
Abstract
Multicomponent phase space contains extended regions of random or near-random multicomponent solid-solution single phases, stabilised by a relatively large configurational entropy of mixing that can often (though not always) suppress compound formation between the different atomic species. The present paper shows that there are very extensive variations of local nanostructure, local atomic clusters and associated local lattice strains within multicomponent high-entropy solid-solution single phases such as the fcc Cantor alloys, bcc Senkov alloys and rock-salt-structured Rost mono-oxides, even when there is no short-range ordering, i.e. even when the solid solution is completely random or ideal. There are, for instance, many billions of different local nanostructures and different local atomic clusters in equiatomic five-component fully random solid-solution single-phase materials such as the original fcc Cantor alloy CrMnFeCoNi and the original bcc Senkov alloy VNbMoTaW, extending over distances of many microns, with associated fluctuating hydrostatic and shear lattice strains of several percent. The number and extent of the variations in local nanostructure, atomic clusters and lattice strains increase dramatically to even higher values with increasing number of components in the material. The present paper also shows that there are similar variations in local nanostructure, local atomic clusters and associated local lattice strains surrounding point defects such as vacancies, line defects such as dislocations and planar defects such as grain boundaries and external surfaces, influencing many important material properties such as diffusion, plastic flow, recrystallisation, grain growth and catalysis. The number and extent of the variations in local nanostructure, atomic clusters and lattice strains make it difficult to have too much confidence in structures and properties of multicomponent high-entropy materials calculated using ab initio and other atomistic computer modelling techniques, since these techniques are restricted to relatively small numbers of atoms and are unable to sample effectively the full range of local structures and properties.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MulticomponentFootnote 1 phase space is characterised by the formation of extended regions of random or near-random multicomponent solid-solution single phases based on well-known binary or ternary crystal structures [1,2,3]. These extended single-phase regions are stabilised in multicomponent phase space because of the relatively large configurational entropy of mixing in a random or near-random multicomponent solid solution, that is often (though not always) large enough to suppress the compound-forming tendency of the interaction energies of mixing between the various component atomic species [1,2,3,4,5]. Unlike single-phase pure materials, the local nanostructure (i.e. the local atomic clustersFootnote 2) and, therefore, the local properties of random or near-random multicomponent solid-solution single phases vary widely from point to point, partly (and fairly obviously) because of the multiplicity of different atomic species at the different lattice points in the crystal, but also (and perhaps somewhat less obviously) because of the even greater multiplicity of local atomic clusters caused by a variety of different near neighbours surrounding different atoms of the same type [1, 3].
The present paper explores the details of the variation of local nanostructure, the variety of local atomic clusters, and the resulting variation of local properties within multicomponent fully random (or ideal) solid-solution single phases. As we will show, these variations are much more extensive and much more significant than has generally been realised in the previous literature [1,2,3], and this is the case even when there is no short-range order in the distribution of the component atoms across the different lattice points in the crystal, i.e. even when the solid solution is completely random or ideal. As well as considering the general point-to-point variation of local nanostructure and local properties in multicomponent fully random solid-solution single phases, this paper also considers the substantial impact of such variations on the structure and behaviour of point, line and planar defects within multicomponent materials (mainly vacancies and dislocations, but also, more briefly, grain boundaries and external surfaces), and the resulting, similarly substantial effects on overall defect-controlled material properties (mainly diffusion and slip, but also, more briefly, recrystallisation, grain growth and catalysis). This paper also refers briefly to the way in which such extensive variations in local nanostructure and local properties make it difficult, if not almost impossible, to use ab initio and other atomistic computer modelling techniques to calculate with any degree of confidence the overall structure and, therefore, many of the properties of multicomponent high-entropy solid-solution single-phase materials.
Multicomponent Solid-Solution Single Phases
Multicomponent Crystal Structures
The first multicomponent high-entropy solid-solution single phase that was discovered was the original single-phase fcc Cantor alloy CrMnFeCoNi,Footnote 3 found during the course of the very first study that launched the field of multicomponent and high-entropy materials [1,2,3,4]. It has been shown many times since then that there are very many different compositions of single-phase fcc Cantor alloy, occupying a large region in multicomponent phase space, made up of a wide range of mixtures of mostly late transition metals [1,2,3,4,5].
Similar large multicomponent high-entropy solid-solution single-phase regions have also been discovered elsewhere in multicomponent phase space for many other metallic and ceramic crystal structures. To give one or two detailed examples, these include for instance: a large single-phase bcc region based on the original Senkov alloy VNbMoTaW, made up of a wide range of mixtures of mostly early transition metals [1, 2, 1,2,6,7,8]; a large single-phase B2 ordered-bcc mono-aluminide region, with the generic formula MAl where M is a mixture of different, mostly transition metals, that was first made by adding substantial amounts of Al to a variety of fcc Cantor and bcc Senkov alloys [1, 2, 4, 5, 1,2,4,5,7,8,9]; and a large single-phase rock-salt-structured mono-oxide region, with the generic formula MO where M is a mixture of different, mostly divalent metal cations, based on the original, first-discovered multicomponent ceramic compound [1, 10], the Rost oxide (MgCoNiCuZn)O. Many other examples of large multicomponent high-entropy solid-solution single-phase regions have also been discovered and have been described and discussed in the literature, including the following: hcp metallic alloys based on mixtures of mostly rare earth metals [1, 2, 11, 12]; intermetallic compounds such as trialuminides [1, 2, 13, 14], Laves phases [1, 2, 15, 16] and topologically close-packed phases [1, 2]; ceramic compounds such as borides, nitrides, perovskites and spinels [1, 2, 1,2,17,18,19] and ionic compounds such as alkali halides [1, 2, 1,2,17,18,19. In this paper, we will concentrate our consideration of the variation of local nanostructure and local properties mainly on the cases of multicomponent high-entropy single-phase fcc Cantor alloys, bcc Senkov alloys, and to a lesser extent, rock-salt-structured Rost mono-oxides, but many of our considerations and conclusions are applicable more widely to all other cases of multicomponent and high-entropy solid-solution single-phase crystal structures.
Number of Components
Early examples of multicomponent high-entropy solid-solution single phases were often based on mixtures of up to five different components [1, 2], including the original single-phase fcc Cantor alloy [3, 4] CrMnFeCoNi, the original single-phase bcc Senkov alloy [6,7,8,9] VNbMoTaW and the original Rost oxide [10, 10,17,18,19] (MgCoNiCuZn)O. It is important to notice that multicomponent random or near-random solid-solution single phases have also been manufactured with many more components, up to as many as 10 or 12 [20, 21]. In this paper, we will, therefore, consider the variations in local nanostructure and properties in multicomponent high-entropy solid-solution single phases with up to 10 or 12 different components. Slightly pre-empting some of the results described later in the paper, we will find, not surprisingly, that the extent of the variation of local nanostructure and local properties increases dramatically with increasing number of components. The range and variety of local nanostructures and local atomic clusters are very substantial indeed even with as few as just four or five components, and become truly enormous when we increase the number of components to as many as 10 or 12.
Thermodynamic Stability
In any single-phase solid solution (including, therefore, all multicomponent high-entropy solid solutions), the molar Gibbs free energy G is given at any temperature T by [1, 3]:
where \({x}_{i}\) is the molar fraction of the i’th component, \({\mu }_{i}\) and \({\mu }_{i}^{ss}\) are, respectively, the chemical potential (or molar free energy) of the i’th component before and after mixing to form the solid solution, and \(\Delta {G}_{mix}\), \(\Delta {H}_{mix}\) and \(\Delta {S}_{mix}\) are, respectively, the molar free energy of mixing, molar heat (or enthalpy) of mixing and molar entropy of mixing. The first term on the right-hand side of Eq. (1) is the free energy of the unmixed components, and the last two terms are the free energy change caused by mixing them to form the single-phase solid solution.
In general, the heat of mixing is not zero and depends on pairwise, three-way, four-way, etc. interaction energies \({\omega }_{ij}\), \({\psi }_{ijk}\), \({\chi }_{ijkl}\), etc. between the different atomic species [1, 3], i.e. \(\Delta {H}_{mix}=f\left({\omega }_{ij},{\psi }_{ijk},{\chi }_{ijkl}\dots \right)\ne 0\). The heat of mixing can be either positive or negative, representing an overall tendency for the solid solution either to separate into pure components (with a positive heat of mixing) or to form interatomic compounds (with a negative heat of mixing). The entropy of mixing is a combination of two terms [1, 3]: the ideal entropy of mixing, associated with a completely random distribution of the different atoms on the lattice points of the solid-solution crystal structure,Footnote 4 given by Boltzmann’s equation as \({\Delta S}_{mix}^{ideal}=-R{\sum }_{i}{x}_{i}\text{ln }{x}_{i}\); and the excess entropy of mixing \(\Delta {S}_{mix}^{xs}={f}{^\prime}\left({\omega }_{ij},{\psi }_{ijk},{\chi }_{ijkl}\dots \right)\), associated with any non-randomness in the distribution of atoms on the lattice points of the solid-solution crystal structure, as well as any changes caused by mixing the different component atoms in their vibrational, electronic or magnetic entropy. Overall, therefore [1, 3]:
According to the third law of thermodynamics, all materials are a mixture of phase-separated perfect single crystals when they are at equilibrium at absolute zero temperature. This can be seen easily in Eq. (1), since the entropy term on the right-hand side goes to zero at absolute zero, so that the minimum Gibbs free energy is given by a macroscopic mixture of pure components when the heat of mixing is positive, and a macroscopic mixture of pure components and/or compounds when the heat of mixing is negative. On the other hand, all materials form a random solid solution when heated to a sufficiently high temperature, as long as they do not melt first. This can also be seen easily in Eq. (1), since the configurational entropy term on the right-hand side becomes dominant at sufficiently high temperatures, suppressing any compound-forming tendency of the interaction energies between the different atomic species (and also suppressing, therefore, a corresponding overall negative heat of mixing), so that the minimum Gibbs free energy is now given by a fully mixed solid solution.
In other words, multicomponent and high-entropy random solid-solution single phases can only be thermodynamically stable and at true equilibrium at elevated temperatures. On cooling below a critical demixing temperature, there must develop a driving force for phase separation by a combination of precipitation, spinodal decomposition and/or ordering and compound formation. The demixing temperature is often, however, low enough to prevent atomic diffusion and suppress demixing. In many cases, therefore, equilibrium high-temperature random solid-solution single phases can continue to be stabilised kinetically even when cooled down to lower temperatures, such as at room temperature.
In fact, several multicomponent high-entropy solid-solution single phases have been shown to be completely stable kinetically at low temperatures, to decompose by precipitation and/or spinodal decomposition when held for extended periods of time at intermediate temperatures, and to be completely thermodynamically stable at high temperatures (but still below the melting point) [1]. For instance, the as-cast and recrystallised single-phase fcc solid solution in the original Cantor alloy CrMnFeCoNi is found experimentally [22, 23] (and in reasonable agreement with thermodynamic calculations [24]) to be kinetically stable below an annealing temperature of approximately 450 °C, to be thermodynamically stable above an annealing temperature of approximately 800 °C, and to exhibit demixing and precipitation only after several years of heat treatment at intermediate annealing temperatures between approximately 450 and 700 °C. Similarly, the original Rost oxide (MgCoNiCuZn)O is thermodynamically stable [10] above a temperature of approximately 800 °C, exhibits phase separation at lower temperatures [10] and can be cycled reversibly between the two structures [10] by heating and cooling above and below 800 °C.
Short-Range Order
Close to but just above the demixing temperature, solid solutions are not, of course, expected to be completely random or ideal, and instead are expected to exhibit short-range ordering [25,26,27]. This is because the configurational entropy at intermediate temperatures just above the critical demixing temperature is sufficient to prevent demixing into separate thermodynamically stable bulk phases, but not yet sufficient to prevent local non-randomness in the distribution of the component atoms on the lattice points of the solid-solution crystal structure.
Indeed short-range order has been seen experimentally in a number of multicomponent solid-solution single phases, with the resulting short-range atomic clusters that form much discussed in the literature [28,29,30,31,32]. This paper will not discuss short-range ordering effects at all, and will only consider the local nanostructure and local atomic clusters that are formed in completely random multicomponent high-entropy solid solutions. In other words, the local nanostructures and atomic clusters that we discuss are characteristic of fully random solid solutions either in an equilibrium multicomponent material at a high temperature or in a kinetically constrained multicomponent material at a low temperature, in which short-range ordering has been suppressed by quenching sufficiently rapidly through the relatively limited intermediate range of temperatures where significant short-range ordering can take place.
It is worth noticing that the single-phase solid-solution structures in the original fcc Cantor alloy CrMnFeNiCo and the original rock-salt-structured Rost mono-oxide (MgCoNiCuZn)O have both been shown by detailed atom-probe measurements [33] and high-resolution energy-dispersive X-ray maps [10], respectively, to exhibit in almost all cases an ideal random distribution of the multicomponent atoms or cations (respectively) on the underlying crystal lattice or cation sublattice (respectively), with undetectable levels of non-randomness [10, 33]. An example of random atomic distributions obtained from atom-probe measurements by Laurent-Brocq et al. [33] for the homogenised and recrystallised original Cantor alloy CrMnFeCoNi is shown in Fig. 1. Similar results have been obtained in several other (though not, of course, in all) multicomponent single-phase solid solutions [10, 34, 35]. Slightly pre-empting again some of the results described later in the paper, we will find that the variety and extent of local atomic clusters in such multicomponent fully random solid solutions are very large, much greater than has generally been recognised in the previous literature. In fact, the previously rather unremarked effects of random atomic clustering in the local nanostructures of single-phase solid-solution materials are almost certainly in many cases more widespread and more significant than the much more extensively discussed effects of atomic clustering caused by short-range ordering.

Atom-probe (AP) measurements from the homogenised and recrystallised single-phase fcc Cantor alloy CrMnFeCoNi, showing a random atomic-scale distribution of all five components: (a) composition maps (each point is a single atom); (b) composition profiles for Mn and Fe; and (c) statistically random composition distributions for Mn and Fe (after Laurent-Brocq et al. [33])
Local Nanostructure
Local Atomic Clusters
Figure 2 shows a schematic random distribution of 10 equiatomic components A–J on a two-dimensional square lattice, demonstrating in very simplified form two important features of multicomponent fully random single-phase solid-solution materials such as the fcc Cantor alloys, bcc Senkov alloys and other close-packed metallic alloys:
-
1.
There is a very large number of different local atomic environments or local atomic clusters with, for example, different A atoms surrounded by a wide range of different distributions of the other A, B, C, D, E, F, G, H, I and J atoms, and similar different distributions around all the other nine types of atom;
-
2.
There are significant local atomic distortions and lattice strains, caused by the different sizes and electronic structures of the different atoms and their varied local atomic environments.

Schematic representation of 10 components distributed randomly on a square lattice
We will deal with the first of these effects, namely the variation of local atomic clusters, in the present section of this paper; and the second of these effects, namely the variation of local lattice strains, in the next section. The same two effects are, of course, also present in the rock-salt-structured Rost mono-oxides and all the other kinds of multicomponent high-entropy single-phase intermetallic and ceramic compounds, though in a slightly modified form, in the sense that the different local atomic structures and local lattice strains are caused by a variety of different atoms distributed on one or more, but not necessarily all the sub-lattices in the compound crystal structure.
It is rather obvious that multicomponent high-entropy solid-solution single-phase materials, such as, for instance, the equiatomic five-component fcc Cantor alloy CrMnFeCoNi, the equiatomic five-component bcc Senkov alloy VNbMoTaW or the equiatomic 10-component schematic model in Fig. 2, are different from corresponding single-component single-phase pure materials such as, for instance, pure fcc Cu or pure bcc Fe (or, indeed pure diamond cubic Si or pure quartz silica SiO2) for the very simple reason that there are 5 or 10 (in the particular cases of the original Cantor and Senkov alloys and Fig. 2) different atoms with 5 or 10 (in these particular cases) different properties distributed in a random or near-random way across the different lattice points, rather than the same atom (or molecule) with identical properties at every lattice point. But the variation in local structure and properties in multicomponent high-entropy single-phase materials is much more extensive and complex than implied just by noticing that there are 5 or 10 (or however many) rather than one different kind of atom and, therefore, 5 or 10 (or however many) rather than one different kind of lattice point. This is because the local structure and properties at any lattice point depend not just on which particular atom is located at that particular lattice point, but also on its interactions with its neighbouring atoms at adjacent and nearby lattice points. In other words, the variation in properties from lattice point to lattice point depends on the range of local atomic environments, i.e. on the range of different local atomic clusters consisting of each atom at that lattice point together with its surrounding near neighbours.
The Number of Local Atomic Clusters
Consider a general lattice with a single atom at each lattice point, and with \({n}_{1}\), \({n}_{2}\) and \({n}_{3}\) first, second and third near neighbours respectively.Footnote 5 The cluster size of each atom together with its first near neighbours is \({n}_{1}+1\) atoms, increasing to \({n}_{2}+{n}_{1}+1\) atoms including second as well as first near neighbours, and to \({n}_{3}+{n}_{2}+{n}_{1}+1\) atoms including third as well as first and second near neighbours. The numbers of different clusters \({N}_{1}\) of \({n}_{1}+1\) atoms, \({N}_{2}\) of \({n}_{2}+{n}_{1}+1\) atoms and \({N}_{3}\) of \({n}_{3}+{n}_{2}+{n}_{1}+1\) atoms in a multicomponent high-entropy equiatomic single-phase solid-solution material with a random (ideal or regular) arrangement of c components are given [36] by the law of permutations with repetitionFootnote 6:
These are orientation-dependent cluster numbers (i.e. equivalent clusters with a different orientation are regarded as different), as is appropriate when we are dealing with properties that depend on orientation, such as dislocation slip, diffusion or magnetisation, when the material is responding to an oriented vector of applied stress, concentration gradient or magnetic field, respectively. If cluster orientation is not significant, however, the number of different clusters is smaller because of the number of self-similarity operations s associated with the crystal symmetry:
with s = 24 for instance for a cubic crystal [37, 38]. These are orientation-independent cluster numbers (i.e. equivalent clusters with a different orientation are regarded as the same), as is appropriate when we are dealing with properties that are independent of orientation, such as density, bulk modulus or chemical potential.
Table 1 shows the numbers of oriented and non-oriented local atomic clusters \({N}_{1}\), \({N}_{2}\) and \({N}_{3}\) out to first, second and third near neighbours, respectively, for multicomponent equiatomic single-phase fcc Cantor alloys with c components, where c is in the range 3–50, taking for fcc \({n}_{1}=12\), \({n}_{2}=6\) and \({n}_{3}=24\), so the cluster sizes are 13, 19 and 43 atoms, respectively. Table 2 shows equivalent numbers for multicomponent equiatomic single-phase bcc Senkov alloys, taking for bcc \({n}_{1}=8\), \({n}_{2}=6\) and \({n}_{3}=12\), so the cluster sizes are 9, 15 and 27 atoms, respectively. The same results are shown graphically in Figs 3 and 4 for fcc and bcc, respectively. It is obvious from Tables 1 and 2 and Figs 3 and 4that the numbers of different local atomic clusters in multicomponent solid-solution single-phase materials are very large indeed.

Number of different oriented (upper lines) and non-oriented (lower lines) local atomic clusters out to first (\({N}_{1}\)), second (\({N}_{2}\)) and third (\({N}_{3}\)) near neighbours for a multicomponent single-phase fcc material with c components

Number of different oriented (upper lines) and non-oriented (lower lines) local atomic clusters out to first (\({N}_{1}\)), second (\({N}_{2}\)) and third (\({N}_{3}\)) near neighbours for a multicomponent single-phase bcc material with c components
Most (though not all) of the important properties of a material are determined by interactions between first and second near-neighbour atoms, and many (though not all) are orientation-dependent. Concentrating, therefore, on the number of oriented first and second near-neighbour local atomic clusters, there are just under twenty trillion (1.9 × 1013) different clusters in the original single-phase fcc Cantor alloy CrMnFeCoNi, and just over thirty billion (3.1 × 1010) different clusters in the original single-phase bcc Senkov alloy VNbMoTaW, as shown in Tables 1 and 2. With non-oriented clusters and first near neighbours only, the numbers of local atomic clusters are, of course, somewhat smaller but still very large, almost fifty million (4.8 × 107) for the original fcc Cantor alloy and eighty thousand (8.0 × 104) for the original bcc Senkov alloy. With six components, the number of oriented first and second near-neighbour local atomic clusters increases to just over six hundred trillion (6.1 × 1014) for fcc Cantor alloys and almost half a trillion (4.7 × 1011) for bcc Senkov alloys; with eight components to well over a quadrillion (1.4 × 1017) for fcc Cantor alloys and over thirty trillion (3.5 × 1013) for bcc Senkov alloys; and with 10 components to well over a quintillion (1019) for fcc Cantor alloys and a quadrillion (1015) for bcc Senkov alloys. The number of local atomic clusters is, of course, very much larger again, and indeed becomes astronomic, if we include third near neighbours (> 1030 or > 1018 for the five-component Cantor and Senkov alloys, respectively) and/or increase the number of components to even more than 10 (> 1019 or > 1015 for Cantor and Senkov alloys, respectively), as shown in Tables 1 and 2 and Figs 3 and 4.
The Extent of Atomic Clustering
The number of local atomic clusters is in all these cases, by any stretch of the imagination, very large indeed, and this has an important effect on the spatial consistency of the properties of multicomponent high-entropy single-phase solid solutions such as the fcc Cantor and bcc Senkov alloys. Taking the cube root of the number of local atomic clusters gives the size of a piece of the material sufficiently large to include all possible local atomic configurations, i.e. a piece of the material big enough to average reasonably over all the variations of local nanostructure and the range of different local atomic structures and, therefore, big enough to represent fully the material and its properties. The linear dimension of such a piece of material is ∛\(\overline{1.9\times {10}^{13}}=2.7\times {10}^{4}\) clusters or ~ 8 \(\mu\) m for oriented, first and second near-neighbour clusters in the original single-phase fcc Cantor alloy, and ∛\(\overline{3.1\times {10}^{10}}=3.1\times {10}^{3}\) clusters or ~ 1 \(\mu\) m in the original single-phase bcc Senkov alloy, in both cases taking the atomic (and, therefore, cluster) separation very roughly as 0.3 nm. In other words, the properties of the original fcc Cantor and bcc Senkov alloys will vary to some extent from grain to grain or from particle to particle if their grain size (in a polycrystalline bulk material) or particle size (in an as-deposited or mechanically alloyed particulate material) is below ~ 8 \(\mu\) m or ~ 1 \(\mu\) m, respectively. To put it another way, the grain size or particle size needs to be above ~ 8 \(\mu\) m or ~ 1 \(\mu\) m, respectively, to have a material with fully consistent properties. To put it yet another way, we can think of individual grains or particles smaller than ~ 8 \(\mu\) m or ~ 1 \(\mu\) m, respectively, as being somewhat like different large macromolecules of the material. For equiatomic 10-component single-phase fcc Cantor or bcc Senkov alloys, the grain size or particle size needs to be even bigger, above ∛\(\overline{{10}^{19}}=2.2\times {10}^{6}\) clusters ≈ 0.7 mm or ∛\(\overline{{10}^{15}}={10}^{5}\) clusters ≈ 30 \(\mu\) m, respectively, to have fully consistent properties. And, if third near neighbours are important, the grain size or particle size needs to be even bigger again, above ∛\(\overline{1.1\times {10}^{30}}={10}^{10}\) clusters ≈ 3 m or ∛\(\overline{7.5\times {10}^{18}}=2\times {10}^{6}\) clusters ≈ 0.6 mm, for the original equiatomic five-component single-phase fcc Cantor or bcc Senkov alloy, respectively, to have fully consistent properties.
This is clearly a very different situation from that which is found in conventional materials consisting of either a single component or a single main component with one or more dilute alloying additions. The extremely wide variation in local nanostructure and the extremely large number of different local atomic environments and local atomic clusters in multicomponent high-entropy single-phase fcc and bcc solid solutions such as the Cantor and Senkov alloys (and indeed in other crystal structures such as multicomponent high-entropy intermetallic and ceramic compounds) play, therefore, an important role in material properties that depend strongly on local atomic interactions, such as vacancy migration and diffusion, dislocation slip and plastic flow, grain-boundary motion and recrystallisation, or surface structure and catalysis, as discussed in more detail in later sections of this paper. The extremely wide variation in local nanostructure and the extremely large number of different local atomic environments and atomic clusters in these materials also make it, as mentioned previously, extremely challenging if not in many cases near impossible to determine their structure and properties with any degree of confidence by fundamental techniques such as ab initio molecular dynamics or quantum mechanical modelling that are typically limited to no more than one or two thousand atoms at best [39, 40].
Individual Atomic Clusters
There is clearly a very varied set of different local nanostructures and local atomic clusters, with the variations extending over very large distances and volumes, within the structure of multicomponent high-entropy solid-solution single-phase materials such as the fcc Cantor alloys, bcc Senkov alloys and rock-salt-structured Rost mono-oxides, even when the distribution of atomic or cationic species is completely random or ideal. It is interesting to consider the probability of finding particular types of cluster within these fully random solid-solution single-phase materials. For instance, consider the oriented first and second near-neighbour clusters in the original equiatomic fcc Cantor alloy CrMnFeCoNi. As discussed above, each cluster consists of \({N}_{2}={n}_{2}+{n}_{1}+1=19\) atoms, there are just under twenty trillion different clusters (1.9 × 1013), and an individual grain or particle of the material containing all possible local atomic clusters is ~ 8 \(\mu\) m in size. Let us consider how many clusters there are consisting of large numbers of the same atom species. Any grain or particle just ~ 8 \(\mu\) m in size contains all the possible 1.9 × 1013 different local atomic clusters. This means that there are 5 clusters containing 19 atoms of the same type, since there is one 19-atom cluster consisting of all Cr atoms, one consisting of all Mn atoms, one consisting of all Fe atoms, one consisting of all Co atoms and one consisting of all Ni atoms. And there are 5 × 4 × 19 = 380 clusters containing 18 atoms of the same type, since there are 4 × 19 = 76 clusters of 19 atoms containing just 18 Cr atoms, and the same number each containing just 18 Mn, 18 Fe, 18 Co or 18 Ni atoms.Footnote 7 Similarly, there are 5 × 4 × 4 × 19 × 18 = 27,360 clusters containing 17 atoms of the same type, since there are 4 × 4 × 19 × 18 = 5,472 clusters of 19 atoms containing just 17 Cr atoms, and the same number each containing just 17 Mn, 17 Fe, 17 Co or 17 Ni atoms.Footnote 8 Similarly there are increasingly large numbers of clusters containing 16 atoms, 15 atoms, etc. of the same type. The numbers of such clusters are, of course, tiny compared to the total number of almost twenty trillion different clusters overall, but are, nevertheless, surprisingly large. In fact, the somewhat surprisingly large numbers of clusters containing many atoms of the same type are an example of the well-known general effect that random arrangements contain a somewhat surprising amount of local order, since a random arrangement is not, as is commonly (mis-)perceived, the same as (and is indeed considerably different from) an evenly distributed arrangement.
Lattice Strain
Lattice Distortions in Pure Materials
At absolute zero, all the atoms or molecules in a single-component single-phase material are located exactly on their respective crystal lattice points.Footnote 9 The lattice is then undistorted. With an undistorted lattice at absolute zero, the variation of diffracted intensity with diffraction angle \(I\left(\theta \right)\) for X-rays, electrons or neutrons consists in each case of a series of perfectly sharp lines at specific Bragg angles \({\theta }_{hkl}\) corresponding to specific diffracting crystal planes {hkl} that are characteristic of the particular crystal structure of the material, as shown schematically for an fcc crystal in Fig. 5. And the radial distribution function or pair-distribution function (RDF or PDF) \(\rho \left(r\right)\), derived by Fourier transformation of the intensity function \(I\left(\theta \right)\), consists similarly of a set of perfectly sharp lines at specific distances \({r}_{n}\) where n = 1, 2, 3, etc. corresponding to the 1st, 2nd, 3rd, etc. near-neighbour shells that are again characteristic of the particular crystal structure of the material, as shown schematically again for an fcc crystal in Fig. 6.

Schematic variation of diffracted intensity I versus diffraction angle \(\theta\), showing the {111}, {200} and {311} diffraction lines in an fcc crystal at absolute zero and at a realistic temperature T

Schematic radial distribution function \(\rho \left(r\right)\) for the probability variation of finding two atoms at a separation r, showing the 1st, 2nd and 3rd near-neighbour shells in an fcc crystal at absolute zero and at a realistic temperature T
A crystal lattice becomes distorted when one or more atoms is displaced from its lattice point. At realistic temperatures above absolute zero, the atoms in a single-component single-phase material have thermal energy and vibrate about their crystal lattice positions, so the lattice expands, the characteristic diffraction angles and near-neighbour distances increase slightly, and the diffraction lines and RDF peaks broaden approximately symmetrically, as the atoms spend time at a variety of different positions around their lattice points, as also shown in Figs. 5 and 6. The atoms are still located on average exactly on their respective crystal lattice points, but their positions deviate stochastically with time as they oscillate elastically about their lattice points. In other words, because of thermal vibrations at realistic temperatures, a snapshot of the atomic structure of the material at any particular time shows a stochastically distorted lattice, with most of the atoms displaced from their lattice points, but the time-averaged structure remains virtually identical to the undistorted lattice.Footnote 10 At the same time, thermally induced crystal defects such as vacancies and dislocations are also formed at realistic temperatures above absolute zero, creating a small number of local regions where the lattice is permanently distorted, with atoms in the near vicinity of the defects deviating permanently from their respective lattice points. Permanent lattice distortions caused by atoms in the vicinity of crystal defects contribute to a further, usually small and, in this case, usually asymmetric broadening of the diffraction lines and the corresponding RDF peaks and near-neighbour shells shown in Figs. 5 and 6.
Lattice Distortions in Dilute Solid Solutions
Addition of solute atoms to a single-component single-phase material provides additional sources of lattice distortion, because the size and electronic structure of the solute atoms are different to those of the solvent atoms. For dilute solutions, the solute atoms are widely separated and are effectively independent and non-interacting. There are then two basic effects: first, the lattice as a whole expands or contracts; and second, each solute atom provides an additional local region of permanent lattice distortion. The first effect, the lattice expansion or contraction, is often linear, and dilute binary solutions usually obey Vegard’s law, with the lattice constant a (or constants a, b and c) proportional to the solute concentration x:
where k is the Vegard’s law constant [41]. And in multicomponent dilute solid solutions with two or more different solutes, the lattice expansion or contraction is usually linearly additive [41]:
where \({k}_{i}\) and \({x}_{i}\) are, respectively, the Vegard’s law constant and the concentration of the i’th solute.
Lattice Distortions in Multicomponent Solid Solutions
Lattice distortions are more complex in concentrated multicomponent high-entropy single-phase solid-solution materials, because the different solute atoms interact with each other, often producing non-linear effects. The overall expansion or contraction of the crystal lattice and the variation of lattice parameter with the concentration of the different components do not obey Vegard’s law so well. Alonso and Alonso [42] have compiled data for a total of 79 different cases of lattice parameter measurements in concentrated multicomponent single-phase fcc and bcc alloys, showing positive and negative deviations from Vegard’s law by up to 4–5%, with an average deviation of ± 0.64%. The original fcc Cantor alloy CrMnFeCoNi and the original bcc Senkov alloy VNbMoTaW deviate relatively little from Vegard’s law, and deviations are generally somewhat smaller in multicomponent alloys restricted to transition metals only, compared with those that contain a mixture of transition and main-group elements.
As well as not obeying Vegard’s law, local lattice distortions around individual solute atoms are also more complex in concentrated multicomponent high-entropy single-phase materials for another reason, because every atom acts effectively as a centre of local lattice distortion, caused by the wide variety of local atomic configurations as discussed in the last section. Every atom i is effectively displaced from its lattice point, by an atom displacement vector \({d}_{i}=\left({d}_{ix},{d}_{iy},{d}_{iz}\right)\), where \({d}_{ix}\), \({d}_{iy}\) and \({d}_{iz}\) are its displacements in the three directions x, y and z; and the sum total of all the atomic displacements causes each atom to be a centre of local stress and strain, as given by local atom stress and strain tensors \({\sigma }_{i}\) and \({\epsilon }_{i}\):
where the individual stress and strain tensor components \({\sigma }_{iab}\) and \({\epsilon }_{iab}\) are individual local stresses and strains on the face of the ith atom normal to axis a = x, y or z in the direction of axis b = x, y or z. There are different stress and strain tensors for each atom i, so concentrated multicomponent high-entropy solid-solution single-phase materials support a complex and very extensive internal spatial distribution of hydrostatic and shear stresses and strains, fluctuating throughout the material from zero at some atom positions undisplaced from their lattice points up to maximum positive values and down to minimum negative values at other atom positions displaced substantially from their lattice points.
Lattice Distortions in Cantor Alloys
Table 3 shows 12-coordinated metallic Goldschmidt atomic radii r for some of the components that have been used to manufacture multicomponent single-phase fcc Cantor alloys [43]. The maximum difference in atomic size \(\Delta r\) amongst the five different components, the corresponding maximum atomic misfit \({\delta }_{max}\) and the root-mean-square (RMS) average atomic misfit \({\delta }_{av}\) in the original Cantor alloy CrMnFeCoNi are as follows:
where summation is over all the components i = 1 to n, \({x}_{i}\) is the mole fraction of the i’th component (\({x}_{i}=0.2\) for all five components in the original Cantor alloy), n is the number of components (n = 5 for the original Cantor alloy) and \({r}_{av}={\sum }_{i}{x}_{i}{r}_{i}\). The values of \(\Delta r\), \({\delta }_{max}\) and \({\delta }_{av}\) remain unchanged when Cu is included in a modified CrMnFeCoNiCux Cantor alloy; but they are somewhat larger when Al instead of Cu is included in a modified AlxCrMnFeCoNi Cantor alloy:
for the maximum Al solubility with \({x}_{\text{Al}}\) = 8% and \({x}_{i\ne {\text{Al}}}\) = 18.4%
We might expect, therefore, to find lattice distortions on a scale of somewhat under approximately 1 pm or 1% on average, rising to a maximum of about 3 pm or 3% in the original Cantor alloy, and quite a bit higher in some of the modified Cantor alloys such as those containing Al. Clearly these calculations are rather rough and simplistic, since they ignore effects such as short-range order, non-ideality, relaxation, and electronic distortions. More detailed analyses of the expected lattice distortion have taken account of measured lattice parameters of the fcc Cantor alloys (instead of \({r}_{av}\)), used a variety of alternative definitions of atomic misfit and lattice distortion \({\delta }_{max}\) and \({\delta }_{av}\) and used ab initio calculations of the single-phase fcc Cantor alloy structures [44,45,46,47] (the latter are more satisfying theoretically but, as mentioned previously, are not able to cover the enormous number of different local environments). Nevertheless they all lead to similar though slightly higher predicted local lattice distortions [44,45,46,47] of \(\Delta r\approx\) 3.3–6.6 pm.
Lattice distortions have been measured in a variety of multicomponent solid solutions (but mainly in Cantor alloys) by a variety of different techniques, including synchrotron X-ray diffraction (XRD) [44], neutron diffraction (ND) [48] and extended X-ray absorption fine-structure analysis (EXAFS) [45, 49]. Measurements are difficult because of the need to separate on the one hand static local atomic displacements caused by lattice distortions and on the other hand dynamic displacements caused by thermal vibrations and bulk lattice strain. Essentially, sophisticated fitting software has to be used with very high-quality diffraction data to separate out these different effects within the overall broadening of diffraction peaks such as those shown schematically in Figs. 5 and 6.
Okamoto et al. [44] used single-crystal synchrotron X-ray diffractometry to measure root-mean-square (RMS) average atomic displacements from the X-ray peaks in high-purity single-crystal specimens of the original Cantor alloy. The lattice distortions were found to be 4.8 and 7.7 pm within an estimated error of ± 0.5 pm at 25 K and 300 K, respectively, indicating an average static atomic displacement (at 25 K) of \(\Delta r\approx\) 4.8 pm and an average room-temperature dynamic (i.e. thermal) displacement of 7.7 – 4.8 = 2.9 pm. Owen et al. [48] used high-precision neutron powder diffractometry to measure partial distribution functions (PDFs) in gas-atomised specimens of the original Cantor alloy. In this case, the difference in PDF full peak widths at half maximum height (FWHM) between the Cantor alloy and pure Ni was attributed to static atomic distortions in the Cantor alloy of \(\Delta r\approx\) 2 pm, with the remaining shared component of peak width representing dynamic (thermal) distortions of ~ 18 pm, again claimed to be within an error of approximately ± 0.5 pm. Oh et al. [45] obtained extended X-ray absorption fine-structure (EXAFS) spectra for each of the components Cr, Mn, Fe, Co and Ni in a recrystallised sample of the original Cantor alloy, indicating mean individual elemental distortions relative to the average interatomic bond length of approximately + 0.1, + 0.5, − 0.1, − 0.4 and − 0.1% for Cr, Mn, Fe, Co and Ni respectively,Footnote 11 with individual fluctuations quoted to be an order of magnitude larger, up to a maximum of 2–3%. In other words, local distortions were greatest for Cr and Mn atoms, intermediate for Fe and Ni atoms and smallest for Co atoms, in rough agreement with the Goldschmidt atomic radii in Table 3, with an overall maximum distortion of 3% of \({r}_{av}\) (= 126.2 pm), i.e. \(\Delta r\approx\) 3.8 pm. Using similar EXAFS methods, Tan et al. [49] measured rather different mean individual elemental distortions of − 0.1, − 0.2, + 0.3%, − 0.9% and + 1.1% for Cr, Mn, Fe, Co and Ni, respectively, correlating poorly with the component Goldschmidt atomic radii (unlike Oh et al.’s results [45]), and with no indication of the maximum size of the measured distortions.
The different experimental and calculated results for the maximum magnitude of local atomic distortions \(\Delta r\) in the original fcc Cantor alloy CrMnFeCoNi are collected together in Table 4. In summary, we can conclude that there are small but significant local lattice distortions in the original fcc Cantor alloy CrMnFeCoNi that fluctuate randomly over very large distances (because of the enormous number of different local atomic cluster configurations discussed previously), with an average value of just under 1 pm (corresponding to just under ~ 1% strain), reaching a maximum value of ~ 2–6 pm (corresponding to ~ 2–6% strain). Similar measurements and calculations indicate, as expected, somewhat smaller lattice distortions in simpler fcc Cantor alloy compositions such as CrFeCoNi [46, 46,49,50,51], but somewhat larger lattice distortions in modified Cantor alloy compositions containing additional components such as VFeCoNi [52] and FeNiCoCrPd [51]. Overall, the fcc Cantor alloys clearly exhibit significant local lattice distortions and strains that are expected to influence many of their material properties, as discussed later in this paper.
Lattice Distortions in Senkov Alloys
Table 5 shows 12-coordinated metallic Goldschmidt atomic radii r for some of the components that have been used to manufacture multicomponent single-phase bcc Senkov alloys [43]. The maximum difference in atomic size \(\Delta r\) amongst the five different components, the corresponding maximum atomic misfit \({\delta }_{max}\) and the root-mean-square (RMS) average atomic misfit \({\delta }_{av}\) in the original Senkov alloy VNbMoTaW are as follows:
The values of \(\Delta r\), \({\delta }_{max}\) and \({\delta }_{av}\) remain unchanged when either Al or Ti is included in modified AlxVNbMoTaW or TixVNbMoTaW Senkov alloys, respectively; but they are somewhat larger again when either Zr or Hf is included in modified VNbZrxMoTaW or VNbMoHfxTaW Senkov alloys, respectively:
for an equiatomic six-component alloy with \({x}_{i}=16.67\text{\%}\).
We might expect, therefore, to find lattice distortions on a scale of approximately 4 pm or 3% on average rising to a maximum of 7–8 pm or 10% in the original Senkov alloy, and still higher in some of the modified Senkov alloys such as those containing Zr or Hf. Clearly these calculations are again rather rough and simplistic, since they ignore effects such as short-range order, non-ideality, relaxation and electronic distortions. Other analyses of the expected lattice distortion have again tried to use more fundamental and ab initio calculations of the single-phase bcc Senkov alloy structures, leading to similar predicted local lattice distortions of \(\Delta r\approx\) 5–15 pm [46].
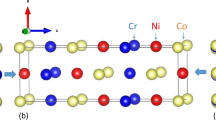
Zou et al. [53] used single-crystal X-ray diffractometry to measure root-mean-square (RMS) average atomic displacements from the X-ray peaks in high-purity single-crystal specimens of the original Senkov alloy. The static lattice distortions were found to be \(\Delta r\approx\) 7.4 pm, with an associated dynamic (thermal) distortion of 6.1 pm, within an error of ± 0.5 pm. Lattice distortions on a scale of tens of pm have also been observed directly by Zou et al. [53] in the same alloy by high-resolution transmission electron microscopy (HRTEM), with [100] zone axis images containing distorted {110} planes, as shown in Fig. 7. Guo et al. [54] used a combination of transmission synchrotron X-ray diffractometry and time-of-flight (TOF) neutron diffractometry to obtain radial distribution functions (RDFs) from a modified ternary Senkov alloy ZrNbHf, with an overlap of the first and second near-neighbour peaks corresponding to local lattice distortions of approximately 9.5 pm. Similar combined XRD and ND results on an equiatomic quaternary modified Senkov alloy ZrNbTaHf have shown static and dynamic (thermal) lattice distortions of 13.7 and 2.4 pm, respectively [55].

HRTEM images of the equiatomic quaternary NbMoTaW bcc Senkov alloy: a bright-field (BF) image with [100] zone axis; b corresponding inverse fast-Fourier transform; c and e enlarged images of the boxes in (b); and d and f traces corresponding to c and e to show the lattice distortions (after Zou et al [53])
The different experimental and calculated results for the maximum magnitude of local atomic distortions \(\Delta r\) are collected together in Table 6. In summary, we can conclude that there are significant local lattice distortions in the bcc Senkov alloys, considerably larger than those found in the fcc Cantor alloys, that again fluctuate randomly over very large distances (because of the enormous number of different local atomic cluster configurations discussed previously), with average values of approximately 4 pm (corresponding to ~ 3% strain), reaching maximum values of ~ 5–20 pm (corresponding to > 5% strain). The bcc Senkov alloys like the fcc Cantor alloys clearly exhibit significant local lattice distortions and strains that are expected to influence many of their material properties, as discussed later in this paper.
Lattice Distortions in Rost Oxides
Table 7 shows ionic radii [56] for some of the metallic cations that have been used to manufacture multicomponent single-phase NaCl-type rock-salt-structured Rost mono-oxide ceramics MO. The maximum difference in ionic size \(\Delta r\) amongst the five different metallic cation components, the corresponding maximum atomic misfit \({\delta }_{max}\) and the root-mean-square (RMS) average atomic misfit \({\delta }_{av}\) in the multicomponent single-phase NaCl-type original Rost mono-oxide (MgCoFeNiCu)O are as follows:
though these values are reduced to \(\Delta r=6{\text{pm}}\), \({\delta }_{max}=7.5\text{\%}\) and \({\delta }_{av}=\) 0.6%, respectively, when the ferromagnetic electronic spins are aligned on the Co and Ni cations. Yet again, multicomponent interatomic and ceramic compounds such as (MgCoFeNiCu)O clearly exhibit significant local lattice distortions and strains that are expected to influence many of their material properties, as discussed later in this paper.
Vacancies and Diffusion
Vacancies and Diffusion in Pure Materials
Vacancies are one of the most common point defects in the structure of a material [57,58,59]. According to the third law of thermodynamics, all crystals are perfect at equilibrium at absolute zero and there are, therefore, no vacancies (or any kind of defect) at absolute zero. At any realistic temperature, however, the excess energy of creating a vacancy can be balanced by the entropy associated with mixing vacancies throughout the crystal, so there is an equilibrium number of vacancies in any crystal that increases with increasing temperature. In a single-component pure material, there is a characteristic and fixed excess energy required to create a vacancy, the vacancy formation energy \(\Delta {E}_{v}\), which is independent of where the vacancy is in the crystal, since all the lattice sites are identical. The probability of finding a vacancy at any particular lattice point \({p}_{v}\) and the equilibrium number of vacancies in the crystal \({N}_{v}\) are given at any temperature T, therefore, by Boltzmann’s equation [57, 58]:
where \({N}_{o}\) is Avogadro’s number and k is Boltzmann’s constant.Footnote 12
The most common form of atomic motion or diffusion in solid crystalline materials is by vacancy migration or vacancy hopping, in which an atom adjacent to a vacancy jumps into the vacant lattice site [57,58,59]. Each time an atom jumps into a vacancy, it has to overcome an energy barrier in order to squeeze between surrounding nearby atoms. In a single-component pure material, there is a characteristic and fixed energy barrier for such atomic jumps and, therefore, for the process of vacancy hopping, the vacancy migration energy \(\Delta {E}_{m}\), which is again independent of where the vacancy is in the crystal, since all the lattice sites are identical. The probability of an atom adjacent to a vacancy jumping into it by acquiring enough energy to overcome the barrier and squeezing through the surrounding atoms, i.e. the probability of vacancy migrationFootnote 13\({p}_{m}\), is given again by Boltzmann’s equation [57,58,59]:
Atomic motion in a single-component single-phase pure material is, then, fairly straightforward. From time to time an atom adjacent to a vacancy acquires enough energy to squeeze past its neighbours and jump into the vacancy. Since the probability of a vacancy being present and the probability of an adjacent atom acquiring enough energy to jump into it are both independent of their position in the lattice (because all lattice sites are identical), the resulting process of vacancy hopping and reverse atomic migration or diffusion is completely random and stochastic, both spatially and temporally. In other words, solute diffusion takes place via a random walk. This means that it obeys Fick’s laws, with the flux of solute atoms J proportional to the solute concentration gradient \(\partial c/\partial x\) and the change of solute concentration with time \(\partial c/\partial t\) proportional to its spatial curvature \({\partial }^{2}c/\partial {x}^{2}\). The constant of proportionality in both cases is the diffusion coefficient D [57,58,59]:
where dn/dt is the rate at which a number of atoms dn migrate in time dt through unit cross-sectional area A. The diffusion coefficient D depends on the frequency of vacancy hopping, i.e. the frequency with which individual solute atoms jump into adjacent vacancies, so it is proportional to the probability \({p}_{v}\) of a vacancy being present and the probability \({p}_{m}\) of a solute atom jumping into it:
where \({D}_{o}\) and \(Q=\Delta {E}_{v}+\Delta {E}_{m}\) are, respectively, the frequency factor and the activation energy for diffusion [57,58,59].
Vacancies in Multicomponent Solid Solutions
Atomic motion in a concentrated multicomponent high-entropy solid solution is (not surprisingly) much more complex, essentially because of the wide variety of different local atomic structures and associated lattice distortions surrounding different vacancies, leading to both a wide range of different vacancy formation energies \(\Delta {E}_{v}\) and a wide range of different vacancy migration energies \(\Delta {E}_{m}\), depending upon where the vacancy is and what exactly are its precise surrounding atoms. Just as there is an enormous number of different local atomic clusters surrounding each individual type of atom in a multicomponent solid solution (as discussed previously), there is a similarly enormous number of different local atomic clusters surrounding the vacancies in a multicomponent solid solution or, to put it in another way, there is an enormous number of different vacancy structures.
Consider a general lattice with a single atom at each lattice point, and with \({n}_{1}\), \({n}_{2}\) and \({n}_{3}\) first, second and third near neighbours, respectively.Footnote 14 The cluster size of atoms surrounding any vacancy is then \({n}_{1}\) if we only consider its first near neighbours, \({n}_{2}+{n}_{1}\) if we also include its second near neighbours, \({n}_{3}+{n}_{2}+{n}_{1}\) if we also include its third near neighbours, and so on. The numbers of different orientation-dependent clusters \({N}_{1}\) of \({n}_{1}\) atoms, \({N}_{2}\) of \({n}_{2}+{n}_{1}\) atoms, and \({N}_{3}\) of \({n}_{3}+{n}_{2}+{n}_{1}\) atoms surrounding a vacancy in a multicomponent equiatomic single-phase material with a random (ideal or regular) arrangement of c components are given [36] again by the law of permutations with repetitionFootnote 15:
These are again orientation-dependent cluster numbers and vacancy structures (i.e. equivalent vacancy structures of a different orientation are regarded as different, similar to atomic clusters discussed previously), as is appropriate when we are dealing with properties or processes that depend on orientation. This is sometimes the case for atomic diffusion, when it takes place driven by a fixed concentration gradient in a fixed direction, such as during oxidation or carburisation at the free surface of the material. If cluster orientation is not significant, however, the number of different clusters is smaller (again similar to atomic clusters discussed previously) because of the number of self-similarity operations s associated with the crystal symmetry:
with s = 24 for instance for a cubic crystal [37, 38]. These are again orientation-independent cluster numbers and vacancy structures (i.e. equivalent vacancy structures of a different orientation are regarded as the same), as is appropriate when we are dealing with properties or processes that are independent of orientation. This is also sometimes the case with atomic diffusion, when it takes place in all directions simultaneously, driven by a general overall reduction in the bulk free energy of the material, such as during the process of precipitation or grain growth, and is also the case when we are dealing with the properties of vacancies themselves, such as their volume or electronic structure.
Table 8 shows the numbers of oriented and non-oriented local atomic clusters surrounding vacancies \({N}_{1}\), \({N}_{2}\) and \({N}_{3}\), i.e. the numbers of oriented and non-oriented vacancy structures, out to first, second and third near neighbours, respectively, for multicomponent equiatomic single-phase fcc Cantor alloys with c components, where c is in the range 3–50, taking for fcc \({n}_{1}=12\), \({n}_{2}=6\) and \({n}_{3}=24\), so the cluster sizes are 12, 18 and 42 atoms, respectively. Table 9 shows equivalent numbers for multicomponent equiatomic single-phase bcc Senkov alloys, taking for bcc \({n}_{1}=8\), \({n}_{2}=6\) and \({n}_{3}=12\), so the cluster sizes are 8, 14 and 26 atoms, respectively. The same results are shown graphically in Figs. 8 and 9 for fcc and bcc, respectively. It is obvious from Tables 9 and 10 and Figs. 8 and 9 that the numbers of different vacancy structures in multicomponent single-phase materials are very large indeed. This is, of course, essentially the same result as discussed previously for the numbers of different local atomic clusters surrounding individual atoms in a multicomponent solid solution, as shown in Tables 1 and 2 and Figs. 3 and 4. The only difference is that the numbers of vacancy structures are a bit smaller, since the cluster sizes are reduced by one, with a fixed vacancy at the centre of each cluster rather than a variety of c different components.

Number of different oriented (upper lines) and non-oriented (lower lines) local atomic clusters around a vacancy out to first (\({N}_{1}\)), second (\({N}_{2}\)) and third (\({N}_{3}\)) near neighbours for a multicomponent single-phase fcc material with c components

Number of different oriented (upper lines) and non-oriented (lower lines) local atomic clusters around a vacancy out to first (\({N}_{1}\)), second (\({N}_{2}\)) and third (\({N}_{3}\)) near neighbours for a multicomponent single-phase bcc material with c components
The number of oriented first and second near-neighbour vacancy structures in the original single-phase fcc Cantor alloy CrMnFeCoNi with five components is, for instance, just under four trillion (3.8 × 10 12), and the equivalent number of vacancy structures in the original single-phase bcc Senkov alloy also with five components is just under 10 billion (6.1 × 109). With non-oriented clusters and first near neighbours only, the numbers of vacancy clusters are, of course, somewhat smaller but still very large, 10 million (107) for the original fcc Cantor alloy and sixteen thousand (1.6 × 104) for the original bcc Senkov alloy. With six components, the number of oriented first and second near-neighbour vacancy clusters increases to a hundred trillion (1014) for fcc Cantor alloys and almost a hundred billion (7.8 × 1010) for bcc Senkov alloys; with eight components to well over a quadrillion (1.8 × 1016) for fcc Cantor alloys and well over a trillion (4.4 × 1012) for bcc Senkov alloys; and with 10 components to a quintillion (1018) for fcc Cantor alloys and a hundred trillion (1014) for bcc Senkov alloys. The number of vacancy clusters is, of course, very much larger again, and indeed becomes astronomic, if we include third near neighbours (> 1029 or > 1018 for the five-component Cantor and Senkov alloys, respectively) and/or increase the number of components to (say) 10 or more (> 1018 or > 1014 for Cantor or Senkov alloys respectively), as shown in Tables 9 and 10 and Figs. 8 and 9.
Diffusion in Multicomponent Solid Solutions
In a multicomponent high-entropy single-phase solid-solution material, it is important to realise that the excess energy required to create a vacancy or the vacancy formation energy \(\Delta {E}_{v}\) is different for each of the enormous number of different vacancy structures, because of the differences in local lattice distortions; and it is also important to realise that the energy barrier to an adjacent atom jumping into a vacancy or the vacancy migration energy \(\Delta {E}_{m}\) is also different for each of these different vacancy structures, again because of the differences in local lattice distortions. In other words, there is a wide spread of values for both the vacancy formation and vacancy migration energies \(\Delta {E}_{v}\) and \(\Delta {E}_{m}\).
There is little or no lattice distortion in a single-component pure material, and the variation of energy as an individual vacancy hops from one lattice site to another consists of a series of identical jumps, as shown schematically in Fig. 10, from identical wells at each of the lattice sites all with the same well energy \({E}_{w}\), over identical saddle points midway between each of the lattice sites all with the same saddle-point energy \({E}_{s}\). The well energy \({E}_{w}\) is constant at all points in the hopping process because the vacancy structure and, therefore, its formation energy \(\Delta {E}_{v}\) are independent of where the vacancy is in the lattice; and, similarly, the saddle-point energy \({E}_{s}\) is constant at all points in the hopping process, because the vacancy structure and, therefore, its migration energy \(\Delta {E}_{m}={E}_{s}-{E}_{w}\) are independent of where the vacancy is in the lattice.

Schematic variation of energy as a vacancy hops from lattice site to lattice site: (a) in a single-component material with all jumps identical; and (b) in a multicomponent material with variable jumps (s = saddle-point energy; w = energy well)
In a concentrated multicomponent high-entropy solid solution, however, the situation is very different, because of the large number of vacancy structures and associated local lattice distortions at different lattice points, the corresponding range of vacancy formation and migration energies \(\Delta {E}_{v}\) and \(\Delta {E}_{m}\), and the corresponding spread of well and saddle-point energies \({E}_{w}\) and \({E}_{s}\). In a concentrated multicomponent high-entropy solid-solution material, the variation in energy of an individual vacancy as it hops from one lattice site to another consists instead, therefore, of a series of variable jumps, as also shown schematically in Fig. 10, from variable wells at the different lattice sites all with different well energies \({E}_{w}\), over variable saddle points midway between each of the lattice sites all with different saddle-point energies \({E}_{s}\).
The overall rate of atomic diffusion and, therefore, the diffusion coefficient in concentrated multicomponent high-entropy solid-solution materials is affected in a variety of ways [60] by the variation in vacancy formation and migration energies \(\Delta {E}_{v}\) and \(\Delta {E}_{m}\), and the corresponding spread of well and saddle-point energies \({E}_{w}\) and \({E}_{s}\). High-energy barriers in some places make it particularly difficult for a vacancy to hop from one site to the next; low-energy barriers in other places make it particularly easy for a vacancy to hop from one site to the next; and a vacancy can be trapped in some places, hopping backwards and forwards between adjacent or near-adjacent lattice points, or going round and round in circles. The overall impact of these different effects is quite complex, and strictly speaking diffusion is no longer truly random spatially, since the lattice points are no longer all identical. Thomas and Patala [60] have shown that, with some simplifying assumptions, the different effects can be separated. Simple Boltzmann expressions for the probability of a vacancy being present \({p}_{v}\) and the probability of an adjacent atom jumping into it \({p}_{m}\) can then be replaced by expectation values \(\langle {p}_{v}\rangle\) and \(\langle {p}_{m}\rangle\) obtained by (rather complex) integrationsFootnote 16 over the range of different formation and migration energies \(\Delta {E}_{v}\) and \(\Delta {E}_{m}\), and their corresponding well and saddle-point energies \({E}_{w}\) and \({E}_{s}\), allowing for correlations between different jumps with different energies, so the diffusion coefficient D is now given by
where \(\langle {F}_{ij}\rangle\) is another expectation value obtained by integration over the series of different correlation factors between different i’th and j’th jumps.
Thomas and Patala [60] calculated the expected spread of well and saddle-point energies \({E}_{w}\) and \({E}_{s}\) for vacancies hopping between adjacent lattice sites at 1000 °C in the original five-component single-phase fcc Cantor alloy CrMnFeCoNi, using a nudged elastic band (NEB) molecular dynamics (MD) method with a modified embedded atom method (MEAM) potential within the Sandia Labs LAMMPS software. They obtained mean well and saddle-point energies of \(\overline{{E}_{w}}=0\) (taken as an arbitrary zero-energy reference point) and \(\overline{{E}_{s}}=0.81\) eV, i.e. the average barrier height and, therefore, vacancy migration energy was \(\overline{\Delta {E}_{m}}=\overline{{E}_{s}}-\overline{{E}_{w}}=0.81\) eV, with standard deviations of \({\sigma }_{w}=0.08\) eV corresponding to a relatively narrow distribution of well energies, and \({\sigma }_{s}=0.31\) eV corresponding to a relatively broad distribution of saddle-point energies. The diffusion coefficient D was found to be fairly sensitive to the spread of energy values during the hopping process, varying between a half and five times a reference diffusion coefficient D* in an equivalent material with the same barrier energy of 0.81 eV but with constant well and saddle-point energies.
The exact results obtained by these calculations should be treated with considerable caution (as with other modelling results), since they average over only about three thousand vacancy hops, well below the total number of almost four trillion different vacancy structures (including first and second near neighbours). In other words, they fail to capture anywhere near the full range of different vacancy structures, local lattice distortions, and corresponding formation and migration energies and well and saddle-point energies, which strictly speaking should all be sampled in order to obtain accurate results. This explains why a range of different results have been obtained by similar ab initio and MD calculations by different investigators, for instance \(\overline{\Delta {E}_{m}}=\overline{{E}_{s}}-\overline{{E}_{w}}=\) 0.60 ± 0.05 eV by Kottke et al. [61] and 0.97 ± 0.28 eV obtained by Choi et al. [62] and Li et al. [63], compared with 0.81 ± 0.32 eV obtained by Thomas and Patala [60]. Nevertheless, the results show clearly that diffusion is slower when there is a wide spread of well energies (high \({\sigma }_{w}\)) because vacancies can become trapped in the lowest energy wells; but is faster when there is a wide spread of saddle-point energies (high \({\sigma }_{s}\)) because vacancies can take migration paths that simply avoid the highest energy barriers.
There has been considerable discussion in the previous literature about whether or not diffusion is slower in multicomponent high-entropy solid-solution single phases relative to pure materials [64,65,66,67,68], somewhat confused by the difficulties of deciding what are appropriate comparison materials, and the complexity of measuring diffusion coefficients from multicomponent material diffusion couples. There is, however, little doubt that diffusion is quite slow in many cases [3, 64, 65], by up to about a half or one order of magnitude, with correspondingly slow diffusion-controlled processes such as precipitation and recrystallisation [1,2,3], almost certainly caused by the wide variety of local atomic structures as described above.
Dislocations and Slip
Dislocations and Slip in Pure Materials
Dislocations are one of the most important kinds of line defect in the structure of a material [57, 69, 70]. The crystal lattice is displaced by the Burgers vector b at all points along the dislocation line l, with edge and screw dislocations corresponding to \(\varvec b\perp \varvec l\) and \(\varvec b\parallel \varvec l,\) respectively. There is an excess energy per unit length of a dislocation \(\Delta {E}_{d}\), arising partly from the substantial displacement of atoms along the dislocation line itself, i.e. in the dislocation core, and partly from much smaller elastic atomic displacements induced throughout the crystal lattice outside the core. The excess energy per unit length or line tension of a dislocation \(\Delta {E}_{d}\) is given by [57, 69, 70]:
where \({E}_{c}\) and \({r}_{c}\) are the energy and radius of the highly distorted dislocation core region, G is the shear modulus, R is the dislocation spacing, the factor \(\alpha =1\text{ or }1-\nu\) for screw or edge dislocations, respectively and \(\nu\) is Poisson’s ratio. The largest atomic displacements are in the core of a dislocation, but most of its excess energy is in the large number of small atomic displacements in the mid-range elastic strain field [57, 69, 70].
According to the third law of thermodynamics, all crystals are perfect at equilibrium at absolute zero and there are, therefore, no equilibrium dislocations (or any kind of defect) at absolute zero. At any realistic temperature, however, the excess energy of creating a dislocation can be balanced by the entropy associated with dislocations being present throughout the crystal, so there is an equilibrium number of dislocations in any crystal that increases with increasing temperature. The equilibrium number of dislocations is rather small, and most materials contain a much higher, non-equilibrium number, usually generated as a consequence of external stresses and strains during manufacturing, handling and use at relatively low temperatures.
In most crystals, the dislocation energy is high and dislocations are both difficult to create and immobile. In close-packed metal crystals, however, particularly fcc (and to a lesser extent hcp and bcc), the dislocation energy is much lower and dislocations are relatively easy both to create and to move [57, 69, 70]. Mobile dislocations move in response to shear stresses, usually (though not exclusively) across the close-packed planes of the crystal lattice, and as they move they carry the displacement of the Burgers vector with them. Continued dislocation slip in this way produces gross plastic flow and results in a permanent set in the crystal. Under an applied load, dislocations in an fcc metal (and to a lesser extent hcp and bcc metals) are created in large numbers and move relatively easily and rapidly throughout the material, so the materials are soft (deform at a low applied stress), ductile (deform extensively) and tough (resistant to fracture), with local dislocation creation and motion preventing the build-up of local high stresses and the formation and growth of cracks [57, 69, 70].
Soft pure metals and conventional alloys can be hardened or strengthened by a variety of different methods, most of which restrict the mobility of the dislocations that would otherwise allow plastic flow to take place easily, i.e. they usually reduce its ductility and toughness [71, 72]. The overall shear flow stress \(\tau\) required to move dislocations on the close-packed planes can usually be treated as a straightforward sum of the different contributions to the material strength [71, 72]:
where \({\tau }_{o}\) is the lattice friction stress or Peierls-Nabarro stress, the intrinsic stress needed to move a dislocation in a perfect piece of the crystal, and \({\tau }_{wh}\), \({\tau }_{ss}\), \({\tau }_{gb}\) and \({\tau }_{ph}\) are strengthening contributions from work hardening, solution hardening, grain boundaries and precipitates, respectively.
Dislocations in Multicomponent Solid Solutions
Although some features are similar, the structure and properties of dislocations are, not surprisingly, more complicated in multicomponent materials such as the fcc Cantor and bcc Senkov alloys. In a multicomponent high-entropy solid-solution single-phase material, there is an enormous number of different atomic structures around different atoms along the length of the core of a dislocation, in the same way as discussed previously for the number of different atomic clusters within the structure generally, as shown in Tables 1 and 2 and Figs 3 and 4, and the number of different atomic clusters surrounding vacancies (or vacancy clusters), as shown in Tables 9 and 10 and Figs. 8 and 9. This means that the atomic structure varies widely from point to point along any dislocation line, and also from time to time at any given point on a dislocation line as it moves from lattice site to lattice site under the action of an applied stress.
From Table 1, there are almost twenty trillion (1.9 × 1013) different oriented first and second near-neighbour atomic clusters and, therefore, different local dislocation structures surrounding each individual atom at the core of a dislocation in the original equiatomic five-component fcc Cantor alloy CrMnFeCoNi; and from Table 2, there are over thirty billion (3.1 × 1010) in the original equiatomic five-component bcc Senkov alloy VNbMoTaW. Including first near neighbours only, the numbers are somewhat smaller but still significant, just over a billion (1.2 × 109) and two million (2 × 106), respectively; but including third near neighbours the numbers are much larger again, 1.1 × 1030 and 7.5 × 1018, respectively. Taking the atomic size as ~ 0.3 nm, the total dislocation length needed to sample all possible first and second near-neighbour dislocation structures is 5.7 km or 9.3 m for the Cantor and Senkov alloys, respectively, corresponding to a volume of 0.57 cm3 or 0.93 mm3 and a grain size of 8 mm or 1 mm, respectively, in a well-annealed material with 1010 dislocation lines/m2, and 5.7 × 106 µm3 or 9.3 × 103 µm3 and a grain size of 178 µm or 21 µm, respectively, in a highly deformed material with 1015 dislocation lines/m2. In other words, there is an enormously extensive variation in local atomic structure along the line of any dislocation in an equiatomic five-component fcc Cantor or bcc Senkov alloy, which persists over very large volumes of material.Footnote 17
Dislocations in fcc Cantor Alloys
There have been quite a large number of detailed studies of the structure of dislocations and the resulting mechanisms of plastic flow in multicomponent solid-solution single-phase materials, notably in a wide range of fcc Cantor alloys and bcc Senkov alloys, mostly using a variety of high-resolution transmission electron microscope techniques. There are several extensive reviews of this work [76,77,78]. Dislocation structures in fcc Cantor alloys are in general found to be very similar to those observed previously in pure fcc metals and dilute fcc binary alloys with a relatively low stacking fault energy [76,77,78]. As seen in all low stacking fault energy fcc alloys, the characteristic 1/2<110 > dislocations dissociate into two 1/6<112 > Shockley partials separated by a stacking fault. The separation between the two partials and the width of the stacking fault d is determined by a balance between a repulsive force proportional to Gb2 resulting from the overlap of the two partial dislocation stress fields, and an attractive force proportional to the stacking fault energy \({\gamma }_{sf}\) trying to reduce the size of the stacking fault between them, so that [[69, 70]:
where \(\alpha \approx\) 0.3–1.6 is a factor depending on the Poisson’s ratio and the dislocation character (screw, edge or mixed). Table 10 shows the relatively low values of stacking fault energy that have been calculated from the measured separation of partials in several single-phase fcc Cantor alloys, typically in the range 10–30 mJ/m2, similar to pure fcc Au, stainless steel and \(\alpha\)-brass [69, 69,73,74,75,76,77,78]. Also as seen in all low stacking fault alloys, the effect of an applied stress is continuously to create new dissociated 1/2<110 > dislocations that slip on close-packed {111} planes and form pile ups at obstacles such as grain boundaries, unable to cross slip because of the dislocation dissociation into partials with their associated stacking faults [69, 69,73,74,75,76,77,78].
Unlike pure fcc metals and dilute binary fcc alloys, however, the dislocations in multicomponent single-phase fcc Cantor alloys are wavy rather than straight on a near-atomic scale, with a wide variation along the dislocation line in the separation of the two Shockley partials (and, therefore, the width of the stacking fault and corresponding stacking fault energy), and a much higher shear stress needed for slip along the {111} planes [76,77,78,79,80,81,82]. An example of a wavy dislocation and measurements of the varying partial separation and corresponding stacking fault energy [79] are shown in Fig. 11 for the original fcc Cantor alloy CrMnFeCoNi. These effects can all be ascribed to the variation in local atomic structures along the dislocation lines as described above, with the associated local lattice strains acting as pinning centres for the dislocations. Okamoto et al. [79] used high-resolution weak-beam imaging to make a large number of measurements of the separation between partials along the length of dislocation lines in the original fcc Cantor alloy CrMnFeCoNi, which were found to be in the range d = 3–8 nm with an average of d = 5.7 nm, corresponding to stacking fault energies in the range \({\gamma }_{sf}=\) 25–35 mJ/m2 with an average of \({\gamma }_{sf}=30\) mJ/m2. They also measured the variation in partial separation and corresponding stacking fault energy along dislocation lines of different character, i.e. with different orientations \(\theta\), where \(\theta\) is the angle between the Burgers vector b and the dislocation line l, finding that the partial separations were in the range d = 3–5 nm for a screw dislocation with \(\theta =0^\circ\), increasing gradually with increasing \(\theta\) to d = 5–8 nm for an edge dislocation with \(\theta =90^\circ\). Similarly, Smith et al. [80] used end-on high-resolution images obtained by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) to measure the separation distances of thirty dislocations in a strained fcc Cantor alloy, all with Burgers vectors b = ½<110> and orientations \(\theta =60^\circ\). The resulting separation distances ranged from d = 1 to 9 nm with an average d = 4.8 nm, similar results to those found by Okamoto et al. [79].

(a) High-resolution weak-beam image of a ½<110> dislocation in the original fcc Cantor alloy CrMnFeCoNi showing a wavy dislocation line and separation into two 1/6<112> Shockley partials; and (b) partial separation distance d in a large number of dislocations as a function of dislocation orientation \(\theta\), ranging from pure screw (\(\theta =0^\circ\)) to pure edge (\(\theta =90^\circ\)) (after Okamoto et al. [79]).
Laplanche et al. [81] and Xu et al. [82] employed similar TEM techniques to those used by Okamoto et al. [79] and Smith et al. [80] to investigate dislocation structures in modified single-phase fcc Cantor alloys CrCoNi and Al0.1CrFeCoNi, respectively. Both alloys were found to have lower stacking fault energies than the original five-component Cantor alloy and, therefore, correspondingly greater separation of the Shockley partials. As in the original Cantor alloy, the separation between the partials was again spread broadly in both of the modified Cantor alloys: in CrCoNi, in the range d = 4–7 nm for a screw dislocation with \(\theta =0^\circ\), increasing gradually with increasing \(\theta\) to d = 8–14 nm for an edge dislocation with \(\theta =90^\circ\); and, in Al0.1CrFeCoNi, in the range d = 5–18 nm for a screw dislocation with \(\theta =0^\circ\), increasing gradually with increasing \(\theta\) to d = 9–33 nm for an edge dislocation with \(\theta =90^\circ\). The corresponding stacking fault energies were also spread broadly: in the range \({\gamma }_{sf}=\) 18–26 mJ/m2, with an average of \({\gamma }_{sf}=22\) mJ/m2 for CrCoNi; and in the range \({\gamma }_{sf}=\) 6–21 mJ/m2, with an average value of \({\gamma }_{sf}=12\) mJ/m2 for Al0.1CrFeCoNi.
Similar wavy dislocations with varying separation of partials along the dislocation line have also been seen in a number of multicomponent single-phase fcc Cantor alloys using a variety of atomistic ab initio modelling techniques [83,84,85].
Dislocations in bcc Senkov Alloys
Dislocation structures in bcc Senkov alloys are in general found to be very similar to those observed previously in pure bcc metals and dilute bcc binary alloys [86,87,88,89]. Characteristic ½<111> dislocations are created and move on a variety of {110}, {112}, {123} and other slip planes [86,87,88,89]. Unlike pure bcc metals and dilute binary bcc alloys, however, the dislocations in multicomponent single-phase bcc Senkov alloys are again, like the fcc Cantor alloys, wavy rather than straight on a near-atomic scale, and again, like the fcc Cantor alloys, require a much higher shear stress for slip along the different planes [86,87,88,89]. These effects can again, like the fcc Cantor alloys, be ascribed to the variation in local atomic structures along the dislocation lines as described above, with the associated local lattice strains acting as pinning centres for the dislocations. For instance, Couzinie et al. [86] and Lilensten et al. [87] both used transmission electron microscopy (TEM) techniques to study conventional ½<111> screw dislocations in the equiatomic five-component single-phase bcc-modified Senkov alloy TiZrNbHfTa after tensile testing at room temperature. And more recent studies by Wang et al. [88] of the modified equiatomic ternary bcc Senkov alloy TiNbMo and by Lee et al. [89] of the equiatomic modified quaternary bcc Senkov alloys TiVNbMo and VCrNbMo have shown a wide range of screw, edge and mixed dislocations gliding on a variety of {110}, {112}, {123} and other slip planes. Similar wavy dislocations have again also been seen in a number of multicomponent single-phase bcc Senkov alloys using a variety of atomistic ab initio modelling techniques [90,91,92,93].
Plastic Flow in Multicomponent Solid Solutions
As already mentioned, a much higher shear stress is needed for dislocation slip in multicomponent high-entropy solid-solution single-phase materials, including both the fcc Cantor alloys and bcc Senkov alloys. This is caused by the variation in local atomic structures along the dislocation lines, with the associated local lattice strains acting as pinning centres for the dislocations. Effectively this corresponds to raising the lattice friction stress because of the dense number of pinning centres created by a high concentration of many different types of solute atoms. The shear flow stress can be rewritten as
where \(\tau_{ph}\) is taken as zero in a material with no precipitates, the solid-solution strengthening \(\tau_{ss}\) is incorporated into an enhanced lattice friction stress \(\tau_{o}\), work hardening is still given by a conventional Taylor parabolic variation with dislocation density \(\rho\), i.e. \(\tau_{wh} \approx 2\alpha Gb\rho^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0pt} 2}}}\), where \(\alpha\) is a constant, and grain-boundary hardening is still given by a conventional Hall–Petch variation with grain size \(d\), i.e. \(\tau_{gb} = \beta d^{{ - {1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0pt} 2}}}\), where \(\beta\) is another constant.
Varvenne et al. [94, 95] have shown that a concentrated multicomponent solid-solution pinning effect can be modelled to give the enhanced lattice friction stress \({\tau }_{o}\) as:
where the first term in the bracket on the right-hand side is the intrinsic lattice flow stress at absolute zero and the second term is its temperature T and strain rate \(\dot{\epsilon }\) dependence at more realistic, higher temperatures. \(\Delta E\) is the average energy barrier to dislocation motion which must be overcome in order to unpin dislocations from the complex arrangement of different atomic clusters and associated lattice distortions along their wavy length, \(\zeta\) and w are the average wavelength and average amplitude, respectively, of the wavy dislocation lines and the corresponding variations in local lattice strain, \({\delta }_{av}\) is the average local lattice strain, \({\dot{\epsilon }}_{o}\) is a reference strain rate and K′ is a constant depending on the elastic constants and Poisson’s ratio. This equation gives good agreement with measurements of yield strength in a range of fcc Cantor alloys and bcc Senkov alloys, fully annealed with a large grain size (i.e. with \({\tau }_{wh}\) and \({\tau }_{gb}\) both zero) [77, 94,95,96,97,98,99]. Examples of this good agreement are shown in Fig. 12 for measurements of the yield stress at a range of different temperatures in two fcc Cantor alloys, CrMnFeCoNi and CrFeCoNi and in a range of other alloys at room temperature, all recrystallised with a large grain size so that the contributions of work hardening and grain size in Eq. (25) can be ignored.

Predicted values and experimental measurements of the initial large-grain-size dislocation-free yield stress for a the four-component single-phase fcc modified Cantor alloy CrFeCoNi and b the original five-component single-phase fcc Cantor alloy CrMnFeCoNi as a function of temperature; and c a range of other binary, ternary and quaternary equiatomic single-phase fcc alloys at room temperature (after Varvenne et al. [94])
Grain Boundaries and Surfaces
Grain Boundaries in Pure Materials and Dilute Solid Solutions
Most materials are polycrystalline and consist of an aggregate of differently oriented individual crystals or grains [71, 72]. Grain boundaries are the surfaces where any two grains in a polycrystalline material meet, and the regular crystalline atomic arrangement is disrupted. In fact, grain boundaries are the main kind of planar defect in crystalline materials, and there is a wide variety of different grain-boundary atomic structures with different amounts of disorganisation, depending on the relative orientation of the two grains either side of the grain boundary, and the orientation of the grain-boundary plane itself [38, 69]. To be more specific, a grain boundary is specified by the orientation relationship between the two grains either side of it as given by a common rotation axis r and a misorientation angle \(\theta\), together with the grain-boundary plane as given by its normal n. Since r and n are both unit vectors, they are defined by two angles relative to any given coordinate system, \(r=\left( {\phi_{r}, \psi_{r} } \right)\) and \(n = \left( {\phi_{n}, \psi_{n} } \right)\), and grain boundaries in any given material have, therefore, five independent degrees of freedom, with each one defined by five independent parameters \(\theta\), \({\phi }_{r}\), \({\psi }_{r}\), \({\phi }_{n}\) and \({\psi }_{n}\). There is, of course, an excess grain-boundary energy associated with the disorganisation of the atoms or molecules at any grain boundary which, not surprisingly, varies even in pure materials and simple dilute binary alloys in a complex way with the orientation of the two grains either side of the grain boundary and the orientation of the grain-boundary plane [38, 69].
Low-angle grain boundaries have misorientation angles \(\theta\) less than approximately \(15^\circ\), and consist of well-defined arrays of separated dislocations [29, 120]. With increasing misorientation angle \(\theta\), the dislocation arrays in the grain-boundary plane become more and more closely spaced until the individual dislocations can no longer be recognised above about \(\theta =15^\circ\). The grain-boundary energy \({\gamma }_{gb}\) of a low-angle grain boundary is equal to the energy of the corresponding dislocation arrays, increasing from \({\gamma }_{gb}=0\) at \(\theta =0^\circ\) (i.e. when there is no grain boundary) and reaching a maximum at about \(\theta =15^\circ\), as given by the Read-Shockley equation:
where \(A\propto Gb\) and \(B\propto {E}_{c}/G{b}^{2}\) are constants depending on the elastic constants of the material and the exact nature of the grain boundary and its particular dislocation arrays, and G, b and \({E}_{c}\) are the shear modulus of the material and the Burgers vector and core energy of the array dislocations, respectively [38, 69].
High-angle grain boundaries have misorientation angles \(\theta\) above approximately \(15^\circ\), usually with substantial atomic or molecular disorder that is tightly confined to a width of no more than one or two crystal planes, and, in almost all cases therefore, a correspondingly high and roughly constant grain-boundary energy [38, 69]. In other words, the plot of grain-boundary energy \({\gamma }_{gb}\) versus rotation angle \(\theta\) shows a plateau of constant high grain-boundary energy, typically in the range ½–1 J/m2, for almost all misorientations \(\theta\) above ~ \(15^\circ\). There are, however, some special high-angle grain boundaries at particular orientation relationships where the two grains share a relatively high number of coincident lattice sites. These particular orientation relationships are said to exhibit a coincident site lattice or CSL, and the corresponding special high-angle grain boundaries are called \(\Sigma n\) boundaries, where n is the inverse ratio of the number of coincident sites to the number of normal lattice sites, which geometrically must always be an odd integer [38, 69]. Depending on the detailed atomic or molecular structure of the material, some (though not all) of these special CSL orientation relationships and corresponding \(\Sigma n\) high-angle boundaries exhibit somewhat reduced atomic or molecular disorganisation, with a correspondingly reduced grain-boundary energy, as manifested by the presence of low-energy dips or cusps at these particular orientation angles on the high-angle grain-boundary energy plateau of the plot of grain-boundary energy \({\gamma }_{gb}\) versus rotation angle \(\theta\). The simplest example of a special grain boundary with a CSL orientation relationship is the \(\Sigma 3\) twin boundary in an fcc pure material, formed by a rotation of \(\theta =70.53^\circ\) about \([01\overline{1}]\) with a symmetric (111) grain-boundary plane [69].
Grain Boundaries in Multicomponent Solid Solutions
As just described, there is a wide range of grain boundaries with a correspondingly wide range of complex structures and energies even in pure materials and simple alloys [38, 69]. Not surprisingly, the complexity of grain-boundary structures is much greater still in multicomponent high-entropy materials. The topological complexity of accommodating atomic and molecular sites midway between two differently oriented crystals is compounded in a multicomponent solid-solution single-phase material by the additional chemical complexity of accommodating a large number of different component atoms across the various grain-boundary sites. Table 1 shows that there are almost twenty trillion different oriented first and second near-neighbour local atomic clusters in a five-component equiatomic single-phase fcc Cantor alloy, and Table 2 shows that there are just over thirty billion in a five-component equiatomic single-phase bcc Senkov alloy. In any polycrystalline Cantor or Senkov alloy, this enormous variety of different local atomic clusters has to be distributed across the many different grain-boundary sites all along the many different grain-boundary planes. The complexity and multiplicity of grain-boundary structures in multicomponent high-entropy solid-solution single-phase materials arising from such a vast range of different local atomic clusters is, of course, similar to the complexity and multiplicity discussed previously, arising in the same way for vacancy and dislocation structures in these materials.
Direct observation of grain-boundary structures is not easy even in pure materials and simple dilute binary alloys, because of the complex imaging conditions created by the atomic or molecular disorganisation in grain-boundary regions [38, 69], complicating the use of standard high-resolution imaging techniques such as transmission electron microscopy (TEM), X-ray diffraction (XRD) and atom-probe tomography (AP). And the problems associated with the direct observation of grain-boundary structures are only made worse by the chemical complexity found in multicomponent high-entropy materials. Not surprisingly, therefore, there have been relatively few attempts to investigate multicomponent high-entropy grain-boundary structures by direct observation [1, 2, 100]. Perhaps more surprisingly, there have also been relatively few attempts to use atomistic modelling techniques [1, 2, 1,2,101,102,103], which are more straightforward technically, but always suffer, as remarked previously, from not being able to handle anywhere near the very large number of different local atomic clusters that are present in multicomponent high-entropy materials.
Grain Size in Multicomponent Solid Solutions
There has, however, been a lot of interest in controlling the grain size of multicomponent high-entropy materials as one of the methods of tuning their properties for potential applications [1, 2]. Single-phase fcc Cantor alloys and bcc Senkov alloys have been manufactured with grain sizes ranging from large single crystals down to very fine-scale nanocrystalline materials [1, 2].
A common method of controlling grain size is by using a mixture of deformation and heat treatment to homogenise and then recrystallise the material [104, 105]. The range of such thermomechanical processing treatments that can be used is enormous: with many different combinations of different types of deformation and heat treatment available. In general, recrystallisation during heat treatment after deformation takes place in three stages: recovery, when the dislocations generated by deformation are reorganised into low-angle grain boundaries by a combination of cross slip and climb; recrystallisation by the nucleation of new undeformed, dislocation-free grains which grow and consume the old high-energy deformed grains and finally, grain growth or Ostwald ripening as large dislocation-free grains continue to grow, increasing the grain size and decreasing the overall grain-boundary energy [104, 105]. The resulting grain size, texture (the range of crystal orientations) and grain-boundary character (the spread of different types of grain boundary) depend in a complex way on the exact details of the thermomechanical processing that is used [104, 105].
There have been quite a few studies of recrystallisation, grain growth and texture in the original equiatomic five-component single-phase fcc Cantor alloy CrMnFeCoNi [106,107,108,109,110,111,112] and its equiatomic single-phase fcc quaternary (CrFeCoNi, MnFeCoNi and CrMnCoNi) and ternary (CrFeNi, CrCoNi, MnFeNi, MnCoNi and FeCoNi) subsystems [111, 111,113,114,115]. Recrystallisation and grain growth in these multicomponent materials are found to be considerably slower than in fcc pure metals and binary alloys, essentially caused by the wide range of local atomic structures and corresponding lattice strains, slowing the rates of atomic diffusion during heat treatment [106, 107, 111, 114]. Recrystallisation temperatures in multicomponent equiatomic single-phase fcc Cantor alloys are, therefore, in the range \({T}_{r}=\) 700–900 °C (973–1,173 K) for one hour anneals, as shown by the variation of hardness versus annealing temperature for several different alloys [114]. These correspond to homologous temperatures relative to the melting point \({T}_{r}/{T}_{m}=\) 0.6–0.75, depending on the particular alloy composition and the extent of the initial deformation treatment and subsequent heat treatment [114], considerably higher than more typical values in conventional pure metals and binary alloys of \({T}_{r}/{T}_{m}=\) 0.3–0.5. The resulting final recrystallised grain sizes are, as a consequence, very small and slow to coarsen, often no larger than 1–5 \(\mu\) m after 1 h at annealing temperatures of 700–800 °C (973–1,073 K) [106, 107, 111, 114] and much smaller than found in fcc nickel [114], independent of the extent of the initial deformation treatment [106, 107, 111, 114], though larger grain sizes can be achieved by annealing at higher temperatures.
Complex recrystallisation microstructures and textures are developed in multicomponent single-phase fcc Cantor alloys, often with a high fraction of twin boundaries, and the exact details depend on the extent of the initial deformation treatment and subsequent heat treatment, again because of the wide range of local atomic structures and associated lattice strains promoting the nucleation of different crystal orientations and twins during grain-boundary migration [106,107,108,109,110,111,112,113,114,115]. In general, similar results to those described above have also been found for recrystallisation and grain growth in other multicomponent high-entropy materials, such as single-phase fcc Al0.1CrFeCoNi [116] and CrFeCoNi2 [115] and two-phase fcc + bcc Al0.3CrFeCoNi [117] and Al0.5CrFeCoNi [118].
Surfaces and Catalysis
All real pieces of any material are bounded by external surfaces, with an excess surface energy caused (fairly obviously) by the surface atoms or molecules not being fully embedded within the crystal lattice and, therefore, not being fully bonded in the same way as the other atoms or molecules in the bulk of the material [71, 72]. Like grain boundaries, the local atomic structure at the surface of a material and the corresponding surface energy depend in a complicated way on the crystallography, orientation and topography of the surface [71, 72]. Many important processes and corresponding properties of a material, such as for instance wear, oxidation, corrosion and catalysis, depend intrinsically on its exposure to the external environment and are controlled, therefore, by the details of the variety of local atomic structures at its external surfaces. These processes and associated properties are, not surprisingly, fairly complex even in relatively simple single-phase single-component pure materials [71, 72] and, also not surprisingly, become much more complex again in multicomponent high-entropy materials, because of the enormous variety of different local atomic structures (similar to those discussed in previous sections of this paper).
From Table 1, there are almost twenty trillion (1.9 × 1013) different oriented first and second near-neighbour atomic clusters in the bulk of the original equiatomic five-component fcc Cantor alloy CrMnFeCoNi; and from Table 2, there are over thirty billion (3.1 × 1010) in the original equiatomic five-component Senkov alloy VNbMoTaW. Surface atoms are surrounded by fewer near neighbours than bulk atoms, the exact number varying from place to place on the surface depending on its crystallographic orientation and topography. We can approximate this reduction in number of near neighbours at the surface to an average of about a half, so the number of different oriented first and second near-neighbour surface atomic clusters is about 10 trillion (9.5 × 1012) and fifteen billion (1.5 × 1010) in the original equiatomic five-component fcc Cantor alloy CrMnFeCoNi and the original equiatomic five-component Senkov alloy VNbMoTaW, respectively. Including first near neighbours only, the numbers of surface atomic clusters are somewhat smaller but still significant, just over half a billion (6 × 108) and a million (106), respectively; but including third near neighbours the numbers are much larger again, 5.5 × 1029 and 3.7 × 1018, respectively. As with local atomic structures at vacancies, dislocations and grain boundaries, these numbers are very large indeed. Concentrating on oriented first and second near-neighbour surface atomic clusters, a square surface area of approximate linear dimension \(\sqrt{9.5\times {10}^{12}}=3.1\times {10}^{6}{\text{ clusters}}\approx 1{\text{mm}}\) and \(\sqrt{1.55\times {10}^{10}}=1.2{\times 10}^{5}{\text{ clusters}}\approx 40\mu {\text{m}}\) is needed for the original Cantor and Senkov alloys, respectively, in order to sample all possible surface atomic clusters, taking the atomic (and therefore cluster) separation as roughly 0.3 nm. To put this another way, there is an enormous range of different atomic structures across the surface of any multicomponent high-entropy solid-solution single-phase material, providing a correspondingly enormous variety of different sites where processes such as surface adsorption, oxidation, corrosion and catalysis can take place.
The variety of different atomic structures across the surface of multicomponent solid-solution single-phase metallic and ceramic materials has been shown to lead to considerable enhancements in catalytic efficiency for many different chemical reactions taking place at the surfaces of a variety of different multicomponent high-entropy materials [119,120,121,122]. As an example of multicomponent metallic catalysis, five-component fast-moving-bed-pyrolised (FMBP) FeCoPdIrPt nanoparticles on graphene oxide (FeCoPdIrPt/GO) were shown by Gao et al. [21] to provide considerably more efficient catalysis as the working electrode for the hydrogen evolution reaction (HER) in a solution of one molar potassium hydroxide (KOH) than either single, binary or ternary nanoparticles (such as Fe/GO, CoPd/GO or PdIrPt/GO) or conventional platinised graphite Pt/C. The multicomponent nanoparticles FeCoPdIrPt/GO operated at a much lower overvoltage than platinised carbon Pt/C, and with 26 times greater mass activity under equivalent conditions. As an example of multicomponent ceramic catalysis, Okejiri et al. [123] manufactured ruthenium-containing multicomponent perovskite nanoparticles based on barium zirconate BaZrO3 using a sonication process to precipitate the nanoparticles from an initial solution of multiple precursors. Ru0.13(BaSrBi)(ZrHfTiFe)O3 nanoparticles achieved ~ 50% conversion of CO to CO2 after < 1 s at 90 °C (363 K) compared with 50% conversion requiring a much higher temperature of 255 °C (528 K) for BaRuO3 even though it contains a much higher concentration of ruthenium.
Conclusions
We have shown that there are very extensive variations in local nanostructure, local atomic clusters and associated local lattice strains in multicomponent high-entropy solid-solution single-phase materials such as the fcc Cantor alloys, the bcc Senkov alloys and the rock-salt-structured Rost mono-oxides, even when there is no short-range ordering, i.e. when the solid solution is completely random or ideal. There are typically many billions of different local nanostructures and different local atomic clusters in well-known equiatomic five-component fully random solid-solution single-phase materials such as the original fcc Cantor alloy CrMnFeCoNi, the original bcc Senkov alloy VNbMoTaW and the original Rost oxide (MgCoNiCuZn)O, extending over distances typically of many microns, and with associated fluctuating hydrostatic and shear lattice strains typically of several percent. Moreover, the number and extent of the variations in local nanostructure and local atomic clusters increase dramatically with increasing number of components in the material. There are (perhaps surprisingly) large numbers of large single-atom clusters present even in fully random solid-solution single-phase materials.
We have also shown that there are similar variations in local nanostructure, local atomic clusters and associated local lattice strains surrounding point defects such as vacancies, line defects such as dislocations and planar defects such as grain boundaries and external surfaces. The variations in local nanostructure, atomic clusters and associated lattice strains surrounding vacancies, dislocation cores, grain boundaries and external surfaces influence many of the overall properties of multicomponent solid-solution single-phase materials, such as diffusion, plastic flow, recrystallisation and catalysis. The number and extent of the variations in the local nanostructure and local atomic clusters make it difficult to have much confidence in using ab initio and other atomistic computer modelling techniques to calculate the overall structure and, therefore, many of the properties of such multicomponent solid-solution single-phase materials, since such modelling techniques are inevitably restricted to relatively small assemblies of atoms that are unable to sample effectively the full range of local material structures and properties.
Figure Permissions
Figure 1: Fig 8 from Laurent-Brocq M, Akhatova A, Perrière L, Chebini S, Sauvage X, Leroy E. Insights into the phase diagram of the CrMnFeCoNi high entropy alloy. Acta Materialia, 2015, vol 88, pp 355-365. https://doi.org/10.1016/j.actamat.2015.01.068. Elsevier CCRL licence no 5761381364800. Permission granted 3/4/24.
Figure 7: Fig 4 from Zou Y, Maiti S, Steurer W, Spolenak R. Size-dependent plasticity in an Nb25Mo25TA25W25 refractory high-entropy alloy. Acta Materialia, 2014, vol 65, pp 85-97. https://doi.org/10.1016/j.actamat.2013.11.049. Elsevier CCRL licence no 5761380311613. Permission granted 3/4/24.
Figure 11: Fig 4 from Okamoto NL, Fujimoto S, Kambara Y, Kawamura M, Chen ZMT, Matsunoshita H, Tanaka K, Inui H, George EP. Size effect, critical resolved shear stress, stacking fault energy, and solid solution strengthening in the CrMnFeCoNi high-entropy alloy. Nature Scientific Reports, 2016, vol 6, paper number 35863. https://doi.org/10.1038/srep35893(2016). Open access – no permission required.
Figure 12: Fig 7 from Varvenne C, Luque A, Curtin WA. Theory of strengthening in fcc high entropy alloys. Acta Materialia, 2016, vol 118 pp 164-176. https://doi.org/10.1016/j.actamat.2016.07.040. Elsevier CCRL licence no 5761390545562. Permission granted 3/4/24.
Data availability
Data sets generated during the current study are available from the corresponding author on reasonable request.
Notes
In this paper, the term multicomponent material will be taken to mean any material containing three or more components, i.e. \(c\ge 3\), where c is the number of components in the material, in line with the normal meaning of multiple as more than one or two [1,2,3]. In this paper, the term high-entropy material will be taken to mean any material containing high concentrations of three or more components, i.e. c \(\ge\) 3, where the components are all in a random or near-random solid-solution single-phase structure, i.e. a high-entropy material is any concentrated single-phase solid-solution multicomponent material [1,2,3]. This is slightly different, but simpler and more logical than the common use of the terms high-entropy material and medium-entropy material to mean any material containing high concentrations of more than five components, i.e. c \(\ge\) 5, and between three and five components, i.e. 5 \(\ge\) c \(\ge\) 3, respectively.
In this paper the terms “local nanostructure” and “local atomic clusters” are used more or less interchangeably and synonymously. What is meant by “local atomic clusters” is explained more precisely later in the paper.
In this paper, we adopt the following conventions [1,2,3] when using chemical formulae to describe multicomponent materials: (1) for metallic alloys, atomic species are listed in order of ascending atomic number and not alphabetically (e.g. CrMnFeCoNi not CoCrFeMnNi for the original Cantor alloy); (2) for compounds, atomic species are again listed in order of ascending atomic number and not alphabetically, but separately for each sublattice, and in brackets where necessary (e.g. (MgCoNiCuZn)O and not (CoCuMgNiZn)O for the original Rost oxide); and (3) atomic ratios or atomic percent are given as subscripts to define the concentration of the different atomic species, with the subscripts left out for equiatomic alloy compositions (e.g. CrMnFeCoNi not Cr20Mn20Fe20Co20Ni20 and (MgFeCoNiCu)O not (Mg20Fe20Co20Ni20Cu20)O).
In a multicomponent material such as an fcc Cantor alloy or a bcc Senkov alloy, the ideal configurational entropy of mixing is, as described in the main text above, associated with a completely random distribution of the different atoms on all the lattice points of the solid-solution crystal structure, and is indeed given by Boltzmann’s Eq. 1, 3 as \({\Delta S}_{mix}^{ideal}=-R{\sum }_{i}{x}_{i}\text{ln }{x}_{i}\). But note that this is, of course, again as described in the main text above, the ideal configurational entropy of mixing per mole of material. In a multicomponent compound material such as a rock-salt-structured Rost mono-oxide, however, the ideal configurational entropy of mixing is not quite the same, and is now associated with a completely random distribution of only the different cations on only the cationic sublattice points of the solid-solution crystal structure. The ideal configurational entropy of mixing is still given by Boltzmann’s equation as \({\Delta S}_{mix}^{ideal}=-R{\sum }_{i}{x}_{i}\text{ln }{x}_{i}\), but note that this is now the configurational entropy of mixing per mole of cations.
Note that \({n}_{1}=z\), the normal coordination number.
We want to calculate how many ways we can pick \({n}_{1}+1\) (or, including second or second and third near neighbours, \({n}_{2}+{n}_{1}+1\) or \({n}_{3}+{n}_{2}+{n}_{1}+1\)) atoms from a total of c components. Let the atomic sites in the cluster be labelled 1, 2, 3, etc. up to \({n}_{1}+1\) (or \({n}_{2}+{n}_{1}+1\vee {n}_{3}+{n}_{2}+{n}_{1}+1\)). Different sets of the c components distributed across the different atomic sites are clearly different, but different arrangements of any given set of c components across the different atomic sites are also different, In other words, we need an ordered permutation rather than a non-ordered combination. And we need a permutation with repetition since each of the different component atoms can be picked more than once [36]. The result is that we can pick the first atom 1 in c ways, the second in c ways, the third in c ways, and so on up to the (\({n}_{1}+1\))th atom (or the (\({n}_{2}+{n}_{1}+1\)) th or (\({n}_{3}+{n}_{2}+{n}_{1}+1\)) th atom), so the number of atomic clusters is \({N}_{1}={c}^{{n}_{1}+1}\) (or \({N}_{2}={c}^{{{n}_{2}+n}_{1}+1}\) or \({N}_{3}={c}^{{{{n}_{3}+n}_{2}+n}_{1}+1}\)).
In any 19-atom cluster, there are 19 ways of selecting 18 atoms of the same type, i.e. 19 different sites that can be selected for one atom to be different; in each case, there are 5 ways of selecting the 18 atoms of the same type; and in each of these cases, there are 4 ways of selecting the different atom; so there are 4 × 19 = 76 clusters of each type and 5 × 4 × 19 = 380 clusters in total.
In any 19-atom cluster, there are 19 × 18 ways of selecting 17 atoms of the same type, i.e. 19 different sites that can be selected for the first atom to be different and 18 different sites that can be selected in each case for the second atom to be different; in each case, there are again 5 ways of selecting the 17 atoms of the same type; and in each of these cases, there are 4 × 4 ways of selecting the two different atoms; so there are 4 × 4 × 19 × 18 = 5,472 clusters of each type and 5 × 4 × 4 × 19 × 18 = 27,360 clusters in total.
Or, for a molecular crystal or a lattice with a basis, exactly on points that are all well defined in geometric relation to the lattice points.
It is not completely identical because of thermal expansion, i.e. the potential field within which the atoms vibrate is not perfectly symmetrical so there is a gradual slight increase in lattice parameter with increasing atomic vibration at increasing temperature.
Positive and negative values represent increased and decreased bond lengths respectively, corresponding to local atomic expansion and compression strains, respectively.
More accurately, the probability of finding a vacancy at any lattice point \({p}_{v}={{N}_{v}/N}_{o}=\text{exp }\left(-\Delta {G}_{v}/kT\right)=\text{exp }\left(-\left(\Delta {H}_{v}-T\Delta {S}_{v}\right)/kT\right)=\text{exp }\left(-\left(\Delta {E}_{v}+p\Delta {V}_{v}-T\Delta {S}_{v}\right)/kT\right),\) where \(\Delta {G}_{v}\), \(\Delta {H}_{v}\), \(\Delta {S}_{v}\) and \(\Delta {V}_{v}\) are the excess Gibbs free energy, enthalpy, entropy and volume of forming a vacancy, respectively, and p is the pressure. But the pressure term \(p\Delta {V}_{v}\) can be neglected as insignificant at atmospheric pressure compared to the formation energy \(\Delta {E}_{v}\), and \(\Delta {S}_{v}/k\approx 1\) so the pre-exponential factor \(\text{exp }\left(\Delta {S}_{v}/k\right)\) is a small numerical factor and can also be neglected (i.e. taken as \(\text{exp }\left(\Delta {S}_{v}/k\right)\approx 1\)) to a first approximation [57,58,59].
Note again that \({n}_{1}=z\), the coordination number.
We want again to calculate how many ways we can pick \({n}_{1}\) (or, including second or second and third near neighbours, \({n}_{2}+{n}_{1}$$or$${n}_{3}+{n}_{2}+{n}_{1}\)) atoms from a total of c components. Let the atomic sites in the cluster be labelled 1, 2, 3, etc. up to \({n}_{1}\) (or \({n}_{2}+{n}_{1}\vee {n}_{3}+{n}_{2}+{n}_{1}\)). Different sets of the c components distributed across the different atomic sites are clearly different, but different arrangements of any given set of c components across the different atomic sites are also different. In other words, we need an ordered permutation rather than a non-ordered combination. And we need a permutation with repetition since each of the different component atoms can be picked more than once [36].
Notice that these integrations are not simply over the range of wells and saddle points that exist in the material, but instead have to be over the somewhat different range of wells and saddle points actually sampled by a vacancy as it hops throughout the material.
It is not, perhaps, entirely fanciful to compare the total information content coded in an array of twenty trillion different local atomic clusters along a 5.7 km length of a dislocation line coiled up in half a cubic centimetre of the original Cantor alloy, with a typical 100 micron diameter human cell containing 46 chromosomes each of which is a DNA molecule with 50–200 million base pairs, making a total of approximately three billion letters in the genetic script describing and controlling all aspects of human life [57].
References
B. Cantor, The Fundamentals of Multicomponent High-Entropy Materials (Oxford University Press, Oxford, 2024)
B.S. Murty, J.W. Yeh, S. Ranganathan, P.P. Bhattacharjee, High-Entropy Alloys, 2nd edn. (Elsevier, Amsterdam, 2019)
B. Cantor, Multicomponent high-entropy Cantor alloys. Prog. Mater. Sci. 120, 100754 (2021)
B. Cantor, I.T.H. Chang, P. Knight, A.J.B. Vincent, Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A375–377, 213–218 (2004)
J.W. Yeh, S.K. Chen, S.J. Lin, J.Y. Gan, T.S. Chin, T.T. Shun, C.H. Tsau, S.Y. Chang, Nanostructured high entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299–303 (2004)
O.N. Senkov, G.B. Wilks, D.B. Miracle, C.P. Chuang, P.K. Liaw, Refractory high-entropy alloys. Intermetallics 18, 1758–1765 (2010)
O.N. Senkov, D.B. Miracle, K.J. Chaput, Development and exploration of refractory high entropy alloys—a review. J. Mater. Res. 33, 3092–3218 (2018)
D.B. Miracle, M.H. Tsai, O.N. Senkov, V. Soni, R. Banerjee, Refractory high entropy superalloys. Scripta Mater. 187, 445–452 (2020)
G.M. Muralikrishna, A.C.M. Esther, K Guruvidyathri, P Watermeyer, C.. Liebscher, K.N. Kulkarni, G. Wilde, S.V. Divinski, B.S. Murty, Novel multicomponent B2-ordered aluminides: compositional design, synthesis, characterization and thermal stability. Metals 10, paper 1411 (2020)
C.M. Rost, E. Sachet, T. Borman, A. Moballegh, E.C. Dickey, D.Hou, J.L. Jones, S. Curtarolo, J.P. Maria. Entropy-stabilized oxides. Nat. Commun. 6, paper number 8485 (2015)
A. Takeuchi, K. Amiya, T. Wada, K. Yubuta, W. Zhang, High-entropy alloys with a hexagonal close-packed structure designed by equi-atomic alloy startegy and binary phase diagrams. J. Metals 66, 1984–1992 (2014)
J.W. Qiao, M.L. Bao, Y.J. Zhao, H.J. Yanh, Y.C. Wu, Y. Zhang, J.A. Hawk, M.C. Gao, Rare-earth high entropy alloys with hexagonal close-packed structure. J. Appl. Phys. 124, paper number 195101 (2018)
A. Takeuchi, T. Wada, Y. Zhang, MnFeNiCuPt and MnFeNiCuCo high-entropy alloys designed based on L10 structure in Pettifor map for binary compounds. Intermetallics 82, 107–115 (2017)
S. Wang, S. Chen, Y. Jia, Z. Hu, H. Huang, Z. Yang, A. Dong, G. Zhu, D. Wang, D. Shu, F. Tian, Y. Dai, B. Sun, Fcc-L12 ordering transformation in equimolar FeCoNiV multi-principal element alloy. Mater Design 168, paper number 107648 (2019)
K.B. Kim, P.J. Warren, B. Cantor, Formation of metallic glasses in novel (Ti33Zr33Hf33)((100-x-y)(Ni50Cu50)(x)Al(y) alloys. Mater. Trans. 44, 411–413 (2003)
K.B. Kim, P.J. Warren, B. Cantor, Crystallization behaviour of novel (TiZrHf)100-x-y (NiCu)x Aly with x = 48–55. J Metastable Nanocrystalline Mater 24–25, 657–660 (2005)
R.Z. Zhang, M.J. Reece, Review of high entropy ceramics: design, synthesis, structure and properties. J Mater Chem A 7, 22148–22162 (2019)
A. Sarkar, B. Breitung, H. Hahn, High entropy oxides: the role of entropy, enthalpy and synergy. Scripta Mater. 187, 43–48 (2020)
H. Xiang, Y. Xing, F.Z. Dai, H. Wang, L. Su, L. Miao, G. Zhang, Y. Wang, X. Qi, L. Yao, H. Wang, B. Zhao, J. Li, Y. Zhou, High-entropy ceramics: present status, challenges, and a look forward. J Adv Ceram 10, 385–441 (2021)
Y. Yao, Z. Huang, P. Xie, S.D. Lacey, R.J. Jacob, H. Xie, F. Chen, A. Nie, T. Pu, M. Rehwoldt, D. Yu, M.R. Zachariah, C. Wang, R.S. Yassar, J. Li, L. Hu, Carbothermal shock synthesis of high-entropy alloy nanoparticles. Science 359, 1489–1494 (2018)
S. Gao, S. Hao, Z. Huang, Y. Yuan, S. Han, L. Lei, X. Zhang, R. Shahbazian-Yassar, J. Lu, Synthesis of high-entropy alloy nanoparticles on supports by the fast moving bed pyrolysis. Nat. Commun. 11, paper number 2016 (2020)
E.J. Pickering, R. Muñoz-Moreno, H.J. Stone, N.G. Jones, Precipitation in the equiatomic high-entropy alloy CrMnFeCoNi. Scripta Mater. 113, 106–109 (2016)
F. Otto, A. Diouhy, K.G. Pradeep, M. Kubenova, D. Raabe, G. Eggeler, E.P. George, Decomposition of the single-phase high-entropy alloy CrMnFeCoNi after prolonged anneals at intermediate temperatures. Acta Mater. 112, 40–52 (2016)
G. Bracq, M. Laurent-Brocq, C. Varvenne, L. Perrière, W.A. Curtin, J.-M. Joubert, I. Guillot, Combining experiments and modelling to explore the solid solution strengthening of high and medium entropy alloys. Acta Mater. 177, 266–279 (2019)
J.M. Cowley, An approximate theory of order in alloys. Phys. Rev. 77, 669–675 (1950)
D. De Fontaine, The number of independent pair-correlation functions in multicomponent systems. J. Appl. Crystallogr. 4, 15–19 (1971)
A.V. Ceguerra, R C. Powles, M.P. Moody, S.P. Ringer, Quantitative description of atomic architecture in solid solutions: a generalized theory for multicomponent short-range order. Phys. Rev. B 82, paper number 132201 (2010)
F.X. Zhang, S. Zhao, K. Jin, H. Xue, G. Velisa, H. Bei, R. Huang, J.Y.P. Ko, D.C. Pagan, J.C. Neuefeind, W.J. Weber, Y. Zhang, Local structure and short-range order in a NiCoCr solid solution alloy. Phys. Rev. Lett. 118, paper number 205501 (2017)
R. Zhang, S. Zhao, J. Ding, Y. Chong, T. Jia, C. Ophus, M. Asta, R.O. Ritchie, A.M. Minor, Short-range order and its impact on the CoCrNi medium-entropy alloy. Nature 581, 283–287 (2020)
D. Shinde, S. Fritze, M. Thuvander, P. Malinovskis, L. Riekehr, U. Jansson, K. Stiller, Elemental distribution in CrNbTaTiW-C high-entropy alloy thin films. Microsc. Microanal. 25, 489–500 (2019)
R. Hu, S. Jin, G. Sha, Application of atom probe tomography in understanding high entropy alloys: 3D local chemical compositions in atomic scale analysis. Progress Mater. Sci. 123, paper number 100854 (2022)
A. Ferrari, B. Dutta, K. Gubaev, Y. Ikeda, P. Srinivasan, B. Grabowski, F. H. W. Körmann, Frontiers in atomistic simulations of high entropy alloys. J. Appl. Phys. 128, paper number 150901 (2020)
M. Laurent-Brocq, A. Akhatova, L. Perrière, S. Chebini, X. Sauvage, E. Leroy, Y. Champion, Insights into the phase diagram of the CrMnFeCoNi high entropy alloy. Acta Mater. 88, 355–365 (2015)
Y. Muniandy, M. He, M. Eizadjou, E.P. George, J.J. Kruzic, S.P. Ringer, B. Gludovatz, Compositional variations in equiatomic CrMnFeCoNi high-entropy alloys. Mater. Characterization 180, paper number 111437 (2021)
M.J. Yao, K.J. Pradeep, C.C. Tasan, D. Raabe, A novel, single phase, non-equiatomic FeMnNiCoCr high-entropy alloy with exceptional phase stability and tensile ductility. Scripta Mater. 72–73, 5–8 (2014)
K. M. Koh, C.C. Chen, Principles and Techniques in Combinatorics. (World Scientific, Singapore, 1992), ch 1, pp 1–50
F.C. Philips, An Introduction to Crystallography (Wiley, New York, 1973)
A. Kelly, K.M. Knowles, Crystallography and Crystal Defects (Wiley, New York, 2012)
Z.H. Barber (ed.), Introduction to Materials Modelling (Maney, London, 2005)
F. Giustino, Materials Modelling Using Density Functional Theory (Oxford University Press, Oxford, 2014)
J.L. Vegard, Die konstitution der mischkristalle und die raumfüllung der atome. Z. Phys. 5, 17–26 (1921)
J.C. Alonso, O.C. Alonso, Derivation of unit cell volume and lattice parameter of cubic high entropy alloys from volume size factors. Intermetallics 137, paper number 107299 (2021)
N.N. Greenwood, A. Earnshaw, Chemistry of the Elements (2nd edition). (Butterworth-Heinemann, 1997).
N.L. Okamoto, K. Yuge, K. Tanaka, H. Inui, E.P. George, Atomic displacement in the CrMnFeCoNi high entropy alloy: a scaling factor to predict solid solution strengthening. Am. Inst. Phys. 6, number 125008 (2016)
H.S. Oh, D. Ma, G.P. Leyson, B. Grabowski, E.S. Park, F. Körmann, D. Raabe, Lattice distortions in the FeCoNiCrMn high entropy alloy studied by theory and experiment. Entropy 18, 321–329 (2016)
H. Song, F. Tian, Q.M. Hu, L. Vitos, Y. Wang, J. Shen, N. Chen, Local lattice distortions in high-entropy alloys. Phys. Rev. Mater. 1, number 023404 (2018)
C.C. Yen, G.R. Huang, Y.C. Huang, H.W. Yeh, D.J. Luo, K.T. Hsieh, E.W. Huang, J.W. Yeh, S.J. Lin, C.C. Wang, C.L. Kuo, S.Y. Chang, Y.C. Lo, Lattice distortion effect on elastic anisotropy of high entropy alloys. J. Alloys Compd 818, number 152876 (2020)
L.R. Owen, E.J. Pickering, H.Y. Playford, H.J. Stone, M.G. Tucker, N.G. Jones, An assessment of the lattice strain in the CrMnFeCoNi high-entropy alloy. Acta Mater. 122, 11–18 (2017)
Y.Y. Tan, M.Y. Su, Z.C.Xie, Z.J. Chen, Y. Gong, L.R. Zheng, Z. Shi, G. Mo, Y. Li, L.W. Li, H.Y. Wang, L.H. Dai, Chemical composition dependent local lattice distortions and magnetism in high entropy alloys. Intermetallics 129, paper number 107050 (2021)
Y. Tong, K. Jin, H. Bei, J.Y.P. Ko, D.C. Pagan, Y. Zhang, F.X. Zhang, Local lattice distortion in NiCoCr, FeCoNiCr and FeCoNiCrMn concentrated alloys investigated by synchrotron X-ray diffraction. Mater. Des. 155, 1–7 (2018)
Y. Tong, S. Zhao, K. Jin, H. Bei, J.P.Y. Ko, Y. Zhang, F.X. Zhang, A comparison study of local lattice distortion in Ni80Pd20 binary alloy and FeCoNiCrPd high entropy alloy. Scripta Mater. 156, 14–18 (2018)
H. Ge, F. Tian, A review of ab initio calculation on lattice distortion in high-entropy alloys. J. Metals 71, 4225–4237 (2019)
Y. Zou, S. Maiti, W. Steurer, R. Spolenak, Size-dependent plasticity in an Nb25Mo25TA25W25 refractory high-entropy alloy. Acta Mater. 65, 85–97 (2014)
W. Guo, W. Dmowski, J.Y. Noh, P. Rack, P.K. Liaw, T. Egami, Local atomic structure of a high-entropy alloy: an X-ray and neutron scattering study. Metallurgical and Materials Transactions 44A, 1994–1997 (2013)
S. Maiti, W. Steurer, Structural disorder and its effect on mechanical properties in single-phase TaNbHfZr high-entropy alloy. Acta Mater. 106, 87–97 (2016)
R.D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallographia 32A, 751–767 (1976)
B. Cantor, The Equations of Materials (Oxford University Press, Oxford, 2020)
R. Abbaschian, L. Abbaschian, R.E. Reed-Hill. Physical Metallurgy Principles (4th edition). (Nelson Engineering, 2009), chapters 7 and 12, pp 194–215 and 348–388
P. Shewmon, Diffusion in Solids, 2nd edn. (Springer, New York, 2016)
S.L. Thomas, S. Patala, Vacancy diffusion in multi-principal element alloys: the role of chemical disorder in the ordered lattice. Acta Mater. 196, 144–153 (2020)
J. Kottke, D. Utt, M. Laurent-Brocq, A. Fareed, D. Gaertner, L. Perrière, L. Rogal, A. Stukowski, K. Albe, S.V. Divinski, Experimental and theoretical study of tracer diffusion in a series of (CoCrFeMn)100-xNix alloys. Acta Mater. 194, 236–248 (2020)
W.M. Choi, Y.H. Jo, S.S. Sohn, S. Lee, B. Lee, NPJ Computing Materials 4, 1 (2018)
Y. Li, R. Li, Q. Peng, S. Ogata, Reduction of dislocation, mean free path, and migration barriers using high entropy alloy: insights from the atomistic study of irradiation damage of CoNiCrFeMn. Nanotechnology 31, paper number 425761 (2020)
K.Y. Tsai, M.H. Tsai, J.W. Yeh, Sluggish diffusion in CoCrFeMnNi high-entropy alloy. Acta Mater. 61, 4887–4897 (2013)
M. Vaidya, K.G. Pradeep, B.S. Murty, G. Wilde, S.V. Divinsky, Bulk tracer diffusion in CoCrFeNi and CoCrFeMnNi high entropy alloys. Acta Mater. 146, 211–224 (2018)
M. Vaidya, S. Trubel, B.S. Murty, G. Wilde, S.V. Divinsky, Ni tracer diffusion in CoCrFeNi and CoCrFeMnNi high entropy alloys. J. Alloy. Compd. 688, 994–1001 (2016)
Q. Li, W. Chen, J. Zhong, L. Zhang, Q. Chen, Z. Liu, On sluggish diffusion in fcc AlCoCrFeNi high-entropy alloys: an experimental and numerical study. Metals 8, article number 16 (2018)
J. Dabrowa, M. Danielewsky, State-of-the-art diffusion studies in the high entropy alloys. Metals 10, 347–391 (2020)
W. Cai, W.D. Nix. Imperfections in crystalline solids. (Cambridge University Press, Cambridge), 2016, part III, pp 209–432.
D. Hull, D.J. Bacon, Introduction to Dislocations (5th edition). (Butterworth-Heinemann, Elsevier, Oxford), 2011, chapters 8–10, pp 157–250
M.F. Ashby, D.R.H. Jones. Engineering Materials: An Introduction to Properties, Application and Design (4th edn.) (Butterworth-Heinemann, Elsevier, Oxford, 2012), chapters 9 and 10, pp 135–156
J.W. Martin, Materials for Engineering. (Institute of Materials, London, 1996), ch 3, pp 69–78.
J. Lu, L. Hultman, E. Holmström, K.H. Antonsson, M. Grehk, W. Li, L. Vitos, A. Golpayegani, Stacking fault energies in stainless steels. Acta Mater. 97, 305–315 (2015)
H.M. Otte, R.P.I. Adler, Formation of stacking faults in polycrystalline brass during tensile deformation. Phil. Mag. 16, 239–252 (1966)
A. Rohatgi, K. Vecchio, G. Gray, The influence of stacking fault energy on the mechanical behaviour of Cu and Cu-Al alloy: deformation twinning, work hardening and dynamic recovery. Metall. Trans. A 32, 135–145 (2001)
Z. Li, S. Zhao, R.O. Ritchie, M.A. Meyers, Mechanical properties of high-entropy alloys with emphasis on face-centered cubic alloys. Prog. Mater. Sci. 102, 296–345 (2019)
E.P. George, W.A. Curtin, C.C. Tasan, High entropy alloys: A focused review of mechanical properties and deformation mechanisms. Acta Mater. 188, 435–474 (2020)
W. Li, D.Xie, D. Li, Y. Zhang, Y. Gao, P. K. Liaw, Mechanical behaviour of high-entropy alloys. Progress Mater. Sci. 118, paper number 100777 (2021)
N.L. Okamoto, S. Fujimoto, Y. Kambara, M. Kawamura, Z.M.T. Chen, H. Matsunoshita, K. Tanaka, H. Inui, E.P. George, Size effect, critical resolved shear stress, stacking fault energy, and solid solution strengthening in the CrMnFeCoNi high-entropy alloy. Nat. Sci. Rep. 6, paper number 35863 (2016)
T.M. Smith, M.S. Hooshmand, B.D. Esser, F. Otto, D.W. McComb, E.P. George, M. Ghazisaeidi, M.J. Mills, Atomic-scale characterization and modelling of 60° dislocations in a high-entropy alloy. Acta Mater. 110, 352–363 (2016)
G. Laplanche, A. Kostka, C. Reinhart, J. Hunfeld, G. Eggeler, E.P. George, Reasons for the superior mechanical properties of medium-entropy CrCoNi compared to high-entropy CrMnFeCoNi. Acta Mater. 128, 292–303 (2017)
X.D. Xu, P. Liu, Z. Tang, A. Hirata, S.X. Song, T.G. Nieh, P.K. Liaw, C.T. Liu, M.W. Chen, Transmission electron microscopy characterization of dislocation structure in a face-centred cubic high-entropy alloy Al0.1CoCrFeNi. Acta Materialia 144, 107–115 (2018)
R. Pasianot, D. Farkas, Atomistic modeling of dislocations in a random quinary high-entropy alloy. Comput. Mater. Sci. 173, paper number 109366 (2020)
S.I. Rao, C. Woodward, T.A. Parthasarathy, O.N. Senkov, Atomistic simulations of dislocation behaviour in a model fcc multicomponent concentrated solid solution alloy. Acta Mater. 134, 188–194 (2017)
X. Liu, Z. Pei, M. Eisenbach, Dislocation core structures and Peierls stresses of the high entropy alloy NiCoFeCrMn and its subsystems. Mater. Design 180, paper number 107955 (2019)
J.P. Couzinie, L. Lilensten, Y. Champion, G. Dirras, L. Perrière, I. Guillot, On the room temperature deformation mechanisms of a TiZrHfNmMo refractory high-entropy alloy. Mater. Sci. Eng. A 645, 255–263 (2015)
L. Lilensten, J.P. Couzinie, L. Perrière, A. Hocini, C. Keller, G. Dirras, I. Guillot, Study of a bcc multi-principal element alloy: tensile and simple shear properties and underlying deformation mechanism. Acta Mater. 142, 131–141 (2018)
F. Wang, G.H. Balbus, S. Xu, Y. Su, J. Shin, P.F. Rottmann, K.E. Knipling, J.C. Stinville, L.H. Mills, O.N. Senkov, I.J. Beyerlein, T.M. Pollock, D.S. Gianola, Multiplicity of dislocation pathways in a refractory multiprincipal element alloy. Science 370, 95–101 (2020)
C. Lee, F. Maresca, R. Feng, Y. Chou, T. Ungar, M. Widom, K. An, J.D. Poplawsky, Y C. Chou, P.K. Liaw, W.A. Curtin, Strength can be controlled by edge dislocations in refractory high-entropy alloys. Nat. Commun. paper number 5474 (2021)
S.I. Rao, B. Akdim, E. Antillon, C. Woodward, T.A. Parthasarathy, O.N. Senkov, Modeling solution hardening in bcc refractory complex concentrated alloys: NbTiZr, Nb1.5TiZr0.5 and Nb0.5TiZr1.5. Acta Materialia 168, 222–236 (2019)
S. Yin, J. Ding, M. Asta, R.O. Ritchie, Ab initio modeling of the energy landscape for screw dislocations in body-centred cubic high-entropy alloys. NPJ Computat. Mater. 6, paper number 110 (2020)
L.T.W. Fey, S. Xu, Y. Su, A. Hunter, I.J. Beyerlein. Transitions in the morphology and critical stresses of gliding dislocations in multiprincipal element alloys. Phys. Rev. Mater. 6, paper number 013605 (2022)
X. Zhou, S. He, J. Marian. Cross-kinks control screw dislocation strength in equiatomic bcc refractory alloys. Acta Materialia vol 211, paper number 116875 (2021)
C. Varvenne, A. Luque, W.A. Curtin, Theory of strengthening in fcc high entropy alloys. Acta Materialia 118, 164–176 (2016)
C. Varvenne, G.P.M. Leyson, M. Ghazisaeidi, W.A. Curtin, Solute strengthening in random alloys. Acta Materialia 124, 660–683 (2017)
C. Varvenne, W.A. Curtin, Strengthening of high entropy alloys by dilute solute additions: CoCrFeNiAlx and CoCrFeNiMnAlx alloys. Scripta Materialia 138 92–95 (2017)
S. Sohn, Y. Liu, J. Liu, P. Gong, S. Prades-Rodel, A. Blatter, B.E. Scanlon, C.C. Broadbridge, J. Schroers, Noble metal high entropy alloys. Scripta Mater. 126, 29–32 (2017)
B. Yin, W.A. Curtin, First-principles-based prediction of yield strength in the RhIrPdPtNiCu high-entropy alloy. NPJ Comput. Mater. 5, paper number 14 (2019)
F. Maresca, W.A. Curtin, Theory of screw dislocation strengthening in random bcc alloys from dilute to high-entropy alloys. Acta Mater. 182, 144–162 (2020)
Q. Lin, X. An, H. Liu, Q. Tang, P. Dai, X. Liao, In-situ high-resolution transmission electron microscopy investigation of grain boundary dislocation activities in a nanocrystalline CrMnFeCoNi high-entropy alloy. J. Alloy. Compd. 709, 802–807 (2017)
P. Wynblatt, D. Chatain, Modeling grain boundary and surface segregation in multicomponent high-entropy alloys. Phys. Rev. Mater 3, paper number 054004 (2019)
D. Farkas, Grain boundary structure in high-entropy alloys. J. Mater. Sci. 55, 9173–9183 (2020)
H. Lee, M. Shabani, G.J. Pataky, F. Abdeljawad, Tensile deformation behavior of twist grain boundaries in CoCrFeMnNi high entropy bicrystals. Nat. Sci. Rep. 11, paper number 428 (2021)
R.D. Doherty, D.A. Hughes, F.J. Humphreys, J.J. Jonas, D.J. Jensen, M.E. Kassner, W.E. King, T.R. McNelley, H.J. McQueen, A.D. Rollett, Current issues in recrystallization: a review. Mater. Sci. Eng. A 238, 219–274 (1997)
J. Humphreys, G.S. Rohrer, A. Rollett, Recrystallization and related annealing phenomena, 3rd edn. (Elsevier, Amsterdam, 2017)
W.H. Liu, Y. Wu, J.¥. He, T.G. Nieh, Z.P. Lu. Grain growth and the Hall–Petch relationship in a high-entropy FeCrNiCoMn alloy. Scripta Materialia 68, 526–529 (2013)
P.P. Bhattacharjee, G.D. Sathiaraj, M. Zaid, J.R. Gatti, C. Lee, C.W. Tsai, J.Y. Yeh, Microstructure and texture evolution during annealing of equiatomic CoCrFeMnNi high-entropy alloy. J. Alloy. Compd. 587, 544–552 (2014)
F. Otto, N.L. Hanold, E.P. George, Microstructural evolution after thermomechanical processing in an equiatomic single-phase CoCrFeMnNi high-entropy alloy with special focus on twin boundaries. Intermetallics 54, 39–48 (2014)
G. Laplanche, O. Horst, G. Eggeler, E.P. George, Microstructural evolution of a CoCrFeMnNi alloy after swaging and annealing. J. Alloy. Compd. 647, 548–557 (2015)
G.D. Sathiaraj, P.P. Bhattacharjee, Analysis of microstructure and microtexture during grain growth in low stacking fault energy equiatomic CoCrFeMnNi high entropy and Ni-60wt%Co alloys. J. Alloy. Compd. 637, 267–276 (2015)
B.R. Chen, A.C. Yeh, J.W. Yeh, Effect of one-step recrystallization on the grain boundary evolution of CoCrFeMnNi high entropy alloy and its subsystems. Nat. Sci. Rep. 6, paper number 22306 (2016)
J. Gu, S. Ni, Y. Liu, M. Song, Regulating the strength and ductility of a cold rolled FeCrCoMnNi high-entropy alloy via annealing treatment. Mater. Sci. Eng. A 755, 289–294 (2019)
P. Shiyamoorthi, P. Asghari-Rad, J.W. Bae, H.S. Kim, Fine tuning of tensile properties in CrCoNi medium entropy alloy through cold rolling and annealing. Intermetallics 113, paper number 106578 (2019)
Z. Wu, H. Bei, F. Otto, G.M. Pharr, E.P. George, Recovery, recrystallization, grain growth and phase stability of a family of fcc-structured multicomponent equiatomic solid solution alloys. Intermetallics 46, 131–140 (2014)
Z. Yang, F. He, Q. Wu, K. Zhang, D. Cui, B. Guo, B. Han, J. Li, J. Wang, Z. Wang, Distinct recrystallization kinetics in NiCoCrFe-based single-phase high-entropy alloys. Metall. and Mater. Trans. A. 52, 3799–3810 (2021)
S.W. Wu, L. Xu, X.D. Ma, Y.F. Jia, Y.K. Mu, Y.D. Jia, G. Wang, C.T. Liu, Effect of annealing temperatures on microstructures and defprmation behavior of Al0.1CrFeCoNi high-entropy alloy. Mater. Sci. Eng. 805, paper number 140523 (2021)
X. Wang, Z. Zhang, Z. Wang, X. Ren, Microstructural evolution and tensile properties of Al0.3CoCrFeNi high-entropy alloy associated with B2 precipitates. Materials 15, paper number 1215 (2022)
T. Guo, J. Li, J. Wang, W.Y. Wang, Y. Liu, X. Luo, H. Kou, E. Beaugnon. Microstructure and properties of bulk Al0.5CoCrFeNi high-entropy alloy by cold rolling and subsequent annealing. Mater. Sci. Eng. A729, 141–148 (2018)
Y. Zhang, D. Wang, S. Wang, High-entropy alloys for electrocatalysis: design, characterization and applications. Small 18, paper number 2104339 (2022)
K. Kusada, M. Mukoyoshi, D. Wu, H. Kitagawa, Chemical synthesis, characterization and properties of multi-element nanoparticles. Angew. Chem. 61, paper number 09616 (2022)
H. Li, J. Lai, Z. Li, L. Wang, Multi-sites electrocatalysis in high-entropy alloys. Adv. Funct. Mater. 31, paper number 2106715 (2021)
B. Wang, Y. Yao, X. Yu, C. Wang, C. Wu, Z. Zou, High entropy alloys: from theory to experiment. J. Mater. Chem. A 9, 19410–19438 (2021)
F. Okejiri, Z. Zhang, J. Liu, M. Liu, S. Yang, S. Dai, Room-temperature synthesis of high-entropy perovskite oxide nanoparticle catalysts through ultrasonication-based method. Chemsuschem 13, 111–115 (2019)
Acknowledgements
The author wishes to thank the Department of Materials at the University of Oxford and the Brunel Centre for Advanced Solidification Technology (BCAST) at Brunel University London for the provision of laboratory and office facilities.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author has no conflicts of interest. He received laboratory and office support from the Department of Materials, University of Oxford and Brunel Centre for Advanced Casting Technology (BCAST), Brunel University London.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cantor, B. Local Nanostructure in Multicomponent High-Entropy Materials. High Entropy Alloys & Materials (2024). https://doi.org/10.1007/s44210-024-00040-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44210-024-00040-4