
Abstract
Purpose
To establish a prognostic model to predict the survival of patients with esophageal cancer (EC).
Methods
We extracted the expression profiles of prognostic-related genes and clinicopathological data from TCGA and GEO databases. Subsequently, a comprehensive bioinformatics analysis was conducted to construct a prognostic model utilizing LASSO and multivariate Cox regression. The stability of the risk signature was validated through Kaplan-Meier and ROC curve analyses on the training, internal testing, and external testing sets. Furthermore, we developed a nomogram that incorporates the risk score and clinical features to predict the suvival. Additionally, a nomogram incorporating the risk score and relevant clinical parameters was developed to enhance survivorship prediction. Furthermore, we delved into exploring the correlation between the risk score and immune cell abundance, expression of cancer checkpoints, as well as responses to immunotherapy and chemotherapeutic agents.
Results
In this study, we successfully identified 19 prognosis-related genes out of a pool of 65 PANoptosis-related genes (PRGs) sourced from existing literature. Through consensus clustering analysis, we classified patients into two distinct groups as PANcluster A and B. Furthermore, the risk score derived from the five PANoptosis-related signatures emerged as an independent prognostic factor among patients with EC. To enhance the prognostic accuracy, we devised a nomogram integrating the risk score with clinical risk characteristics, enabling the prediction of 1-year, 2-year, and 3-year overall survival (OS) rates. Notably, individuals classified in the high-risk group demonstrated poorer prognoses compared to their low-risk counterparts. Furthermore, the risk score displayed substantial correlations with immune cell abundance, expression levels of cancer checkpoints, and responses to immunotherapy and chemotherapeutic agents. These pivotal findings underscore the significance of considering PANoptosis-related patterns in improving prognostic assessment and predicting treatment responses in patients diagnosed with esophageal cancer.
Conclusion
We constructed a reliable prognostic risk model for EC utilizing five PRGs. The developed nomogram serves as a valuable tool in predicting patient outcomes, offering crucial insights that can inform and guide treatment decisions for individuals diagnosed with EC.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introdution
Esophageal cancer (EC) is a highly prevalent malignant tumor worldwide, with a significant impact on global health. According to the 2020 Global Epidemiology of Cancer report, there were 604,000 new cases and 544,000 deaths attributed to esophageal cancer [1]. Significant advancements in diagnosis, surgery, radiotherapy, chemotherapy, and immunotherapy have led to substantial improvements in the clinical efficacy for EC patients. Consequently, the overall survival rate of patients with EC has increased compared to the past. In recent years, tumor immunotherapy and the application of immune checkpoint inhibitors have shown promising results in cancer treatment. Programmed cell death protein 1 (PD-1), a 288 amino acid protein expressed on the surface of T-cells and involved in apoptosis, was first discovered in 1992 [2]. Despite these advancements, the 5-year survival rate for EC remains relatively low.
Cell death is a fundamental physiological process that occurs in organisms, serving various roles such as embryonic development, organ maintenance, aging, and the coordination of immune responses and self-immunity [3]. Among the different forms of programmed cell death, namely apoptosis, necroptosis, and necrosis, recent research has focused on elucidating their redundancy and crosstalk [4].A newly discovered concept called PANoptosis emphasizes the interactions and coordination between apoptosis, necroptosis, and necrosis [5]. Dysregulation of cell death and inflammatory responses has been associated with tumor development. Jiang et al. reported that CASP8 plays a pivotal role as a key protein in the signaling pathway of tumor PANoptosis crosstalk [6]. Previous studies have emphasized the significance of patient characteristics, clinicopathologic tumor features, molecular markers, and treatment strategies in determining prognosis. Clinicopathologic factors, molecular markers, and treatment approaches have been linked to prognosis [7, 8]. However, the current prognostic assessment may not adequately capture these potential survival differences due to the intricate relationship among prognostic factors, leading to significant prognostic heterogeneity. Therefore, there is a compelling need to integrate these diverse factors into a predictive model that can effectively guide treatment decisions.
Numerous studies have focused on constructing tumor classifications and prognostic signatures by analyzing gene or non-coding RNA expression levels to predict the survival and immune landscape of cancer patients [9,10,11,12]. In our study, we demonstrate the effectiveness of molecular clustering and prognostic features based on PANoptosis in predicting the prognosis and intratumoral immune landscape of patients diagnosed with EC. Additionally, we have calculated risk scores and established prognostic markers that accurately predict overall survival (OS) and response to immunotherapy in these patients. Furthermore, we have developed a comprehensive prognosis model specifically related to PANoptosis in patients with esophageal cancer. Our findings strongly support a significant association between PANoptosis impacts on EC patient prognosis, biological behavior, and response to anticancer drugs. These results contribute to a deeper understanding of the underlying mechanisms of PANoptosis and provide valuable insights for the development of novel treatment strategies for patients with esophageal cancer.
Meterials and methods
Data acquisition
Gene expression data and relevant clinical information of EC patients were retrieved from the TCGA (https://portal.gdc. cancer.gov)and GEO (https://www.ncbi.nlm.nih.gov/geo/, ID: GSE53625) databases. Sixty-five PANoptosis-related genes (PRGs) were identified from prior studies [13]. TCGA and GEO data were combined and batch effects were eliminated using the Combat algorithm of the sva R package (R software, version 4.3.2). After matching the transcriptome data from both databases with eligible survival data, 183 samples from TCGA EC cohort were used for modeling and internal validation. Meanwhile, 179 samples of the GEO ESCC cohort were used for external validation. Patients with no follow-up data or incomplete clinical information were excluded from the study. Finally, 362 patients were included in the study.
Selection and consensus clustering analysis of PANoptosis-related genes
We identified PANoptosis-associated differentially expressed genes by using the “limma” package, screening for genes meeting criteria: “|log2FC| > 1” and “adjusted P < 0.05” in different EC subtypes. Prognosis-related DEGs (PRDEGs) were then determined through Kaplan-Meier analysis and univariate Cox regression. Consensus clustering, based on PRDEGs, revealed different expression patterns within esophageal cancer. The optimal clustering was selected, and its prognosis was assessed using Kaplan-Meier analysis. Principal component analysis (PCA) assessed expression patterns. Clinical features were compared using the Wilcoxon test, and DEGs between clusters were identified (|log fold change (FC)| >1, p-value < 0.05). Gene set variation analysis (GSVA) assessed biological process disparities. Single-sample gene set enrichment analysis (ssGSEA) evaluated immune cell infiltration and immune-related function activity. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses explored biological functions and pathways associated with PANDEGs, all considering a significance threshold of p-values and q-values < 0.05.
Construction and verification of the PANoptosis-related prognostic signature
To construct the PANoptosis-related gene prognostic signature, we identified significant differentially expressed genes between Cluster A and Cluster B. Utilizing these genes, we randomly allocated TCGA patients in a 5:5 ratio into training and internal testing sets. Subsequently, a prognostic signature was developed by incorporating five genes, determined through least absolute shrinkage and selection operator (LASSO) regression analysis and multivariate Cox regression analysis. The survival, survminer, and glmnet R packages facilitated this process. Patients from GEO served as the external verification set. The risk score was computed using the expression levels of the five genes, subsequently categorizing patients into high and low-risk groups based on the median risk score. K-M analysis assessed survival disparities between the high-risk and low-risk groups. The prediction efficiency of the risk score was evaluated using receiver operating characteristic (ROC) curves and the area under the curve (AUC).
Establishing a predictive nomogram
An nomogram model that integrates risk scores derived from the prognostic signature and relatively complete information about clinicopathologic or theraputic factors was meticulously formulated. Calibration graphs were meticulously generated to visually delineate the congruence or disparities between the prognostic estimations offered by the nomogram model and the actual survival rates observed in esophageal cancer patients.
Evaluation of PANoptosis-related genes in immune microenvironment
To elucidate the association between the risk score and the tumor microenvironment (TME), CIBERSORT was employed to quantify the abundance of infiltrating immune cells in heterogeneous samples from both high- and low-risk groups. TME scores, encompassing stromal, immune, and ESTIMATE scores, were compared between the high- and low-risk groups using the Wilcoxon signed-rank test. Response to immunotherapy and chemotherapeutic drugs, the tumor mutation burden (TMB) score of patients was computed, and the correlation between the risk score and TMB was scrutinized using the Spearman method. The Tumor Immune Dysfunction and Exclusion (TIDE, https://tide.dfci.harvard.edu/) database assesses the potential for tumor immune escape based on the gene expression profile of tumor samples. Additionally, the "OncoPredict" R package was employed to predict drug responses in vivo or in cancer patients using data screened through cell lines. Finally, the "OncoPredict" R package was utilized to assess differences in drug sensitivity among patients in distinct riskscore groups.
Results
Identification and analysis of PANoptosis related genes in esophageal cancer Patients
The expression data of 183 EC patients were obtained from the TCGA database, and the expression levels of PRGs were compared across 13 normal and 181 tumor samples. A total of 65 PRGs were considered from previous studies, as indicated in Fig. 1a. Out of these, 42 Differentially Expressed Genes (DEGs) exhibited differential expression in EC samples compared to normal tissue, shown in Fig. 1b. All 42 DEGs demonstrated upregulation in tumor samples (p < 0.05). To elucidate the prognostic significance of these genes, Kaplan-Meier curves depict the relationship between DEGs and the prognosis of EC patients, there were 19 DEGs with statistically different survival rates between the high and low expression groups (Fig. 1c).

a The PANoptosis gene list contains 65 genes, of which 25 are from pyroptosis, 32 are from apoptosis, and 8 are from necroptosis; (b) The expression of PANoptosis-related genes in normal and tumor tissues; (c) 19 DEGs with statistically different survival rates between the high and low expression groups
Identification of prognostic PANoptosis-related geneclusters in esophgeal cancer
PRDEGs were identified using KM analysis and univariate Cox regression. The Cox regression revealed two genes associated with overall survival: CASP5 (HR 1.246, 95%CI:1 .033~1.502, P=0.0210) and IRF1 (HR 1.332, 95%CI: 1.035~1.714, P=0.0261) . We investigated the correlation of consensus clustering for prognostic PRDEGs with the characteristics and survival of EC patients. By incrementing the clustering variable (k), we determined that when k = 2, the classification met the standard (Fig. 2a, Figure S1). A total of 159 patients were categorized into two clusters (A and B) based on PRDEGs expression levels, cluster A (n = 132) and cluster B (n = 50). As shown in Fig. 2b, patients in cluster A demonstrated significantly longer survival compared to those in cluster B (p = 0.014). Principal Component Analysis (PCA) exhibited a clear separation between cluster A and B (Fig. 2c). To assess differences in immune cell infiltration between the two clusters, ssGSEA was conducted, revealing that cluster B exhibited higher levels of immune cell infiltration, including activated B cells, activated CD4 + T-cells, activated CD8 + T-cells, activated dendritic cells, macrophages, mast cells, monocytes, natural killer T cells, and neutrophils (Fig. 2d). GSVA highlighted that cluster B was significantly enriched in immune-related pathways, including natural killer cell-mediated cytotoxicity, antigen processing and presentation, and primary immunodeficiency signaling pathways (Fig. 2e). Based on these findings, GO and KEGG analyses illustrated the relevant biological processes (BP), cellular components (CC), molecular functions (MF), and pathways (Fig. 2f, g, h). The identified genes in dfferent cluster were associated with BP including skin development, embryonic organ morphogenesis, and humoral immune response, correlating with CC, such as the apical plasma membrane, external side of the plasma membrane, cluster of actin-based cell projections, and T cell receptor complex. These genes were involved in MF such as cytokine activity, carboxylic acid binding, and immune receptor activity. According to KEGG analysis, these genes participated in certain cancer-related pathways, including the cytokine and cytokine receptor interaction, Cell adhesion molecules, Th1 and Th2 cell differentiation, and Chemical carcinogenesis-DNA adducts.

a According to the similarity displayed by the expression levels of prognstic PANoptosis-related genes and the proportion of ambiguous clustering measure identified the optimal custer (k=2); (b) Survival of patients in cluster A and cluster B (p = 0.014); (c) PCA exhibited a clear separation between cluster A and B; (d) Differences in immune cell infiltration between the two clusters were analyzed by ssGSEA; (e) GSVA highlighted that cluster B was significantly enriched in immune-related pathways; (f, g) GO analyses illustrated the relevant BP, CC, MF, and pathways; (h) KEGG analyses illustrated the relevant BP, CC, MF, and pathways
Construction and Validation of Prognostic Signatures for PANoptosis-related genes
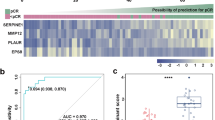
We intersected the data of DEGs with varied expression in the two clusters (|log fold change (FC)| > 1, p-value < 0.05), identifying 1809 genes. Subsequently, LASSO and Cox regression analyses were executed to narrow down these intersected genes (Fig. 3a). This method can effectively discern the most available forecast markers and produce a prognostic indicator to predict clinical results. The dashed perpendicular line illustrates the first-rank value of logλ with the minimum segment likelihood bias. The risk score was calculated based on the coefficients for each intersected gene by the LASSO algorithm, and the equation is as follows: risk score = ( 0.5113 × IRF1 expression level) + ( 0.4222 × NEUROG3 expression level) + ( 0.4871 × TMIGD2 expression level) - ( 0.3701 × SHOX2 expression level) - ( 0.5441 × TRBJ1-2 expression level). Boxplots illustrated higher risk scores from cluster B compared to those in cluster A (Fig. 3b). A Sankey diagram depicted the associations among clusters, risk groups, and survival status (Fig. 3c). In Figure 3d, patients in the high-risk group exhibited a significantly lower probability of survival than those in the low-risk group in both TCGA and GEO databases (p < 0.05). Subgroup analysis indicated that the risk model effectively distinguished survival prognosis in N +, IIB-IIIA, and T3-4 groups (Fig. 3e), suggesting that this risk classification may be more predictive value for the prognosis of locally advanced esophageal cancer. In the case data from TCGA database and GEO database( GSE53625, 179 ESCC patients from China), ROC curves were generated to assess the predictive efficiency of the risk score, with AUCs for 1, 2, and 3-years survival at 0.730, 0.768, and 0.725, respectively, outperforming other clinical features (Fig. 3f). The AUC values of 1, 2, and 3-year survival of patients in TCGA training set, internal TCGA validation set, the external GEO validation set were shown in Figure S2.

a The LASSO regression analysis and partial likelihood deviance on the prognostic genes; (b) Association between risk score and molecular classifications; (c) Sankey plot showed the correlation between molecular subtpye, risk groups and survival status; (d) KM curve showed that patients in high-risk group had a worse prognosis in both TCGA and GEO databases (p < 0.05); (e) Subgroup analysis indicated that the risk model effectively distinguished survival prognosis in N+, IIB-IIIA, and T3-4 groups; (f) ROC methods were used to evaluate the efficiency of the risk score at predicting patient survival
Construction and evaluation of the prognostic nomogram of esophageal cancer
We performed univariate and multivariate Cox regression analysis to evaluate whether the risk model of five genes and other clinical information (such as pathological stage,adjuvant radiation therapy, alcohol consumption, etc.) had independent prognostic characteristics. Univariate analysis showed that clinical stage (p< 0.001), N stage (p< 0.001), and risk score (p= 0.005) were considerably associated with the OS (Fig. 4a). Multivariate Cox regression analysis showed that clinical stage (p< 0.001) and risk score (p< 0.002) remained closely correlated with the OS (Fig. 4a). revealing that clinical stage and risk score remained closely correlated with OS . C-Index suggested that risk score is a better predictor of survival than other clinical factors (Fig. 4b). The risk score and additional clinical features were integrated into a nomogram model (Fig. 4c). Calibration plots demonstrated close alignment between the nomogram-predicted and actual survival probabilities of esophageal cancer patients (Fig. 4d), confirming the accurate predictive capability of this nomogram model for the survival of esophageal cancer patients.

a Univariate and multivariate Cox regression analysis revealed the risk model and clinical stage independently correlated with OS; (b) C-Index suggested that risk score is a better predictor of survival than other clinical factors; (c) Nomogram using risk score and other clinical features were constructed for predicting survival; (d) Calibration graph investigated that then omogrampredicted survival rates were close to the actual survival rates of EC patients
Comparative evaluation of the tumor microenvironments and response to immunotherapy and chemotherapeutic drugs
In evaluating differences in immune cell infiltration between the two risk groups, the findings indicated elevated levels of activated mast cells in the high-risk group (Fig. 5a, c). Calculation of TME scores unveiled that the low-risk group displayed a higher stromal and lower immune score (Fig. 5b). Additionally, the tumor mutation burden (TMB) demonstrated a significant association with the risk score (Fig. 5d). Assessing the predictive potential for checkpoint blockade therapy, boxplots illustrated variations in immune checkpoint gene expression between the two risk groups . Notably, checkpoint genes such as CTLA4, TNFRSF18, CD44, TNFSF18, CD276, and PDCD1LG2ID exhibited higher expression levels in the low-risk group (Fig. 5e). Violin plots further indicated that the low-risk group had a higher TIDE, representing a more favorable response to PD-1 and CTLA-4 blockers (Fig. 5f). We also found that IC50 indicated that the high-risk group was more sensitive to chemotherapeutic drugs, Oxaliplatin, Gemcitabine Crizotinib, and Camptothencin; while low-risk group was more sensitive to Paclitaxel (Fig. 5g).

a, c Differences in immune cell infiltration were shown between the two risk groups; (b) Correlation between risk score and immunerelated scores. *p < 0.05; **p < 0.01; and ***p < 0.001; (d) Tumor mutation burden (TMB) in two risk groups; (e) The differences of immune checkpoint gene expression in two risk groups; (f) ) Violin plots showed the relationship between TIDE scores and risk groups; (g) IC50 differences of six anticancer drugs in two risk group. *p < 0.05; **p < 0.01; and ***p < 0.001
Dicussion
Despite notable improvments in thraputic strategies have been showed, the 5-year survival rate of esophageal cancer patients remains relatively low. The intricate relationship among prognostic factors for esophageal cancer contributes to significant prognostic heterogeneity [1].Therefore, it is particularly important to search for new predictive models to forecast patient survival and enable early intervention. PANoptosis is recently discovered as a novel inflammatory programmed cell death mechanism [14]. It combines and regulates cell death pathways, such as apoptosis, pyroptosis, and necroptosis, by forming a PANoptosome as part of the innate immune responses of the host [15, 16]. Studies have underscored the pivotal role of PANoptosis in both tumorigenesis and anti-tumor therapy by modulating key regulatory elements to thwart tumorigenesis [15, 17]. PANoptosomes can influence inflammatory and immune responses as well as tumorigenesis by regulating PANoptosis.
Past reports have highlighted the potency of markers based on PANoptosis genes in effectively predicting outcomes for esophageal cancer patients [18]. Thus, our study embarks on establishing and validating a risk model utilizing the TCGA and GEO databases to prognosticate esophageal cancer outcomes grounded in the expression patterns of PANoptosis-related genes. In our study, 183 esophageal cancer patients were downloaded from the TCGA database to explore the prognostic function of PRGs. Firstly, we confirmed the prognostic value of 21 PRGs from 65 PRGs. Subsequently, we performed consensus clustering analysis to explore the PANoptosis-related molecular subtype, and the results showed that two distinct subtypes, cluster A and cluster B, where a significant difference in prognosis and enriched pathways/functions was discerned between the two subtypes. Notably, patients within cluster B exhibited markedly poorer prognosis.
The construction of a prognostic signature involved the utilization of LASSO and multivariate Cox regression analyses to meticulously select genes. Patients were subsequently stratified into high and low-risk groups based on their calculated risk scores. Patients in the low-risk group had a superior prognosis compared to those in the high-risk group. Rigorous assessment of predictive efficiency was carried out through ROC analyses. Subgroup analysis indicated that the risk model effectively distinguished survival prognosis in N+ , IIB-IIIA, and T3-4 groups, suggesting that this risk classification may be more predictive value for the prognosis of locally advanced esophageal cancer. Since the tumor heterogeneity and treatment complexity of locally advanced esophageal cancer [19], effective molecular predictive model is needed to guide therapy stratgies. A nomogram model was established according to the risk score and other clinical characteristics to accurately predict the survival of patients. Comparing it with clinical factors, we found that the nomogram predicted survival closely correlated with optimal predictive performance. This result indicates that our predictive model outperforms solely clinical factors in predicting the prognosis of esophageal cancer.
The tumor microenvironment is a complex structure consisting of stromal, cancer, endothelial, and immune cells. Dynamic interactions and crosstalk within the TME involve innate and adaptive immune cells, extracellular immune factors, and cell surface molecules, playing a crucial role in tumorigenesis [20,21,22]. However, our results from GSVA and ssGSEA demonstrated that cluster B was significantly enriched in immune-related pathways, including NK cell-mediated cytotoxicity, antigen processing and presentation, and primary immunodeficiency signaling pathways. Furthermore, cluster B exhibited higher immune cell infiltration levels, involving activated B cells, activated CD4 + T-cells, activated CD8 + T-cells, activated dendritic cells, MDSC, monocytes, natural killer T cells, and T helper cells. Studies have highlighted that the impact of tumor promoting immune cells, such as macrophages, dendritic cells, neutrophils, myeloid suppressor cells, innate lymphocytes, and cytokines in the TME would influence immune function, suppress anti-tumor immune responses by T cells, promote angiogenesis, and enhance cancer cell proliferation, invasion, and metastasis [23,24,25]. NK cells, M1 cells, N1 cells, dendritic cell 1 (DC1 cells), T helper cell 1 (Th1 cells) and CD8 + T cells are tumor suppressive infiltrating immune cells [26, 27]. These findings suggest that cluser B subtypes may respond better to immunotherapy despite poor prognosis.
The examination of immune checkpoint gene expression in both high-risk and low-risk groups included an assessment of IRF1, NEUROG3, TMIGD2, SHOX2, and TRBJ1-2. Several of these genes have established associations with diverse malignant tumors. IRF1, a transcription factor responding to viral infection or interferon stimulation, was notably downregulated by FOXM1c insufficiency in esophageal cancer cell lines, impacting invasion and migration [28]. Wu et al [29] revealed a key role of KAT8-IRF1 condensates in PD-L1 regulation and provide a competitive peptide to enhance antitumor immune responses. TMIGD2 sharing around 10% amino acid sequences with CD28, PD-1, and CTLA, is a transmembrane protein expressed on naive T cells, memory T lymphocytes, tissue-resident T cells, NK cells, plasmacytoid dendritic cells, and innate lymphoid cells [30]. Its expression correlates negatively with angiogenesis, hypoxia, G2/M checkpoint, and epithelial to mesenchymal transition signaling pathways. TMIGD2's positive correlation with immune cell infiltration, including dendritic cells, monocytes, NK cells, gd T cells, and naive CD8 T cells, suggests its potential as a target for developing agonistic bispecific antibodies against tumor resistance to PD-1/PD-L1 blockade therapy [31, 32].Additionally, SHOX2's methylation emerges as a critical event in lung cancer tumorigenesis and progression, presenting itself as a potential biomarker for early or advanced-stage lung cancer screening [33]. In breast cancer, SHOX2 collaborates with STAT3 to promote metastasis [34]. These collective findings underscore the potential of IRF1, TMIGD2, and SHOX2, among others, as valuable biomarkers in cancer diagnosis and therapeutic endeavors.
The analysis of the correlation between the risk score and immune cells revealed no significant expression differences in immune-infiltrated cells between high-risk and low-risk groups [35].Underscored the relevance of an immune score derived from immunogenomic analysis as an indicator of the efficacy of immunotherapy and chemotherapy. In the TCGA dataset, the high-risk group displayed higher stromal and lower immune scores, suggesting a potentially better response to antitumor therapy. Significant differences in immune checkpoint gene expression, including TNFRSF18,CD44, TNFSF18, CD276, and PDCD1LG2ID, were found to be higher in the low-risk group. TNFRSF18, a key regulator of inflammatory and immune response factors, exhibited consistent high expression in NK-T cells within tumor tissues compared to normal tissue. It was highly expressed in TREG cells in lung cancer tissues that did not respond to anti-PD-1 therapy [36]. CD276, identified as a cancer stem cell marker, was highly expressed in cancer stem cells, and CD276 inhibitors demonstrated efficacy in eliminating cancer stem cell formation through enhanced T-cell-mediated anti-tumor immunity [37].Positive CD44 expression in malignant tumor tissues correlated with vascular invasion, distant metastasis, short tumor-free survival, low survival rates, and poor prognosis [38].
Two distinct immune escape mechanisms in tumors involve immunosuppressive factors hindering T cell invasion and functional inactivation of cytotoxic T cells despite high infiltration levels. TIDE scores, which predict immune escape ability by evaluating these mechanisms [39], indicated that low-risk groups had higher TIDE scores, aligning with previous conclusions drawn from TME-related analyses and checkpoint gene analyses. This result contrasts with the prognosis for high-risk group patients. Upon reviewing the TCGA database patients, it was noted that they were included earlier and likely did not receive immunotherapy, contributing to their poor prognosis. Detection of the immune microenvironment and timely immunotherapy might substantially improve the prognosis of high-risk group patients.
Furthermore, IC50 values highlighted differential sensitivity to chemotherapeutic drugs, with the high-risk group exhibiting more sensitivity to Oxaliplatin, Gemcitabine, Crizotinib, and Camptothecin, while the low-risk group demonstrated greater sensitivity to Paclitaxel. This finding aligns with clinical practice at that time, suggesting increased prognosis for esophageal cancer patients in the low-risk group after Paclitaxel treatment. Notably, Oxaliplatin, Gemcitabine, and Camptothecin were also more sensitive in the high-risk group, indicating potential efficacy for patients choosing these drugs after Taxol resistance. For patients unable to tolerate chemotherapy, Crizotinib could be a consideration.
Currently, some scholars have also done PANoptosis for certain cancer types, including colon cancer [40], hepatocellular carcinoma [41], gastric cancer [42] and pancreatic cancer [43]. However, there is still a lack of research on the construction of predictive prognostic model for esophageal cancer and PANoptosis. To optimize this prognostic nomogram, we included relatively complete clinicopathologic and therapeutic information(such as histology, radiation therapy, alcohol consumption, etc.) for anylisis. What's more, 272 of 362 patients from two databases were esophageal squarmous cell carcinoma, indicating that the model might be more suitable for domestic situation. Nevertheless, our study has some limitations. Most analyses were based on data from public datasets, and all samples were obtained retrospectively, which may have caused an inherent case selection bias. In addition, limited molecular biology experiments were performed in the study, and further in vitro and in vivo experiments are needed to validate our findings. Finally, some valuable clinical features such as surgery methods, adjuvant chemotherapy regimes, and tumor markers were not totally available in database. As such, clinical cases are needed to confirm our conclusions.
In conclusion, we constructed a PANoptosis-based molecular clustering and prognostic signature that plays a vital role in predicting survival, TMB, and guiding clinical therapy. The findings of this study may improve our understanding of PANoptosis in esophageal cancer and help develop more effective treatment strategies. However, this study has some limitations, and additional experiments and clinical cases are needed to validate our findings.
Availability of data and materials
The data presented in the study are deposited in the TCGA EC project and GEO database (accession number: GSE53625)
References
Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209–49. https://doi.org/10.3322/caac.21660.
Kouo T, Huang L, Pucsek AB, et al. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res. 2015;3(4):412–23. https://doi.org/10.1158/2326-6066.Cir-14-0150.
Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–21. https://doi.org/10.1038/s41423-020-00630-3.
Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. https://doi.org/10.1038/s41418-017-0012-4.
Malireddi RKS, Tweedell RE, Kanneganti TD. PANoptosis components, regulation, and implications. Aging. 2020;12(12):11163–4. https://doi.org/10.18632/aging.103528.
Jiang M, Qi L, Li L, Wu Y, Song D, Li Y. Caspase-8: A key protein of cross-talk signal way in “PANoptosis” in cancer. Int J Cancer. 2021;149(7):1408–20. https://doi.org/10.1002/ijc.33698.
Miyata H, Yamasaki M, Kurokawa Y, et al. Prognostic value of an inflammation-based score in patients undergoing pre-operative chemotherapy followed by surgery for esophageal cancer. Exp Therapeutic Med. 2011;2(5):879–85. https://doi.org/10.3892/etm.2011.308.
Smyth EC, Lagergren J, Fitzgerald RC, et al. Oesophageal cancer. Nat Rev Dis (Primer). 2017;3:17048. https://doi.org/10.1038/nrdp.2017.48.
Wang Y, Lin K, Xu T, et al. Development and validation of prognostic model based on the analysis of autophagy-related genes in colon cancer. Aging. 2021;13(14):19028–47. https://doi.org/10.18632/aging.203352.
Zhao Z, Liu H, Zhou X, et al. Necroptosis-Related lncRNAs: Predicting Prognosis and the Distinction between the Cold and Hot Tumors in Gastric Cancer. J Oncol. 2021;2021:6718443. https://doi.org/10.1155/2021/6718443.
Song W, Ren J, Xiang R, Kong C, Fu T. Identification of pyroptosis-related subtypes, the development of a prognosis model, and characterization of tumor microenvironment infiltration in colorectal cancer. Oncoimmunology. 2021;10(1):1987636. https://doi.org/10.1080/2162402x.2021.1987636.
Nie J, Shan D, Li S, et al. A Novel Ferroptosis Related Gene Signature for Prognosis Prediction in Patients With Colon Cancer. Front Oncol. 2021;11:654076. https://doi.org/10.3389/fonc.2021.654076.
Pan H, Pan J, Li P, Gao J. Characterization of PANoptosis patterns predicts survival and immunotherapy response in gastric cancer. Clin Immunol (Orlando, Fla). 2022;238:109019. https://doi.org/10.1016/j.clim.2022.109019.
Orning P, Lien E. Multiple roles of caspase-8 in cell death, inflammation, and innate immunity. J Leukoc Biol. 2021;109(1):121–41. https://doi.org/10.1002/jlb.3mr0420-305r.
Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. 2021;597(7876):415–9. https://doi.org/10.1038/s41586-021-03875-8.
Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: Master Regulators of NLRP3 Inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis). Front Cell Infect Microbiol. 2019;9:406. https://doi.org/10.3389/fcimb.2019.00406.
Karki R, Sharma BR, Lee E, et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI insight. 2020;5(12):136720. https://doi.org/10.1172/jci.insight.136720.
Liu LX, Heng JH, Deng DX, et al. Sulconazole Induces PANoptosis by Triggering Oxidative Stress and Inhibiting Glycolysis to Increase Radiosensitivity in Esophageal Cancer. Mol Cell Proteomics. 2023;22(6):100551. https://doi.org/10.1016/j.mcpro.2023.100551.
Shah MA, Kennedy EB, Catenacci DV, et al. Treatment of Locally Advanced Esophageal Carcinoma: ASCO Guideline. J Clin Oncol. 2020;38(23):2677–94. https://doi.org/10.1200/jco.20.00866.
Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–22. https://doi.org/10.1038/ni.2703.
Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50. https://doi.org/10.1038/s41591-018-0014-x.
Greten FR, Grivennikov SI. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity. 2019;51(1):27–41. https://doi.org/10.1016/j.immuni.2019.06.025.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. https://doi.org/10.1016/j.cell.2010.01.025.
Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79(18):4557–66. https://doi.org/10.1158/0008-5472.Can-18-3962.
Fu T, Dai LJ, Wu SY, et al. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol. 2021;14(1):98. https://doi.org/10.1186/s13045-021-01103-4.
Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol. 2021;18(4):842–59. https://doi.org/10.1038/s41423-020-00565-9.
Loi S, Michiels S, Adams S, et al. The journey of tumor-infiltrating lymphocytes as a biomarker in breast cancer: clinical utility in an era of checkpoint inhibition. Ann Oncol. 2021;32(10):1236–44. https://doi.org/10.1016/j.annonc.2021.07.007.
Zhou Y, Wang Q, Chu L, et al. FOXM1c promotes oesophageal cancer metastasis by transcriptionally regulating IRF1 expression. Cell Proliferat. 2019;52(2):e12553. https://doi.org/10.1111/cpr.12553.
Wu Y, Zhou L, Zou Y, et al. Disrupting the phase separation of KAT8-IRF1 diminishes PD-L1 expression and promotes antitumor immunity. Nat Cancer. 2023;4(3):382–400. https://doi.org/10.1038/s43018-023-00522-1.
Li Y, Lv C, Yu Y, et al. KIR3DL3-HHLA2 and TMIGD2-HHLA2 pathways: The dual role of HHLA2 in immune responses and its potential therapeutic approach for cancer immunotherapy. J Adv Res. 2023;47:137–50. https://doi.org/10.1016/j.jare.2022.07.013.
Boulhen C, AitSsi S, Benthami H, et al. TMIGD2 as a potential therapeutic target in glioma patients. Front Immunol. 2023;14:1173518. https://doi.org/10.3389/fimmu.2023.1173518.
Mortezaee K. HHLA2 immune-regulatory roles in cancer. Biomed Pharmacother. 2023;162:114639. https://doi.org/10.1016/j.biopha.2023.114639.
Zhou X, Lu X, Wu H, Liu J, Huang H. Diagnostic performance of SHOX2 promoter methylation as biomarker for lung cancer identification: A meta-analysis update. Thoracic Cancer. 2021;12(24):3327–32. https://doi.org/10.1111/1759-7714.14206.
Teng Y, Loveless R, Benson EM, Sun L, Shull AY, Shay C. SHOX2 cooperates with STAT3 to promote breast cancer metastasis through the transcriptional activation of WASF3. J Exp Clin Cancer Res. 2021;40(1):274. https://doi.org/10.1186/s13046-021-02083-6.
Dai GP, Wang LP, Wen YQ, Ren XQ, Zuo SG. Identification of key genes for predicting colorectal cancer prognosis by integrated bioinformatics analysis. Oncol Letters. 2020;19(1):388–98. https://doi.org/10.3892/ol.2019.11068.
Wang Y, Chen D, Liu Y, et al. Multidirectional characterization of cellular composition and spatial architecture in human multiple primary lung cancers. Cell Death Dis. 2023;14(7):462. https://doi.org/10.1038/s41419-023-05992-w.
Wang C, Li Y, Jia L, et al. CD276 expression enables squamous cell carcinoma stem cells to evade immune surveillance. Cell Stem Cell. 2021;28(9):1597-1613.e7. https://doi.org/10.1016/j.stem.2021.04.011.
Zhang H, Brown RL, Wei Y, et al. CD44 splice isoform switching determines breast cancer stem cell state. Genes Dev. 2019;33(3–4):166–79. https://doi.org/10.1101/gad.319889.118.
Jiang P, Gu S, Pan D, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24(10):1550–8. https://doi.org/10.1038/s41591-018-0136-1.
Zhu JF, Huang Q, Peng XY, et al. Identification of molecular subtypes based on PANoptosis related genes and construction of a signature for predicting the prognosis and response to immunotherapy response in hepatocellular carcinoma. Front Immunol. 2023;14:1218661. https://doi.org/10.3389/fimmu.2023.1218661.
Wang X, Sun R, Chan SX, et al. PANoptosis-based molecular clustering and prognostic signature predicts patient survival and immune landscape in colon cancer. Front Gene. 2022;13:955355. https://doi.org/10.3389/fgene.2022.955355.
Pan HD, Pan JX, Li P, et al. Characterization of PANoptosis patterns predictssurvival and immunotherapy response in gastric cancer. Clin Immunol. 2022;238:109019. https://doi.org/10.1016/j.clim.2022.109019.
Zhang B, Huang BQ, Zhang XN, et al. PANoptosis-related molecular subtype and prognostic model associated with the immune microenvironment and individualized therapy in pancreatic cancer. Front Oncol. 2023;13:1217654. https://doi.org/10.3389/fonc.2023.1217654.
Acknowledgments
We acknowledged TCGA and GEO database for providing their platform and contributors for uploading their meaningful datasets.
Funding
No funding was recieved.
Author information
Authors and Affiliations
Contributions
Chi Zhang, Jing Wang and Wei Li conceived the work. Chi Zhang and Fuben Liao performed the literature review. Data extraction was carried out by Fuben Liao, Jiahui Liu, Zheming Liu, and Chi Zhang, Jing Wang and Wei Li verified the data. Zheming Liu was responsible for data analysis and manuscript writing. Jiahui Liu provided input on data analysis. The final manuscript draft received approval from all authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent for publication was obtained from all participants.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
44272_2024_21_MOESM2_ESM.jpg
Additional file 2: Figure S2. The AUC values of riskcore and other clinical features, in TCGA training set (a, b), internal TCGA validation set (c, d), the external GEO validation set (e, f).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
Liu, Z., Liu, J., Liao, F. et al. PANoptosis-based molecular subtype and prognostic model predict survival and immune landscape in esophageal cancer. CCB 3, 16 (2024). https://doi.org/10.1007/s44272-024-00021-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44272-024-00021-z