. https://doi.org/10.1093/aob/mcw060.
Ufimov R, Zeisek V, Píšová S, Baker WJ, Fér T, van Loo M, et al. Relative performance of customized and universal probe sets in target enrichment: a case study in subtribe Malinae. Appl Plant Sci. 2021;9:e11442. https://doi.org/10.1002/aps3.11442.
Wang HX, Morales-Briones DF, Moore MJ, Wen J, Wang HF. A phylogenomic perspective on gene tree conflict and character evolution in Caprifoliaceae using target enrichment data, with Zabelioideae recognized as a new subfamily. J Syst Evol. 2021;59:897–914. https://doi.org/10.1111/jse.12745.
Weitemier K, Straub SCK, Cronn RC, Fishbein M, Schmickl R, McDonnell A, et al. Hyb-seq: combining target enrichment and genome skimming for plant phylogenomics. Appl Plant Sci. 2014;2:apps.1400042. https://doi.org/10.3732/apps.1400042.
Wickham H. Elegant graphics for data analysis. In: Wickham H, editor. ggplot2. New York: Springer Cham; 2016. p. 1–260.
Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, et al. Welcome to the Tidyverse. J Open Source Softw. 2019;4:1686. https://doi.org/10.21105/joss.01686.
Wilkinson MJ, Szabo C, Ford CS, Yarom Y, Croxford AE, Camp A, et al. Replacing Sanger with next generation sequencing to improve coverage and quality of reference DNA barcodes for plants. Sci Rep. 2017;7:46040. https://doi.org/10.1038/srep46040.
Winter DJ. rentrez: an R package for the NCBI eUtils API. Peer J Preprints 5:e3179v2 [Preprint]. 2017:8. https://doi.org/10.7287/peerj.preprints.3179v2. Cited 2017 Aug 25.
Wolf PG, Robison TA, Johnson MG, Sundue MA, Testo WL, Rothfels CJ. Target sequence capture of nuclear-encoded genes for phylogenetic analysis in ferns. Appl Plant Sci. 2018;6:e01148. https://doi.org/10.1002/aps3.1148.
Xie P, Guo Y, Zhou W, Zhang Z, Yu Y. GeneMiner: a tool for extracting phylogenetic markers from next-generation sequencing data. 2023. https://doi.org/10.22541/au.168172406.69677221/v1.
Yardeni G, Viruel J, Paris M, Hess J, Groot Crego C, de La Harpe M, et al. Taxon-specific or universal? Using target capture to study the evolutionary history of rapid radiations. Mol Ecol Resour. 2021;22:927–45. https://doi.org/10.1111/1755-0998.13523.
Zhang C, Rabiee M, Sayyari E, Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics. 2018;19:153. https://doi.org/10.1186/s12859-018-2129-y.
Zhang Z, Xie P, Guo Y, Zhou W, Liu E, Yu Y, et al. Easy353: a tool to get Angiosperms353 genes for phylogenomic research. Mol Biol Evol. 2022;39:msac261. https://doi.org/10.1093/molbev/msac261.
Zhao L, Jiang XW, Zuo YJ, Liu XL, Chin SW, Haberle R, Potter D, Chang ZY, Wen J. Multiple events of allopolyploidy in the evolution of the racemose lineages in Prunus (Rosaceae) based on integrated evidence from nuclear and plastid data. PLoS One. 2016;11(6):e0157123. https://doi.org/10.1371/journal.pone.0157123.
Zhao Y, Chen YP, Yuan JC, Paton AJ, Nuraliev MS, Zhao F et al. Museomics in Lamiaceae: resolving the taxonomic mystery of Pseudomarrubium. Curr Plant Biol. 2023;35–6. https://doi.org/10.1016/j.cpb.2023.100300.
Zhou W, Soghigian J, Xiang QY, Eaton D. A new pipeline for removing paralogs in target enrichment data. Syst Biol. 2022;71:410–25. https://doi.org/10.1093/sysbio/syab044.
Zuntini AR, Frankel LP, Pokorny L, Forest F, Baker WJ. A comprehensive phylogenomic study of the monocot order Commelinales, with a new classification of Commelinaceae. Am J Bot. 2021;108:1066–86. https://doi.org/10.1002/ajb2.1698.
Acknowledgements
We want to thank the members in SunLab for their insightful comments on the software and manuscript.
Funding
This work is supported by the National Natural Science Foundation of China (grant No. 32270222 to M.S.), Priority Research Programme of the National Key Laboratory for Germplasm Innovation & Utilization of Horticultural Crops to M.S, as well as startup funds from Huazhong Agricultural University.
Author information
Authors and Affiliations
Contributions
MS conceived and coordinated this project; XW wrote the manuscript under the supervision of and with contributions from MS; TX plotted all the graphics in the analyses with XZ’s assistant; YW sorted out the tables; XZ revised the manuscript together with other authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Summary of GenBank seed plant nucleotide sequence statistics and literature related to Angiosperms353 since 2008.
Additional file 2: Table S1.
NCBI accessions for 375-taxa genomic data entries from diverse angiosperm groups used in this study. Table S2. Summary table for the performance of Easy353 and HybPiper across three sequencing strategies (Genome skimming, WGS, and RNA-Seq) and 18-taxa dataset with NCBI accession. Table S3. Statistics of Angiosperms353 captured by Easy353 across four sequencing strategies and 343-taxa dataset with NCBI accession from diverse angiosperm groups used in this study. Table S4. NCBI accessions for 155-taxa genomic data entries and corresponding Angiosperms353 count from eight representative angiosperm groups used in this study. Table S5. Statistics of Angiosperms353 captured by Easy353 in RAD-seq and 30-taxa genomic data entries with NCBI accession from diverse angiosperm groups used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Xiong, T., Wang, Y. et al. Integrating genomic sequencing resources: an innovative perspective on recycling with universal Angiosperms353 probe sets. HORTIC. ADV. 2, 4 (2024). https://doi.org/10.1007/s44281-023-00026-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44281-023-00026-z