
Abstract
Red leaves in autumn are characteristic of a very early-maturing ripening variety of peach (Prunus persica). Analysis of the genetic factors and molecular mechanisms associated with the red-leaf phenotype can help breed peach very early maturing peach varieties. This study investigated the mechanisms underlying the red- and green-leaf phenotypes in autumn. Red compounds accumulated in the older but not younger leaves in the extremely early-maturing peach variety, ‘99-30-33’, and the leaves of the medium-maturing variety, ‘Zhongtao5 (CP5)’. Metabolic analysis showed that cyanidin-3-O-glucoside was the most abundant anthocyanin in the red leaves. The segregation of the progenies obtained from crossing revealed that the red leaves are a unique hereditary phenomenon not in line with Mendel’s law. BSA-seq and RNA-seq analyses suggest that PpNAC1 was essential for enhancing anthocyanin biosynthesis and was highly upregulated in red than in green leaves. Similar to PpNAC1, the anthocyanin activator, PpMYB10.1, was the only gene highly expressed in red leaves. Moreover, the functional genes involved in anthocyanin biosynthesis, such as Prunus persica Flavonoid 3'-hydroxylase (PpF3'H), Prunus persicaDihydroflavonol reductase (PpDFR), Prunus persica Leucoantho-cyanidin dioxygenase (Pp LDOX), Prunus persica Glutathione S-transferase (PpGST), and Prunus persica UDP-glucose, flavonoid-3-O-glucosyltransferase (PpUFGT), were upregulated in the older red leaves of 99-30-33 but downregulated in the younger 99-30-33 and green CP5 leaves. Yeast one-hybrid and dual-luciferase assays further confirmed that PpNAC1, which refers to 'Prunus persica NAC (NAM、A TAF1/2、CUC1/2)' bound to the promoter of PpMYB10.1, PpMYB10.1 stands for 'Prunus persica MYB (v-myb avian myeloblastosis viral) 10.1' and activated its expression along with those of PpGST and PpUFGT. These results provide insights into the mechanisms responsible for the development of red color in peach leaves in autumn.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Peach (Prunus persica) is extensively used in functional studies due to the small size of its genome, ~ 250 Mb, and a short juvenile growth phase of 2 – 3 years (Verde et al. 2013; Shulaev et al. 2008). Leaf color is influenced by a combination of genetic factors and the environment. The composition, content, and distribution of pigments in the leaves change, forming different colors. The leaves of most improved varieties in peaches were generally green, but some were red (Zhou et al. 2014). Some early-maturing peach varieties exhibit an red-colored-autumn leaf phenotype, with the leaves usually remaining green before fruit harvest. After the harvest of fruits in the summer, anthocyanins gradually accumulate in the mature leaves, and the leaves begin to discolor, starting from the petiole to the major veins and gradually turning purplish-red along the veins. The redness of leaves in the autumn mainly occurs in the extremely early- and early-maturing varieties of peaches. Mid- and late-maturing peach varieties do not exhibit an red-colored-autumn leaf phenotype. However, few studies have been conducted on the molecular mechanisms underlying the red-colored-autumn leaf phenomenon associated with the fruit maturation period.
Anthocyanins are the most common secondary metabolites produced by higher plants (Holton and Cornish 1995) and play essential roles in various physiological processes. Anthocyanin biosynthesis occurs in the cytoplasm and the endoplasmic reticulum before being transported to the vacuoles (Winkel et al. 2004). They are glycosides and exhibit light absorption properties, such as pink, purple, red, and blue (Winkel-Shirley 2001). The understanding of anthocyanin biosynthesis in fruit trees has improved rapidly in recent years, and the pathways in apples and grapes have been clarified (Feng et al. 2013; Boss et al. 1996). Anthocyanins are the final products of the flavonoid biosynthetic pathway (Tanaka et al. 2008). The first stage is a pathway common to the metabolism of the secondary metabolite, phenylpropanoic acid in plants (Koes et al. 2005), which involves the conversion of phenylalanine to 4-coumarin-CoA, which is catalyzed by phenylalanine lyase, cinnamate hydroxylase, and 4-coumarin CoA ligase. The second stage is the early stage of flavonoid metabolism, which mainly involves the synthesis of dihydroflavonols by 4-coumarinyl-CoA and malonyl-CoA. Chalcone synthase (CHS), chalcone isomerase (CHI), and flavanone-3-hydroxylase (F-3-H) play a catalytic role at this stage and are referred to as the early synthesis-associated genes of anthocyanin metabolism. The third stage is the late stage of the anthocyanin metabolic pathway, which mainly involves three enzymes. First, dihydroflavonol was catalyzed by dihydroflavonol reductase (DFR) to produce the colorless anthocyanin, dihydroflavonol, which is then converted into the colored anthocyanins by leucoantho-cyanidin dioxygenase (LDOX) and flavonoid 3-glucosyltransferase (UFGT). LDOX catalyzes the oxidation and dehydration of the colorless anthocyanins to form the colored anthocyanins, and UFGT promotes the formation of anthocyanins to maintain their structural stability.
In addition to the functional genes, transcription factors (TFs) are also crucial for the metabolism of anthocyanins (Gonzalez et al. 2008; Shin et al. 2007). Regulatory genes mainly encode three types of TFs: MYB, bHLH, and WD40, which can form the MYB-bHLH-WD40 (MBW) complex (Albert et al. 2014; Xie et al. 2016). In addition, TF-encoding and regulatory genes, such as NAC (NAM,ATAF1/2, and CUC1/2) (Liu et al. 2022), bZIP (basic leucine zipper) (Wang et al. 2022), WRKY (Li et al. 2020), HY5 (Shin et al. 2013), and RL1 (RED LEAF1) (Lv et al. 2022), also affected anthocyanin biosynthesis. These genes interact with the MBW complex or act as candidates directly regulating anthocyanin biosynthesis. NAC TFs are unique to plants (Aida et al. 1997) and play critical regulatory roles in physiological processes, such as plant growth and development, stress responses, and the biosynthesis of secondary metabolites (Olsen et al. 2005; Nuruzzaman et al. 2013).
Certain members of the NAC family of TFs are known to function as regulators of anthocyanin biosynthesis. MdNAC52, which refered to ‘Malus domestica NAC (NAM、ATAF1/2、CUC1/2) 52’, could activate MdMYB9 which refered to ‘Malus domestica MYB (v-myb avian myeloblastosis viral) 9’ and MdMYB11 by binding to their promoters and enhance the accumulation of procyanidins and anthocyanins during color formation in apple fruits. Thus, MdNAC52, which acted downstream of Malus domestica ELONGATED HYPOCOTYL 5 (MdHY5) and responded to light signals, can be used as a target gene (Sun et al. 2019). The expression patterns of MdNAC42 were very similar to those of MdMYB10 and Malus domestica TRANSPARENT TESTA GLABRA1 (MdTTG1) throughout the development of red-fleshed fruits. MdNAC42 bound directly to the promoter of MdMYB10 and activated its expression (Zhang et al. 2020).
A NAC TF, BLOOD (BL), controlled the blood-flesh trait of the landrace ‘Dahongpao’. BL formed a homodimer with Prunus persica NAC (NAM、ATAF1/2、CUC1/2) (PpNAC1), which activated the expression of Prunus persica MYB (v-myb avian myeloblastosis viral) 10.1 (PpMYB10.1) and resulted in the accumulation of anthocyanins in the blood-flesh peach fruits (Zhou et al. 2015). PpNAC25 (Prupe.4G040900) was highly homologous to the positive regulator of fruit ripening in tomatoes, Solanum lycopersicum NAC (NAM、ATAF1/2、CUC1/2)-non-ripening (SlNAC-NOR). The overexpression of PpNAC25 in poplar resulted in deep red-colored shoot tips. Additionally, PpNAC25 could bind directly to the promoter regions of PpMYB10.1 and 10.2, which activated their expression and promoted anthocyanin biosynthesis. PpNAC25 also interacted with PpNAC1 (Geng et al. 2022). Furthermore, NAC TFs play essential regulatory roles in fruit ripening. PpNAC1 and PpNAP6 directly regulate Prunus persica 1-aminocyclopropane-1-carboxylic acid (ACC) ynthase PpACS1), a crucial gene involved in ethylene biosynthesis (Li et al. 2016; Cao et al. 2021), while PpNAC.A59 was involved in fruit maturation by regulating the expression of Prunus persica ethylene responsive factor A16 (PpERF.A16) (Guo et al. 2021).
In this study, a comparative analysis of the leaves of two peach cultivars, 99-30-33 and Zhongtao5, during autumn was performed. 99-30-33 exhibits red-colored leaves in autumn, ripens in late May, and demonstrates a fruit development period of ~ 60 days. Zhongtao5 (CP5) exhibits green-colored leaves in autumn, ripens in mid-July, and has a fruit development period of ~ 100 days. The critical genes associated with the manifestation of red color in the leaves in autumn, PpNAC1 and PpMYB10.1, were further investigated using BSA-seq, RNA-seq, subcellular localization analysis, yeast one-hybrid (Y1H) assays, yeast two-hybrid (Y2H) assays, dual-luciferase assays, and BiFC (Biomoldcule Fluorescence Complement) assays. PpNAC1 and PpMYB10.1 were observed to play essential roles in anthocyanin biosynthesis. Taken together, the results obtained in this study provide evidence that PpNAC1 is a positive regulator of anthocyanin biosynthesis in red-colored-autumn leaves of the highly early-maturing varieties of peach, which was dependent on its interaction with PpMYB10.1.
Materials and methods
Plant materials
Peach plants were grown in the peach garden of the Zhengzhou Fruit Research Institute, Chinese Academy of Agricultural Sciences, Henan Province, China. The two cultivars, 99-30-33 with red-colored-autumn leaves and CP5 with green leaves were used as the experimental material. All the selected plants were grown under conditions of adequate light and moisture (Fig. 1). The samples from 99-30-33 were collected in August 2021 with RY indicating the sample of the young green type and RO of the old red type. The samples from CP5, including the young green type (GY) and the old green type (GO), were also collected simultaneously. Three biological replicates were obtained for each line by pooling the samples collected from five plants. In total, 12 samples from the two cultivars, of which two tissues were in triplicate, were rapidly frozen in liquid N2 upon collection and subsequently stored at –80°C until anthocyanin determination and transcriptomic analysis.

Comparison of leaf color of the trees and branches of 99-30-33 (an extremely early-maturing variety) and CP5 (a medium-maturing variety) in autumn. (a) Comparison of the phenotypes of the whole trees of 99-30-33 and CP5. Bar = 20 cm. (b) Comparison of branches with red or green leaves. Bar = 10 cm. CP5, a kind of peach cultivar, Zhongtao5; RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5; GO, old green leaves of CP5
An F1 population was generated through a cross conducted between 10–7 and 6915 in 2014 to identify the candidate interval. In the spring of 2015, the F1 population consisting of 325 individuals was planted at the test site of the Chinese Academy of Agricultural Sciences in Xinxiang.
Anthocyanin measurement
Qualitative and quantitative analysis of the metabolome was conducted using ultra-performance liquid chromatography-tandem mass spectrometry (Di et al. 2004). Characterization of the compounds was achieved with reference to the second-order spectral information, utilizing the self-built MetWare database. The relative content of the various metabolites was standardized and centralized, followed by K-means clustering analysis. Cluster plotting and Principal Component Analysis (PCA) were performed using MetWare Cloud (https://cloud.metware.cn).
BSA-seq
Genomic DNA was extracted from the young leaves of 28 red-leaf individuals, 28 green-leaf individuals, and their parents, forming two extreme trait mixing pools (R pool for red and G pool for green type). DNA libraries were then constructed and sequenced by the Beijing Genomics Institute, Beijing, China. The reference genome, Lovell v2.1 (https://www.rosaceae.org/gb/gbrowse/prunus_persica_v2.0.a1/) was used. The mutation frequency of the polymorphic sites in the offspring pool, the single nucleotide polymorphism (SNP) index, was calculated. A 1 Mb window was chosen to calculate the sliding window and the mean value of the polymorphic sites was calculated within the window in unit steps of 20 Kb each. The absolute value of the difference in the gene frequency, △SNP-index, between the two offspring pools was obtained, and finally, an SNP-index diagram of the mixed offspring pool was generated (Altschul et al. 1997). The initial 1% of the △SNP-index was selected as the screening threshold, and the regions outside this threshold were considered potential candidate localization regions of regulatory genes.
Transcriptomic analyses
Transcriptomic analyses were carried out by the Wuhan MetWare Biotechnology Corporation (Wuhan, China). RNA from the different parts of the leaves of 99-30-33 and CP5 was extracted. For the preparation of the sequencing library, a NEBNext®Ultra™ RNA Library Prep Kit (Illumina®, New England Biolabs, MA, USA) was utilized following the instructions of the manufacturer. Cluster plotting and PCA were performed using MetWare Cloud (https://cloud.metware.cn). The differentially expressed genes (DEGs) were identified by the DESeq2 software (Liu et al. 2021). The TBtools software was used to draw a heatmap (Chen et al. 2020).
Subcellular localization of PpNAC1
The stop codon in the coding sequence (CDS) of PpNAC1 was deleted, and the CDS was cloned between the SalI and BamHI sites of the 16318-hGFP vector. After amplification of the DNA of the solution containing the positive bacteria was obtained by sequencing. The recombinant plasmids 16318-hGFP and Prunus persica NAC (NAM、ATAF1/2、CUC1/2) 1-Green Fluorescent Protein (PpNAC1-GFP) were extracted using an endotoxin-free plasmid extraction kit (TIANGEN®, Sichuan, China) and were used to transform the protoplasts of Arabidopsis. Subcellular localization analysis was conducted according to a previous study (Yoo et al. 2007).
Yeast one-hybrid (Y1H) assay
The full-length CDSs of the TFs were cloned and inserted using the EcoRI site of the pGADT7 vector. The promoter of the gene was cloned between the HindIII and XhoI sites of the pABAi vector. Y1H analysis was performed according to the method described in a previous study (Wang et al. 2019).
Yeast two-hybrid (Y2H) assay
The entire CDS of PpNAC1 was cloned and inserted using the EcoRI site of the pGADT7 vector. Likewise, the complete CDS of PpMYB10.1 was cloned and inserted between the EcoRI and BamHI sites of the pGBKT7 vector. Y2H analysis was performed following the protocol described in a previous study (Wang et al. 2019).
Dual-luciferase assay
The CDSs of the TFs were cloned between the BamHI and EcoRI sites of the pGreenII 62-SK vector. The promoter regions (2 kb upstream of the start codon, ATG) of the genes were cloned between the KpnI and NcoI sites of the pGreenII 0800-LUC vector. Agrobacterium tumefaciens GV3101 cells (Tolo Biotech, Shanghai, China) carrying the plasmid constructs were cultured overnight and then resuspended in the infiltration buffer containing 10 mM MES at pH ± 5.8, 10 mM MgCl2, and 100 µM acetosyringone for 3 h in the dark. Following incubation, the leaves of 4-week-old Nicotiana benthamiana (N. benthamiana) plants were co-cultivated with the A. tumefaciens cells. The luciferase activity of nine transformed leaves derived from three different tobacco plants was measured using a dual-luciferase reporter assay system (Promega, WI, USA). The entire process was repeated thrice to obtain biological replications.
BiFC assay
PpMYB10.1 and PpNAC1 were cloned and inserted into the pUC-SPYNE and pUC-SPYCE vectors, respectively. A. tumefaciens GV3101 cells were transformed with the two vectors separately, and the leaves of 4-week-old tobacco plants were co-cultivated. The samples were collected 72 h after injection, and the GFP signals were detected using laser confocal fluorescence microscopy (Walter et al. 2004).
Statistical analyses
The data were analyzed using Origin 2022 (Moberly JG et al. 2018). Student’s t-test was used to determine the statistical significance of the differences between the two datasets (*P < 0.05, **P < 0.01).
Results
The color phenotypes of peach autumn leaves
The red-leaf cultivar, 99-30-33, and a green-leaf one, Zhongtao5 (CP5), were selected to observe the leaf phenotype in autumn. The old and young leaves of CP5 remained green and did not turn red. The old leaves of 99-30-33 turned red 150 days after flowering (DAF) (Fig. 1a), while the young leaves remained green (Fig. 1b).
Quantitative analyses of anthocyanins
Metabolomic analyses of leaves from the different parts of the branches in autumn were conducted to identify the anthocyanin(s) responsible for the manifestation of color (Fig. 1b). PCA was employed to categorize the metabolites into four distinct groups. The biological replicates of RY, RO, GY, and GO exhibited a strong consistency, validating the reliability of the metabolomic data. The PCA plot indicated significant variations between the mature and young leaves, with a more pronounced difference in the R group compared with the G group. PC1 and PC2, the two principal components, accounted for 62.17% and 23.73% of the total variance for the R and G groups of both mature and young leaves, respectively (Fig. 2a). Cluster analysis indicated the relative relationships between the samples. The correlation coefficient (R2) between the biological replicates was ≥ 0.9, and that between the red type (RO) and the green type (RY, GY, and GO) was > 0.9 (Supporting Information, Fig. S1a).

Analysis of anthocyanins levels in different leaves of two peach cultivars. (a) PCA chart of the samples used for metabolomic analysis. (b) K-means clustering of all metabolites. (c) Accumulation of six anthocyanins, including cyanidin-3-O-glucoside, cyanidin-3-O-(6-O-p-coumaroyl)-glucoside, cyanidin-3-O-xyloside, cyanidin-3,5-O-diglucoside, peonidin-3-O-glucoside, and peonidin-3-O-rutinoside, in the four tissues of the two cultivars. PC1, principal component 1; PC2, principal component 2; RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5; GO, old green leaves of CP5. (Student’s t-test, **P < 0.01)
A comprehensive analysis revealed the presence of 51 anthocyanins in the entire sample set (Table S1). By employing the K-means clustering methodology, six distinct clusters were identified in the samples, each containing 2–16 different metabolites (Fig. 2b). Notably, cluster 4, comprising 16 metabolites, displayed a pattern of accumulation consistent with the metabolites linked to the red leaf phenotype. Specifically, this cluster exhibited a relatively higher metabolite content in the red leaves than in the other three green leaf sections. Cluster heatmap analysis demonstrated that all the detected anthocyanins and biological replicates clustered into the same group (Fig. 3). Of the 16 anthocyanins in cluster 4, the six most abundant that were enriched in the anthocyanin biosynthesis pathway were cyanidin-3-O-glucoside, cyanidin-3-O-(6-O-p-coumaroyl)-glucoside, cyanidin-3-O-xyloside, cyanidin-3,5-O-diglucoside, peonidin-3-O-glucoside, and peonidin-3-O-rutinoside. Six of them were accumulated to significantly different levels in RO (Table S2). The accumulation patterns of the six anthocyanins in the leaves from the different parts of the branches suggested that the redness of peach leaves might be due to the presence of anthocyanins (Fig. 2c), predominantly cyanidin.

Heatmap-based representation of all the accumulated metabolites for 51 anthocyanins. RY, young green leaves of 99–30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5; and GO, old green leaves of CP5
BSA-seq analysis of the leaf color of peach in autumn
The leaf color phenotypes in autumn of the 325 hybrids of the 10-7 × 6915 cross population were analyzed. The female parent, 10-7, had green leaves, and the male parent 6915 had red leaves. The old and young leaves of 10-7 were green and did not turn red. However, the old leaves of 6915 turned red 150 DAF, whereas its young leaves were green (Supporting Information, Fig. S2). The segregation rate of red and green phenotype was 28:297 (30.8242 > χ20.05, 1) in the F1 population and was inconsistent with Mendel’s law.
To form two distinct pooled groups, the red and the green pool, 28 red and 28 green individuals were selected (Table S3). The DNA from these two pooled groups and the parental samples was utilized for library construction and subsequent resequencing. After barcode trimming, the clean sequence data for the red pool comprised ~ 14,387,287,500 bp, while the green pool contained ~ 12,899,321,100 bp. Both met high-quality standards with a Q20 ≥ 97.17% and a GC content ranging from 38.91% to 39.10%.
The average sequencing depths for the red and green pools were 63.747-fold and 57.154-fold, respectively. This resulted in a total of 95,915,250 reads for the red, and 85,995,474 reads for the green pool, which were subsequently mapped to the Lovell genome v2.1. The overall data exhibited adequate coverage and sound quality (Table S4).
Next, the clean reads were aligned to the reference peach genome, and SNPs calling was conducted, identifying 1,634,937 SNPs. By analyzing the read counts, the SNP-index and the △SNP-index for each SNP position were calculated, applying a confidence level of 95% as the screening threshold. This analysis derived the △SNP-index for the entire genome, spanning chromosomes 1 to 8.
Based on the SNP-index analysis, a candidate interval on chromosome 4 between the positions 11,070,375 and 11,994,184 were identified, where the genes associated with the target trait were suspected to be located (Fig. 4). Utilizing the annotation of the peach genome v2.1, the candidate interval was narrowed down to 922 kb and 95 annotated genes (Table S7) were identified within this region.

Identification of candidate intervals based on BSA-seq. Chromosome is indicated on the horizontal axis, and SNP-index data on the vertical. The blue line indicates the topmost 1% threshold; the pink line, the topmost 5% threshold; the black dot, the △SNP-index of each SNP; and the red line, the result of the sliding window (△SNP-index) analysis. The red circle indicates the candidate region involved in the red-colored-autumn leaves phenotype
Identification of PpNAC1 by RNA-seq
To gather additional insights into the genes located within the mapping interval associated with red-colored-autumn leaf phenotype, RNA-seq and metabolomic analysis of the leaves of 99-30-33 and CP5 at the same stage of color formation were conducted. RNA-seq yielded 90.56 gigabytes (Gb) of clean data across all 12 samples, with an average of 7.55 Gb per sample after filtering. The mean percentage of Q30 bases was 92.54%, and a Guanine Cytosine (GC) content > 45% in each sample for quality control of the transcriptome sequencing data (Table S5).
An assessment of the correlation between the biological replicates showed a minimum R2 value of ≥ 0.9 (Supporting Information, Fig. S1b). Furthermore, PCA demonstrated the clustering of biological replicates within the same group, while the samples from different groups exhibited noticeable dissimilarities (Fig. 5a).

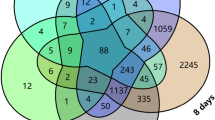
Identification of the crucial gene PpNAC1. (a) PCA chart of the samples used for RNA-seq analysis. (b) Venn diagram indicating the differentially expressed genes. (c) Heatmap representation of the expression of 95 genes within the mapping interval. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5; and GO, old green leaves of CP5; PC1, principal component 1; PC2, principal component 2
To pinpoint the genes potentially responsible for red pigmentation in RO, 10,900 DEGs were analyzed. Among these, 3,132 DEGs were identified in 99-30-33. In comparison to the green type RY, 2,384 DEGs were found to be downregulated, while 748 DEGs were upregulated in the red type RO (Supporting Information, Fig. S3a). In total, 2,490 DEGs were identified from CP5, among which 496 were downregulated and 1,994 were upregulated in the old green leaf type GO (Supporting Information, Fig. S3b). Additionally, 3,000 downregulated and 1,011 upregulated DEGs were identified in GY compared with RO; and 747 downregulated and 520 upregulated DEGs in GO compared with RO (Supporting Information, Fig. S3c, d). Analysis of the Venn diagrams revealed that 204 DEGs were common to the four groups (Fig. 5b). A total of 551 DEGs were identified between RO and the other three groups of green leaves (Table S6), which included Prupe.4G187100 among the genes identified within the mapping interval. Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis revealed that 551 DEGs were distributed into 20 pathways. The largest categories in which the DEGs were significantly enriched included “Anthocyanin biosynthesis”, “Photosynthesis-antenna proteins”, and “Flavone and flavonol biosynthesis” (Supporting Information, Fig. S4). In addition, the expression levels of the genes in the candidate interval were analyzed using the RNA-seq datasets (Table S7). Consistent with the red leaf phenotype in RO, Prupe.4G187100 was significantly highly expressed in the red leaves relative to the green leaves (Fig. 5c).
Expression patterns of the candidate DEGs related to anthocyanin biosynthesis
The structural genes related to anthocyanin biosynthesis were generally more highly expressed in RO than in the other three green leaf parts (Fig. 6). These included Prunus persica Chalcone synthase (PpCHS) (Prupe.1G002900), Prunus persica Flavonoid 3'-hydroxylase (PpF3'H) (Prupe.5G203600), Prunus persica Dihydroflavonol reductase (PpDFR) (Prupe.1G376400), Prunus persica Leucoantho-cyanidin dioxygenase (PpLDOX) (Prupe.5G086700), Prunus persica UDP-glucose, flavonoid-3-O-glucosyltransferase (PpUFGT) (Prupe.2G324700), and Prunus persica Glutathione S-transferase (PpGST) (Prupe.3G013600). PpF3'H, PpLDOX, PpDFR, and PpUFGT facilitated the biosynthesis of anthocyanins, specifically cyanidin and peonidin. Therefore, the upregulation of these genes in RO may explain the higher anthocyanin production in the red leaves in autumn. Interestingly, the expression of PpGST and PpUFGT was 245-fold and 123-fold higher in red than in green leaves, respectively, indicating that they may be the crucial structural genes determining the red leaf color. The transcriptomes of 99-30-33 and CP5 were examined to analyze the TF encoding genes associated with red autumn leaves (RO). The analysis revealed the upregulation of specific TFs in RO, including a NAC (Prupe.4G187100) and a MYB TF (Prupe.3G163100), as depicted in Fig. 6.

Expression levels of the anthocyanin biosynthesis genes, regulatory genes, and the critical gene PpNAC1 in the different autumn season leaves in peach fruit. Data are the mean of three independent biological replicates ± SD. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; and GO, old green leaves of CP5. (Student’s t-test, **P < 0.01)
Regulation of PpMYB10.1 by PpNAC1
To determine the subcellular localization of PpNAC1, a plasmid carrying PpNAC1 fused with green fluorescent protein (GFP) was constructed and then introduced into the Arabidopsis protoplasts. After 72 h, the subcellular localization of the fusion protein PpNAC1-GFP was examined and was detected exclusively in the nucleus (Fig. 7), thus confirming the role of PpNAC1 as a TF involved in regulating anthocyanin biosynthesis. Y1H and dual-luciferase assays were performed to verify whether PpNAC1 directly regulated PpMYB10.1. During the Y1H assay, the transformants containing pGADT7-PpNAC1/pLacZi-PpMYB10.1 grew normally on SD/-L-U + /200 ng/ml AbA medium (Fig. 8a, b). The pGreenII62-SK vector carrying PpNAC1 served as an effector, and the pGreenII0800-LUC vector carrying the promoter of PpMYB10.1 served as a reporter (Fig. 8c). Following the co-infiltration of the TF- and promoter-containing constructs into the leaves of N. benthamiana, the LUC/REN (Luciferase/Renilla) ratios were measured. As illustrated in Fig. 8d, the PpNAC1 or PpMYB10.1 constructs exhibited significantly higher LUC/REN ratios than the corresponding controls, indicating that PpNAC1 could activate the promoter of PpMYB10.1. The potential of PpNAC1 for activating PpMYB10.1 was determined to be 4.91-fold higher. These findings suggest that PpNAC1 can bind to the promoter of PpMYB10.1, thus leading to an activation of the transcription of PpMYB10.1, possibly facilitated through this binding.

Subcellular localization of PpNAC1. The scale bar = 25 μm

PpNAC1 directly interacts with the promoter of PpMYB10.1. (a) Diagram of the plasmid constructs used for yeast one-hybrid assays. (b) The binding ability of PpNAC1 to the promoter of PpMYB10.1 was revealed by yeast one-hybrid assays. (c) Diagram of the plasmid constructs used for the dual-luciferase assays. (d) PpNAC1 activated the promoter of PpMYB10.1, as revealed by dual-luciferase assays. Student’s t-test, **P < 0.01
Interaction between PpNAC1 and PpMYB10.1 promotes the expression of PpUFGT and PpGST
To validate the interaction between PpNAC1 and PpMYB10.1, a Y2H assay was conducted. The CDSs of PpNAC1 and PpMYB10.1 were cloned and inserted into the yeast pGADT7 and pGBKT7 vectors, respectively. Yeast cells containing the pGBKT7-PpMYB10.1 construct and the empty activation domain (AD) constructs displayed robust growth in the quadruple dropout (QDO) medium, indicating that PpMYB10.1 possesses transcriptional, self-activation activity. However, upon the addition of 3-AT (3-amino-1,2,4-triazole) to the medium, this activity was effectively inhibited. The yeast cells containing PpNAC1 and PpMYB10.1 grew normally and turned blue when cultured in the QDO + 3-AT + X-α-gal medium (Fig. 9a). Consequently, the results of the Y2H assay demonstrated that PpNAC1 interacted with PpMYB10.1. This interaction was further investigated by BiFC assays. Functional GFP was reconstituted in the nucleus when PpMYB10.1-nYFP and PpNAC1-cYFP were co-expressed in the leaves of N. benthamiana. However, no fluorescence signal was detected when PpMYB10.1-nYFP or PpNAC1-cYFP were co-expressed with only cYFP or nYFP, respectively (Fig. 9b).

The PpNAC1-PpMYB10.1 interaction could activate the promoters of Prunus persica UDP-glucose, flavonoid-3-O-glucosyltransferase (PpUFGT) and Prunus persica Glutathione S-transferase (PpGST). (a) Prunus persica NAC (NAM、ATAF1/2、CUC1/2) (PpNAC1) could interact with Prunus persica MYB (v-myb avian myeloblastosis viral) 10.1 (PpMYB10.1), as revealed by yeast two-hybrid assays. (b) The physical interactions between PpNAC1 and PpMYB10.1 as ascertained by the BiFC assay. The scale bar = 25 μm. (c) Determination of the activities of the promoters of PpUFGT and PpGST induced by the PpNAC1-PpMYB10.1 interaction using luciferase (LUC)/Renilla luciferase (REN) assay. Student’s t-test, **P < 0.01
Next, dual-luciferase assays were used to determine whether PpNAC1 or PpMYB10.1 enhanced the expression of PpUFGT and PpGST. The leaves of N. benthamiana were co-infiltrated with vectors containing PpNAC1 or PpMYB10.1, together with the promoters of PpUFGT or PpGST. Compared with the empty vector (control), the expression of either PpNAC1 or PpMYB10.1 alone did not enhance the activities of the promoters of these genes. However, when the leaves of N. benthamiana were co-infiltrated with PpNAC1 and PpMYB10.1 combined with the promoters of PpUFGT or PpGST, the PpNAC1-PpMYB10.1 interaction enhanced the activities of the promoters of PpUFGT and PpGST (Fig. 9c). The activation of PpUFGT and PpGST by PpNAC1-PpMYB10.1 was 2.64-fold and 2.86-fold higher than that by the control, respectively. Thus, the coexpression of PpNAC1 with PpMYB10.1 increased the expression of the reporter gene. This result indicated that PpNAC1 positively regulated anthocyanin biosynthesis in the red-colored-autumn leaves of peaches.
Discussion
Peaches, being attractive fruits, are popular with consumers and growers because they are continuously available throughout the summer and autumn, particularly the highly early-maturing varieties that have been launched in recent years. In plants, leaves are commonly green, and the occurrence of red leaves is relatively infrequent, especially among fruit trees. In this study, the leaves of some early-maturing peaches turned red in autumn, which was consistent with the results of previous studies (Zhou et al. 2014). The anthocyanin content of leaves was positively correlated with the expression of red color in the leaves, indicating that a change in the anthocyanin content was an essential factor in determining the leaf color in peach. Interestingly, cyanidin-3-O-glucoside was the most abundant compound, consistent with the blood-flesh peach trait of the cultivar ‘Dahongpao’ (Zhou et al. 2015). These studies suggested that the mechanisms responsible for the accumulation of red pigments in peach leaves and fruits were similar.
In many plant species, anthocyanin accumulation was mainly controlled by the transcriptional regulation of the R2R3 MYB TF. In apple (Malus domestica), MdMYB1 (An et al. 2018), MdMYBA (Mahmoudi et al. 2012), MdMYB10 (Espley et al. 2007), and MYB110a (Sato et al. 2019) were the crucial positive regulators of anthocyanin biosynthesis. MdMYB1 and MdMYBA were mainly involved in anthocyanin biosynthesis in the peel, MdMYB10 in the fruits and leaves, and MYB110a in the outer cortex of the fruit. The homologs of MdMYB10 have been identified in Rosaceae, including Prunus avium MYB (v-myb avian myeloblastosis viral) 10 (PavMYB10) from sweet cherry (Prunus avium) (Lin et al. 2010), PdmMYB10 from European plum (P. domestica L.), PcfMYB10 from cherry plum (P. cerasifera) (Takos et al. 2006), and PyMYB10 from pear (Pyrus sorotina) (Feng et al. 2010). All of these homologs positively regulated anthocyanin biosynthesis in the fruits by inducing the expression of the downstream DFRs. Thus, MYB10 may be a key factor governing the fruit color in Rosaceae. In a previous study, six PpMYB10s were identified in peach (Zhou et al. 2015). In this study, PpMYB10.1 was found to be most highly expressed in the red-colored-autumn leaves (Fig. 6), and the encoded protein binds to the promoters of PpUFGT and PpGST (Supporting Information, Fig. S5). This suggests the probable involvement of PpMYB10.1 in anthocyanin biosynthesis in the red-colored-autumn leaves in the very early-maturing variety of peach. Other TFs are also involved in anthocyanin biosynthesis, such as the NAC TFs. An NAC TF-encoding gene, Prunus persica BLOOD (PpBL), was identified through a comparative transcriptomic analysis of the blood-fleshed cultivar ‘Dahongpao’ with the white-fleshed cultivar ‘Baifeng’ and has been determined to be pivotal for the blood-fleshed trait in peaches. Moreover, during fruit ripening, PpBL could activate PpMYB10.1 (Zhou et al. 2015). A comparison of the expression patterns of the genes between the two varieties has been proven to be an important method for studying the key TFs that may be involved in the regulation of anthocyanin biosynthesis. In this study, PpNAC1 was more highly expressed in red than in green leaves in autumn. Moreover, PpNAC1 could bind strongly to the promoter of PpMYB10.1, confirming that PpNAC1 was involved in anthocyanin biosynthesis in the red peach leaves in autumn.
Similarly, NACs have been reported to regulate anthocyanin biosynthesis in the other species. MdNAC42 was identified by screening the differentially expressed NAC TFs and was more dramatically expressed in red-flesh than in white-flesh apples; and promoted the expression of the genes associated with the anthocyanin biosynthesis pathway and accumulation when overexpressed in the calli of apple (Zhang et al. 2020). Notably, the specific interaction between MdNAC42 and MdMYB10 demonstrated that MdNAC42 was necessary for regulating anthocyanin-based pigmentation in red-fleshed apples (Zhang et al. 2020). Interestingly, Litchi chinensis NAC (NAM、ATAF1/2、CUC1/2) 13 (LcNAC 13), which showed the most significant upregulation when fruits turn red, could act as an activator for the regulation of the structural genes such as Litchi chinensis Chalcone synthases Litchi chinensis Chalcone isomerase (LcCHI), Litchi chinensis Flavonoid 3-hydroxylase (LcF3H), Litchi chinensis Flavonoid 3’-hydroxylase (LcF3'H), Litchi chinensis Dihydroflavonol reductase (LcDFR), Litchi chinensis MYB (v-myb avian myeloblastosis viral) 1 (LcMYB1) associated with the anthocyanin biosynthesis pathway and interacted with LcMYB1 during the ripening of litchi fruits (Jiang et al. 2019). In this study, the mechanisms by which PpNAC1 controls the accumulation of anthocyanins in the red peach leaves in autumn were explored. PpNAC1 not only activated the expression of PpMYB10.1 but also interacted with PpMYB10.1. In addition, PpNAC1 activated the expression of PpUFGT and PpGST by interacting with PpMYB10.1, suggesting that PpNAC1 is a critical element in the regulatory network controlling the anthocyanin-based pigmentation of red peach leaves in autumn. Based on these results, a model explaining the accumulation of anthocyanins in the red leaves of peaches in autumn has been proposed (Fig. 10).

A regulatory model explaining anthocyanin biosynthesis in peach autumn red leaves. High expression of PpNAC1 activated PpMYB10.1, and the PpNAC1-PpMYB10.1 interaction could activate the expression of PpUFGT and PpGST, thereby promoting anthocyanin biosynthesis in autumn red leaves. PpNAC1, Prunus persica NAC (NAM、ATAF1/2、CUC1/2); PpMYB10.1, Prunus persica MYB (v-myb avian myeloblastosis viral) 10.1; PpUFGT/GST, Prunus persica UDP-glucose, flavonoid-3-O-glucosyltransferase/Glutathione S-transferase
The segregation ratio of the red-colored-autumn leaf phenotype in the F1 population indicated that this trait was not governed by a single dominant gene. In this investigation, the F1 population was utilized for mapping, leading to the identification of a 922 kb region housing 95 genes. Based on the results of RNA-seq analysis, it can be proposed that PpNAC1 was most probably responsible for the red-colored-autumn leaf trait. Although PpNAC1 played a role in the development of red leaves in autumn and the anthocyanins accountable for red coloration in peach leaves have been identified, the exact reason behind the elevated expression levels of PpNAC1 in red leaves compared with green leaves remains unclear. Thus, further research is required to elucidate this aspect. The expression of PpNAC1 might be regulated by sequence variations and contributed to the development of the red-colored-autumn leaf phenotype. As the appearance of red leaves is probably not controlled by a single dominant gene, and no sequence variations have been associated with red leaves, the factors such as epigenetic modifications may affect the expression of PpNAC1 (Zhou et al. 2023). In addition, it is also necessary to compare the methylation levels of the promoter regions of PpNAC1 among the different varieties to provide new clues behind the causes determining the formation of red color in leaves in autumn.
Conclusions
Anthocyanins are responsible for the manifestation of red color in the leaves of peach trees during autumn. In this study, a NAC TF, PpNAC1, was found to be highly expressed in red-colored-autumn leaves in peach. PpNAC1 targeted the anthocyanin activator PpMYB10.1 and enhanced anthocyanin biosynthesis. Moreover, PpNAC1 and PpMYB10.1 in synergy, activated the expression of PpUFGT and PpGST associated with anthocyanin biosynthesis. These results provide evidence that PpNAC1 is a positive regulator of anthocyanin biosynthesis in the red-colored-autumn leaves of the extremely early-maturing variety of peach.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Aida M, Ishida T, Fukaki H, Fujisawa H, Tasaka M. Genes involved in organ separation in Arabidopsis: an analysis of the cup-shaped cotyledon mutant. Plant Cell. 1997;9:841–57. https://doi.org/10.1105/tpc.9.6.841.
Albert NW, Davies KM, Lewis DH, Zhang H, Montefiori M, Brendolise C, et al. A conserved network of transcriptional activators and repressors regulates anthocyanin pigmentation in eudicots. Plant Cell. 2014;26:962–80. https://doi.org/10.1105/tpc.113.122069.
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. https://doi.org/10.1093/nar/25.17.3389.
An JP, Wang XF, Li YY, Song LQ, Zhao LL, You CX, et al. EIN3-LIKE1, MYB1, and ETHYLENE RESPONSE FACTOR3 act in a regulatory loop that synergistically modulates ethylene biosynthesis and anthocyanin accumulation. Plant Physiol. 2018;178:808–23. https://doi.org/10.1104/pp.18.00068.
Boss PK, Davies C, Robinson SP. Analysis of the expression of anthocyanin pathway genes in developing Vitis vinifera L. cv Shiraz grape berries and the implications for pathway regulation. Plant Physiol. 1996;111:1059–66. https://doi.org/10.1104/pp.111.4.1059.
Cao X, Wei C, Duan W, Gao Y, Kuang J, Liu M, et al. Transcriptional and epigenetic analysis reveals that NAC transcription factors regulate fruit flavor ester biosynthesis. Plant J. 2021;106:785–800. https://doi.org/10.1111/tpj.15200.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, et al. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202. https://doi.org/10.1016/j.molp.2020.06.009.
Di Paola-Naranjo RD, Sánchez-Sánchez J, González-Paramás AM, Rivas-Gonzalo JC. Liquid chromatographic-mass spectrometric analysis of anthocyanin composition of dark blue bee pollen from Echium plantagineum. J Chromatogr A. 2004;1054:205–10. https://doi.org/10.1016/j.chroma.2004.05.023.
Espley RV, Hellens RP, Putterill J, Stevenson DE, Kutty-Amma S, Allan AC. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007;49:414–27. https://doi.org/10.1111/j.1365-313X.2006.02964.x.
Feng S, Wang Y, Yang S, Xu Y, Chen X. Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta. 2010;232:245–55. https://doi.org/10.1007/s00425-010-1170-5.
Feng F, Li M, Ma F, Cheng L. Phenylpropanoid metabolites and expression of key genes involved in anthocyanin biosynthesis in the shaded peel of apple fruit in response to sun exposure. Plant Physiol Biochem. 2013;69:54–61. https://doi.org/10.1016/j.plaphy.2013.04.020.
Geng M, Shao Y, Zhang M, Zheng X, Tan B, Wang W, et al. Overexpression of peach NAC25 promotes anthocyanin biosynthesis in poplar shoots. Fruit Res. 2022;2:21. https://doi.org/10.48130/FruRes-2022-0021.
Gonzalez A, Zhao M, Leavitt JM, Lloyd AM. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008;53:814–27. https://doi.org/10.1111/j.1365-313X.2007.03373.x.
Guo ZH, Zhang YJ, Yao JL, Xie ZH, Zhang YY, Zhang SL, et al. The NAM/ATAF1/2/CUC2 transcription factor PpNAC.A59 enhances PpERF.A16 expression to promote ethylene biosynthesis during peach fruit ripening. Hortic Res. 2021;8:209. https://doi.org/10.1038/s41438-021-00644-6.
Holton TA, Cornish EC. Genetics and biochemistry of anthocyanin biosynthesis. Plant Cell. 1995;7:1071–83. https://doi.org/10.1105/tpc.7.7.1071.
Jiang G, Li Z, Song Y, Zhu H, Lin S, Huang R, et al. LcNAC13 physically interacts with LcR1MYB1 to coregulate anthocyanin biosynthesis-related genes during litchi fruit ripening. Biomolecules. 2019;9:135. https://doi.org/10.3390/biom9040135.
Koes R, Verweij W, Quattrocchio F. Flavonoids: a colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005;10:236–42. https://doi.org/10.1016/j.tplants.2005.03.002.
Li F, Li J, Qian M, Han M, Cao L, Liu H, et al. Identifification of peach NAP transcription factor genes and characterization of their expression in vegetative and reproductive organs during development and senescence. Front Plant Sci. 2016;7:147. https://doi.org/10.3389/fpls.2016.00147.
Li C, Wu J, Hu KD, Wei SW, Sun HY, Hu LY, et al. PyWRKY26 and PybHLH3 cotargeted the PyMYB114 promoter to regulate anthocyanin biosynthesis and transport in red-skinned pears. Hortic Res. 2020;7:37. https://doi.org/10.1038/s41438-020-0254-z.
Lin-Wang K, Bolitho K, Grafton K, Kortstee A, Karunairetnam S, McGhie TK, et al. An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biol. 2010;10:50. https://doi.org/10.1186/1471-2229-10-50.
Liu S, Wang Z, Zhu R, Wang F, Cheng Y, Liu Y. Three differential expression analysis methods for RNA sequencing: limma, EdgeR, DESeq2. J Vis Exp. 2021;175:e62528. https://doi.org/10.3791/62528.
Liu GS, Li HL, Grierson D, Fu DQ. NAC transcription factor family regulation of fruit ripening and quality: a review. Cells. 2022;11:525. https://doi.org/10.3390/cells11030525.
Lv Y, Chen J, Zhu M, Liu Y, Wu X, Xiao X, et al. Wall-associated kinase-like gene RL1 contributes to red leaves in sorghum. Plant J. 2022;112:135–50. https://doi.org/10.1111/tpj.15936.
Mahmoudi E, Soltani BM, Yadollahi A, Hosseini E. Independence of color intensity variation in red flesh apples from the number of repeat units in promoter region of the MdMYB10 gene as an allele to MdMYB1 and MdMYBA. Iran J Biotechnol. 2012;10:153–60.
Moberly JG, Bernards MT, Waynant KV. Key features and updates for origin 2018. J Cheminform. 2018;10:5. https://doi.org/10.1186/s13321-018-0259-x.
Nuruzzaman M, Sharoni AM, Kikuchi S. Roles of NAC transcription factors in the regulation of biotic and abiotic stress responses in plants. Front Microbiol. 2013;4:248. https://doi.org/10.3389/fmicb.2013.00248.
Olsen AN, Ernst HA, Leggio LL, Skriver K. NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 2005;10:79–87. https://doi.org/10.1016/j.tplants.2004.12.010.
Sato H, Otagaki S, Ono Y, Shiratake K, Matsumoto S. Upregulation of MdMYB110a is responsible for ABA-mediated colouration of type 2 red-fleshed apples. J Hortic Sci Biotech. 2019;94:33–40. https://doi.org/10.1080/14620316.2018.1452638.
Shin J, Park E, Choi G. PIF3 regulates anthocyanin biosynthesis in an HY5-dependent manner with both factors directly binding anthocyanin biosynthetic gene promoters in Arabidopsis. Plant J. 2007;49:981–94. https://doi.org/10.1111/j.1365-313X.2006.03021.x.
Shin DH, Choi M, Kim K, Bang G, Cho M, Choi SB, et al. HY5 regulates anthocyanin biosynthesis by inducing the transcriptional activation of the MYB75/PAP1 transcription factor in Arabidopsis. FEBS Lett. 2013;587:1543–7. https://doi.org/10.1016/j.febslet.2013.03.037.
Shulaev V, Korban SS, Sosinski B, Abbott AG, Aldwinckle HS, Folta KM, et al. Multiple models for Rosaceae genomics. Plant Physiol. 2008;147:985–1003. https://doi.org/10.1104/pp.107.115618.
Sun Q, Jiang S, Zhang T, Xu H, Fang H, Zhang J, et al. Apple NAC transcription factor MdNAC52 regulates biosynthesis of anthocyanin and proanthocyanidin through MdMYB9 and MdMYB11. Plant Sci. 2019;289:110286. https://doi.org/10.1016/j.plantsci.2019.110286.
Takos AM, Jaffé FW, Jacob SR, Bogs J, Robinson SP, Walker AR. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006;142:1216–32. https://doi.org/10.1104/pp.106.088104.
Tanaka Y, Sasaki N, Ohmiya A. Biosynthesis of plant pigments: anthocyanins, betalains and carotenoids. Plant J. 2008;54:733–49. https://doi.org/10.1111/j.1365-313X.2008.03447.x.
Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45:487–94. https://doi.org/10.1038/ng.2586.
Walter M, Chaban C, Schütze K, Batistic O, Weckermann K, Näke C, et al. Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. Plant J. 2004;40:428–38. https://doi.org/10.1111/j.1365-313X.2004.02219.x.
Wang X, Zeng W, Ding Y, Wang Y, Niu L, Yao JL, et al. Peach ethylene response factor PpeERF2 represses the expression of ABA biosynthesis and cell wall degradation genes during fruit ripening. Plant Sci. 2019;283:116–26. https://doi.org/10.1016/j.plantsci.2019.02.009.
Wang S, Zhang X, Li B, Zhao X, Shen Y, Yuan Z. Genome-wide identification and characterization of bZIP gene family and cloning of candidate genes for anthocyanin biosynthesis in pomegranate (Punica granatum). BMC Plant Biol. 2022;22:170. https://doi.org/10.1186/s12870-022-03560-6.
Winkel BSJ. Metabolic channeling in plants. Annu Rev Plant Biol. 2004;55:85–107. https://doi.org/10.1146/annurev.arplant.55.031903.141714.
Winkel-Shirley B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001;126:485–93. https://doi.org/10.1104/pp.126.2.485.
Xie Y, Tan H, Ma Z, Huang J. DELLA proteins promote anthocyanin biosynthesis via sequestering MYBL2 and JAZ suppressors of the MYB/bHLH/WD40 complex in Arabidopsis thaliana. Mol Plant. 2016;9:711–21. https://doi.org/10.1016/j.molp.2016.01.014.
Yoo SD, Cho YH, Sheen J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc. 2007;2:1565–72. https://doi.org/10.1038/nprot.2007.199.
Zhang S, Chen Y, Zhao L, Li C, Yu J, Li T, et al. A novel NAC transcription factor, MdNAC42, regulates anthocyanin accumulation in red-fleshed apple by interacting with MdMYB10. Tree Physiol. 2020;40:413–23. https://doi.org/10.1093/treephys/tpaa004.
Zhou Y, Zhou H, Lin-Wang K, Vimolmangkang S, Espley RV, Wang L, et al. Transcriptome analysis and transient transformation suggest an ancient duplicated MYB transcription factor as a candidate gene for leaf red coloration in peach. BMC Plant Biol. 2014;14:388. https://doi.org/10.1186/s12870-014-0388-y.
Zhou H, Lin-Wang K, Wang H, Gu C, Dare AP, Espley RV, et al. Molecular genetics of blood-fleshed peach reveals activation of anthocyanin biosynthesis by NAC transcription factors. Plant J. 2015;82:105–21. https://doi.org/10.1111/tpj.12792.
Zhou H, Zhang W, Sheng Y, Qiu K, Liao L, Shi P, et al. A large-scale behavior of allelic dropout and imbalance caused by DNA methylation changes in an early-ripening bud sport of peach. New Phytol. 2023;239:13–8. https://doi.org/10.1111/nph.18903.
Acknowledgements
Not applicable.
Funding
This work was supported by the Excellent Youth Foundation of Henan Scientific Committee of China (212300410094), the Agricultural Science and Technology Innovation Program (ASTIP) (CAAS-ASTIP-2023-ZFRI), and the Special Fund of Henan Province for Agro-scientific Research in the Public Interest (NO. 201300110500).
Author information
Authors and Affiliations
Contributions
LN, WZ and ZW conceived and coordinated this project; JM and SS performed the experiments with contributions from AL, LP, WD, GC and JX; WZ and LN provided the study materials; JM and SS wrote the manuscript under the supervision of and with contributions from LN, WZ and ZW. All of the authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Cluster plots of metabolome and RNA-seq samples. (a) Cluster plots of metabolome samples; (b) Cluster plots of RNA-seq samples. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; GO, old green leaves of CP5
Additional file 2: Figure S2.
The autumn leaf color of the male parent 6915 had a fruit development period of approximately 60 days, and that of the female parent 10-7 had a fruit development period of approximately 80 days. Bar = 15 cm
Additional file 3: Figure S3.
Volcano map of differentially expressed genes (DEGs) between the green and red leaves. (a) Volcano map of DEGs in RY vs RO; (b) Volcano map of DEGs in GO vs GY; (c) Volcano map of DEGs in GY vs RO; (d) Volcano map of DEGs in GO vs RO. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; GO, old green leaves of CP5
Additional file 4: Figure S4.
KEGG enrichment analysis of 551 differentially expressed genes (DEGs) between RO and other green leaves (RY, GY and GO). RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; GO, old green leaves of CP5
Additional file 5: Figure S5.
Yeast one-hybrid assays of the binding of PpMYB10.1 to the promoters of PpGST and PpUFGT. PpMYB10.1, Prunus persica MYB (v-myb avian myeloblastosis viral) 10.1; PpGST, Prunus persica Glutathione S-transferase; PpUFGT, Prunus persica UDP-glucose, flavonoid-3-O-glucosyltransferase
Additional file 6: Table S1.
List of accumulated anthocyanins in all samples. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; GO, old green leaves of CP5
Additional file 7: Table S2.
List of differentially accumulated anthocyanins in RO vs other green leaves. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; GO, old green leaves of CP5
Additional file 8: Table S3.
Materials used for BSA-seq.
Additional file 9: Table S4.
Resequencing data of parental and pooled genomic samples.
Additional file 10: Table S5.
Overview of the transcriptome sequencing dataset. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; GO, old green leaves of CP5
Additional file 11: Table S6.
List of differentially expressed genes in the RO vs the other three groups of green leaves. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; GO, old green leaves of CP5
Additional file 12: Table S7.
List of the 95 genes expressed in the 922 kb interval. RY, young green leaves of 99-30-33; RO, old red leaves of 99-30-33; GY, young green leaves of CP5, a kind of peach cultivar, Zhongtao5; GO, old green leaves of CP5
Additional file 13: Table S8.
Primer pairs used in this study. Y1H, yeast one-hybrid; Y2H, yeast two-hybrid; PpNAC1-pGADT7: Prunus persica NAC (NAM、ATAF1/2、CUC1/2) 1-pGADT7-AD; PpMYB10.1pro-pABAi: Prunus persica MYB (v-myb avian myeloblastosis viral) 10.1 promoter-pABAi; PpMYB10.1-pGBKT7: Prunus persica MYB (v-myb avian myeloblastosis viral) 10.1-pGBKT7 DNA-BD; PpNAC1-16318-GFP: Prunus persica NAC (NAM、ATAF1/2、CUC1/2) 1-16318-Green Fluorescent Protein; PpUFGTpro-pGreenII0800-LUC: Prunus persica UDP-glucose, flavonoid-3-O-glucosyltransferase promoter-pGreenII0800-LUC; PpGSTpro-pGreenII0800-LUC: Prunus persica Glutathione S-transferase promoter-pGreenII0800-LUC; PpMYB10.1-NYFP: Prunus persica MYB (v-myb avian myeloblastosis viral) 10.1-N-terminal Yellow Fluorescent Protein; PpNAC1-CYFP: Prunus persica NAC (NAM、ATAF1/2、CUC1/2) 1-C-terminal Yellow Fluorescent Protein
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meng, J., Sun, S., Li, A. et al. A NAC transcription factor, PpNAC1, regulates the expression of PpMYB10.1 to promote anthocyanin biosynthesis in the leaves of peach trees in autumn. HORTIC. ADV. 1, 8 (2023). https://doi.org/10.1007/s44281-023-00012-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44281-023-00012-5