
Abstract
Glioblastoma accounts for almost half of all intracranial primary malignancies and has the worst prognosis. Because of its high malignancy and frequent recurrence after standard therapy, it is of great significance to explore new therapy options. Recently immune therapy has taken remarkable progress in a variety of tumors, among which peptide vaccines utilize peptide sequences based on tumor-specific antigens or tumor-associated antigen targets to activate self-immune response against tumor cells. However, due to the particularity of intracranial central nervous system tumors, the application of peptide vaccines in glioblastoma still faces challenges. This article mainly reviews the immune basis and important clinical trial results of peptide vaccine therapy for GBM, analyzes the reasons for its poor efficacy, and proposes the development direction of peptide vaccines for the unique challenges of immunotherapy in GBM. An in-depth understanding and elaboration of the application and related issues of peptide vaccine in the treatment of GBM will help to formulate relevant treatment strategies in future clinical and basic research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Glioblastoma multiforme (GBM) accounts for 48.6% of all intracranial primary malignancies and is the most common primary central nervous system malignancy in adults [1]. The current standard treatment regimen for GBM (Stupp regimen) includes maximal safety resection, concurrent chemoradiotherapy with temozolomide (TMZ), and adjuvant chemotherapy with TMZ. However, after standard treatment, the prognosis of patients remains poor, with a median overall survival of approximately 15 months, a 5-year survival rate of less than 10%, and a recurrence rate close to 100% [2]. In the past 10 years, although there have been more in-depth studies on the occurrence and development mechanism of glioblastoma, the efficacy of clinical therapy remains poor. Therefore, it is of great significance to explore new treatment options.
In recent years, there has been remarkable research progress in immunotherapy in a variety of tumor fields. Immunotherapy refers to mobilizing the body’s immune system through active or passive methods, inhibiting tumor cell proliferation and inducing tumor cell apoptosis, thereby exerting a therapeutic effect. It mainly includes oncolytic viruses, peptide vaccines, cellular immunotherapy (dendritic cell vaccines and adoptive immune cell therapy) and immune checkpoint inhibitors. Although breakthroughs have been made in immunotherapy in clinical research, the immunotherapy of glioma still faces many challenges due to the particularity of intracranial central nervous system tumors [3].
Peptide vaccines utilize peptide sequences based on tumor-specific antigens or tumor-associated antigen targets, which are highly specific and easy to generate antigens but less immunogenic. To improve immunogenicity, it can be combined with immune adjuvants, or combined with other immune and non-immunotherapy treatments. Polypeptide vaccines include single-target vaccines targeting, multi-target composite vaccines, and individualized vaccines. This article mainly reviews the immune basis and important clinical trial results of peptide vaccine therapy for GBM, analyzes the reasons for its poor efficacy, and proposes the development direction of peptide vaccines for the unique challenges of immunotherapy in GBM. An in-depth understanding and elaboration of the application and related issues of peptide vaccine in the treatment of GBM will help to formulate relevant treatment strategies in future clinical and basic research.
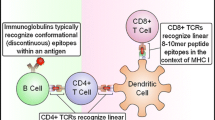
1 Immune microenvironment of GBM
GBM has an inhibitory immune microenvironment, and tumor-infiltrating immune cells mainly include lymphocytes and myeloid cells (Fig. 1). Glioblastoma-associated macrophages (GAM) are the most infiltrating myeloid immune cells, accounting for approximately 30% of the total tumor volume [4]. GAM exhibits an M2 phenotype, and the expression of IL-6 receptor and Fas receptor ligands on its cell surface is up-regulated, which can maintain its M2 phenotype and induce apoptosis of tumor-infiltrating lymphocytes. At the same time, GAM also secretes a large amount of macrophage colony-stimulating factor (M-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) to stimulate the division and proliferation of tumor cells [5] and secretes a large number of inhibitory cytokines such as IL − 6 and TGF-β, inhibiting effective immune response [6], thereby promoting the proliferation and development of tumor cells.

The immune micronenvironment of GBM (Treg: T regular cell, TCR: T cell receptor)
In addition, numerous studies have demonstrated the presence of lymphocytic infiltration in glioblastoma, including CD4+ helper T cells, CD8+ cytotoxic T cells, and regulatory T cells, where helper T cells are more numerous than cytotoxic T cells and cytotoxic T cells are in a state of exhaustion [7]. Depleted cytotoxic T cells upregulate the expression of cell surface co-inhibitory receptors PD-1 and CTLA-4, thereby inhibiting the activation of T cells by MHC antigen complexes and hindering T cell targeting to tumor antigens [8]. On the contrary, a large number of active regulatory T cells are recruited around the tumor by cytokines in the immune microenvironment, and secrete large amounts of TGF-β and IL-10, thereby inhibiting pro-inflammatory factor secretion of cytotoxic T cells and preventing the antigen presentation process [9, 10]. At the same time, glioblastoma cells themselves secrete a large number of immunosuppressive factors, such as IDO, to recruit and activate amounts of regulatory T cells [11]. Tumor cells also up-regulate the expression of co-inhibitory molecules on their surface, such as PD-L1 and HLA-G [12], thereby inhibiting the immune-killing effects of cytotoxic T cells and NK cells. In addition, tumor cells also down-regulate their own MHC expression levels to reduce the likelihood that tumor antigens will be presented [5].
Therefore, immunotherapy is to relieve the inhibitory immune microenvironment of glioblastoma, enhance the immunogenicity of tumor cells, and activate the patient’s effective immune response to tumor cells, thereby inhibiting the growth and reproduction of tumor cells, generating killing effects. Among them, the peptide vaccine is an active immunotherapy method, which constructs 8–25 amino acid polypeptides according to the special antigen sequence of tumor cells, which is used to activate the immune response of the body to achieve the purpose of killing tumor cells. Immune presenting cells present antigenic peptides through the major histocompatibility complex (MHC) on the surface, which mainly activates the cellular immune response and has little effect on humoral immunity.
2 Peptide vaccine targets and current status of drugs in clinical trials
The most important issue in the design of peptide vaccines is to select appropriate target antigens. The ideal target antigen is tumor-specific antigen, which is only expressed in tumor cells and not expressed in normal tissues, also called neoantigen, such as epidermal growth factor receptor variant III (EGFRvIII). The other is tumor-associated antigen, which is abundantly expressed in tumor cells, but at low levels in normal tissues, such as cellular anti-apoptotic protein (survivin) overexpressed in GBM [13]. Since a single peptide molecule has almost no immunogenicity and requires carrier protein binding, keyhole limpet hemocyanin is often used as a carrier protein, which has the effect of an immune agonist. The use of polypeptide vaccines alone can induce immune tolerance to antigenic epitopes, and immune adjuvants are also required to improve the immunogenicity of the vaccines. However, there is no standard adjuvant for glioma polypeptide vaccines, and further research is needed. GM-CSF, a granulocyte-macrophage colony-stimulating factor that can recruit antigen-presenting cells, is currently commonly used [14]. Targets of peptide vaccines commonly used in GBM include EGFRvIII, survivin and heat shock protein [15], while some clinical trials considered other strategies [16,17,18] (Published and ongoing clinical trials are shown in Tables 1 and 2).
2.1 Rindopepimut targeting EGFRvIII
EGFR is a transmembrane protein receptor that can regulate normal cell differentiation, proliferation and apoptosis, and is involved in tumor cell invasion, metastasis and angiogenesis [32]. EGFRvIII is the most common type of EGFR mutation and is found in approximately 20% of newly diagnosed IDH wild-type glioblastoma patients [33]. Compared with the intact EGFR molecule, the extracellular part of exons 2–7 is deleted, which leads to its continuous activation and constitutively activates tyrosine kinases and downstream pathways. Current studies have shown that the presence of EGFRvIII can enhance the resistance of tumor tissue to chemotherapy and radiotherapy, increase the tumorigenicity and invasiveness of cells, and tend to develop early metastasis [34]. Since it is a tumor-specific antigen, it is only expressed on tumor cells, and generates a new amino acid sequence due to gene rearrangement, it is considered to be an ideal immune antigen target. Therefore it is widely used in peptide vaccines and chimeric antigen receptor T-cell immunotherapy (CAR-T) as a therapeutic target [35].
2.2 Survax targeting survival protein
Survivin is an anti-apoptotic protein and previous studies have shown that it is expressed in gliomas. Survivin can be detected in about 85% of cells in GBM specimens, but no survivin-positive cells could be detected in normal brain tissue [36]. Survivin is an intracellular protein, and the antigenic epitopes generated after being interpreted by the proteasome are presented on the surface of tumor cells through MHC-I molecules. Survivin-specific cytotoxic T cells and survivin-specific antibodies have been found in patients with tumors [24], suggesting that survivin is immunogenic and tumor-related, and might be used as a potential vaccine target.
2.3 HSPPC-96 targeting heat shock protein
Heat shock proteins (HSPs) refer to a class of proteins that are widely present in bacteria, plants, and animals that respond to temperature changes. They have molecular chaperone activity and can inhibit the denaturation of biological macromolecules caused by temperature, oxygen content, and ions. The expression of HSPs is regulated by a variety of pathways, and is overexpressed in a variety of cancers, associated with cancer cell proliferation, differentiation, infiltration, and metastasis, thus becoming therapeutic targets and prognostic indicators for several tumors. When HSPs form complexes with autologous tumor antigen polypeptides (HSP-peptide complexes, HSPPCs), they can mediate endocytosis and antigen presentation through binding to APC membrane receptors, activate CD4+, CD8+ T cells, and activate APC’s signaling pathways that generate immune responses to tumor antigen peptides. Compared with peptide vaccines with specific antigens, HSPs vaccines can carry a variety of tumor antigens, which can cope with the poor effect of immunotherapy in the later stage of immunotherapy caused by tumor heterogeneity and immune editing, However, its specificity against tumor antigens is behind that of antigen-based peptide vaccines [37].
2.4 Multi-target and personalized polypeptide vaccine
Further research found that antigenic peptides obtained directly from tumor tissue could better activate the body’s tumor-specific immune response [38], and multi-peptide vaccines use this finding to screen out HLA-A2 restricted antigen peptides from peptides eluted directly from the tumor surface, and they are formulated to form multi-peptide vaccines. Several groups have reported that the safety and efficacy are possible for bispecific or tri-specific peptide vaccines in glioblastoma [39]. Mutations occurring in tumor can generate novel self-antigen epitopes called neoantigens. Basic research has demonstrated that these antigens are overexpressed in glioblastoma, with little or no expression in normal tissue, and present at the peptide level in glioblastoma tissue, warranting the existence of vaccine targets of elicited T cell immune responses [40]. Therefore, a vaccine composed of multiple antigenic peptides can ensure that a wide range of tumor-specific T-cell immune responses can be stimulated in the body, thereby avoiding the generation of tumor escape and preventing the immune attack on normal tissues. Personalized peptide vaccines are highly specific to individuals, and targeting neoantigens, can effectively stimulate immune response highly specific to cancer cells [41]. Furthermore, patients then receive clinical and immunological monitoring to determine the therapeutic effect and immunogenicity [42]. Multi-peptide vaccine and a personalized vaccine can reduce the escape of tumor cells since immune activation, which have stronger immune stimulation potential.
3 Phase I and II clinical studies of peptide vaccines in GBM
3.1 Phase II clinical studies of rindopepimut
Rindopepimut (CDX-110) is a polypeptide vaccine targeting EGFRvIII, consisting of EGFRvIII-specific peptide and keyhole limpet hemocyanin. Phase II clinical trial ACTIVATE enrolled 18 patients with newly diagnosed EGFRVIII-positive GBM [19], following temozolomide concurrent radiotherapy and vaccine maintenance therapy instead of TMZ, with median progression-free survival (14.2 m) and median overall survival (26 m) better than historical controls. Later, the ACT-II [20] and ACT-III [21] clinical trials used CDX-110 combined with TMZ concurrent chemoradiotherapy. The results showed that chemotherapy-induced lymphopenia may increase the immunostimulatory activity of the vaccine, and no dose limit toxicity and serious adverse events of CDX-110 were observed. As for recurrent GBM, Reardon [22] conducted a study enrolling bevacizumab-naïve patients with recurrent EGFRvIII-positive glioblastoma, who were randomized to receive rindopepimut or a control injection of keyhole limpet hemocyanin, each concurrent with bevacizumab. The result showed the potential of survival benefit, however, the therapeutic benefits require validation due to the small sample size and heterogeneity of bevacizumab response among participants. The survival benefit of the patients in these four clinical trials was better than that of the historical control, which provided the possibility for the development of its phase III clinical study.
3.2 Phase I and II clinical studies of SurVax vaccine
SurVaxM is the world’s first peptide-mimicking tumor vaccine that can target cell survival proteins such as survivin, and previous basic research has proven that it has a dual mechanism of action. On the one hand, it can stimulate T cell immunity, and on the other hand, it can inhibit the survivin pathway [43, 44]. Currently, it has been approved by the US FDA orphan drug. The phase I clinical trial completed by Robert et al. included 9 patients with survivin-positive recurrent glioma, 8 of which were primary or secondary glioblastoma [24]. The results showed that 6 patients had responses at a cellular or hormonal level, 3 patients had a partial clinical response and their tumors remained stable for 6 months. Overall, mPFS was 17.6 weeks, mOS was 86.6 weeks, and 7 patients had an overall survival greater than 12 months. In terms of safety, SurVaxM was well tolerated in patients with no dose-limiting toxicities.
The results of a phase II clinical trial (NCT02455557) announced in 2020 showed that a total of 63 patients with newly diagnosed GBM were enrolled and treated with SurVaxM combined with TMZ. The 6-month PFS of the treatment group was 96.3% (54% of the historical control), 12-month OS was 90.9% (61% in historical controls). Overall, the combined treatment group had an mOS of 30.5 months, compared with 14.8 months in the standard-of-care control group, and 13 patients were progression-free at 12 months. However, the results were only available on the clinicaltrials.gov website without a peer-reviewed article published, while no further findings from this study have been published, and more data are still needed to support its efficacy in glioblastoma. The 2020 ASCO meeting abstract announced the design of another Phase II clinical trial of SurVaxM [25]. The study plans to enroll patients with first relapsed GBM, using Pembo monoclonal antibody combined with SurVaxM treatment (NCT04013672), pending further study results.
3.3 Phase II clinical study of HSPPC-96 targeting heat shock protein
The vaccine HSPPC-96 targeting heat shock protein utilizes the patient’s autologous tumor lysate to couple with heat shock proteins, and activates the immune response targeting tumor-specific antigens through the immune-mediated effect of HSP [45]. A phase II clinical study led by Bloch et al. included patients with relapsed GBM [26], and the results showed that the mOS was 42.6 weeks, the 6-month OS was 90.2%, and the 12-month OS was 29.3%. Further research found that lymphocyte count was significantly correlated with prognosis, suggesting that it could indeed activate the body’s immune response to kill tumor cells, but its overall survival benefit was not significant, which may be due to the fact that heat shock proteins are not tumor-specific so that its specific tumor-targeting effect is reduced.
Another single-arm phase II study conducted by Bloch enrolled adult patients with GBM who has underwent surgical resection followed by radiation and chemotherapy and participants received vaccinations of HSPPC-96 after completion of radiation [27]. The results showed that the mOS was 23.8 months and patients with low expression of PD-L1 has a better prognosis than those with high expression, with mOS of 44.7 months and 18.0 months respectively. Further analysis suggested that PD-L1 was the independent predictor of survival. However, more studies are still needed to validate the current findings.
3.4 Multi-target polypeptide vaccine and personalized polypeptide vaccine
The multi-target polypeptide vaccine contains a variety of GBM antigen peptides, which can induce CD4/CD8-T cell responses after combining with a variety of MHC molecules and enhance the probability of anti-tumor immune response. Vaccines such as IMA950 contain 11 antigenic peptides of glioblastoma. A phase I clinical trial published in 2016 explored the effect of IMA950 combined with temozolomide standard treatment [28]. In patients with newly diagnosed glioblastoma, at least 30% produced a immune response, 90% of patients had a T-cell response to at least one antigenic peptide, and progression-free survival in the trial was 74% at 6 months and 31% at 9 months. The subgroup of patients with an injection site reaction was found to have longer survival (median overall survival of 26.7 months), compared with a median overall survival of 13.2 months for those without an injection site reaction. The current results show that the mixed antigen peptide vaccine targeting tumor-specific antigens or tumor-associated antigens is safe and could generate an immune response, but the survival benefit after treatment remains to be ensured by further clinical trial results.
In 2019, a phase I/II clinical study led by Migliorini et al. explored the safety and immunogenicity of IMA950 combined with poly-ICLC in newly diagnosed adult patients with malignant astrocytoma after standard therapy [46]. The results of the study showed that the combination of IMA950 and poly-ICLC was safe and tolerable, and the proportion of CD8+ T cell immune responses against single peptides was 63.2%, while the proportion of immune responses against polypeptides was 36.8%. The median overall survival in the cohort of patients with GBM was 19 months. The study also suggested that vaccine components and immune adjuvants mixed before injection could achieve better immune responses, which indicates that combining vaccines and immune checkpoint inhibitors could make patients have better survival benefits in the future.
The latest study of mixed peptide vaccine is the individualized neoantigen vaccine. The immune targets are designed according to the different mutation sites, types and specific expression of tumor neoantigens in the patient, and then individualized activation of the immune system reduces off-target effects and adverse effects. The European-led GAPVAC trial [29] and the US-led NEOVAX trial [30] both use whole-exome sequencing to design individualized vaccines against tumor neoantigens in patients. GAPVAC in patients with newly diagnosed glioblastoma had a median progression-free survival of 114.2 months and a median overall survival of 29 months, while results from the NEOVAX study of newly diagnosed, methyl guanine methyl transferase (MGMT)-unmethylated GBM patients showed mPFS was 7.6 months and mOS was 16.8 months, suggesting a survival benefit and further analysis found that both can stimulate a strong immune response.
4 Phase III clinical studies of peptide vaccines
4.1 ACTIV study of rindopepimut vaccine
Phase III clinical trial ACTIV is aimed at patients with newly diagnosed GBM and expression of EGFR vIII. The experimental group received Rindopepimut + TMZ + GM-CSF regimen, and the control group was only treated with TMZ + keyhole limpet hemocyanin (without peptide vaccine). The results showed that the overall OS was not prolonged, the treatment group and the control group were 20.1 months and 20.0 months, respectively; in patients with minimal residual (MRD, residual tumor after surgery and chemotherapy < 2 cm2 on enhanced images), the OS was 17.4 months in both groups, and the PFS was 17.4 months. The OS was 14.8 months and 14.1 months, respectively, and the PFS was 3.7 months in both groups in patients with large residual (SRD, residual tumor ≥2 cm2 on enhanced images after surgery and chemotherapy), respectively. It can be seen that Rindopepimut did not prolong the survival time regardless of the residual tumor volume [23].
Data analysis showed that patients with larger tumor remnants may have a potential long-term survival benefit. Since other baseline characteristics of patients with MRD and SRD are consistent, it is speculated that it may not be the smallest tumor burden that makes immunotherapy the most effective. The mOS in the ACT IV control group was longer than that in the historical control. From this, one might speculate that the survival benefit of the control group may be related to the use of keyhole limpet hemocyanin (rather than the placebo), which activates non-EGFRvIII-specific immune responses. In the small phase II clinical trial REACT [22], some patients also used keyhole limpet hemocyanin as a control, and their survival data were not better than historical data. This suggests that the “abnormal” survival data of the control group in ACT IV may be due to the lower risk and better prognosis of the recruited patients. However, the inclusion criteria and baseline characteristics of patients in the ACT IV study were similar to those in the previous phase II trial of rindopepimut. Overall, the prolonged survival in the control group cannot be definitively explained and further exploratory studies are needed. In addition, the researchers found that about 60% of patients lost EGFRvIII expression at relapse, regardless of whether they received rindopepimut. Loss of EGFRvIII is not uncommon even in standard-of-care patients. The loss rate of EGFRvIII in patients in this trial can reach 50% at relapse, suggesting that only half of the patients may benefit from rindopepimut. Therefore, in the subsequent GBM study with relapse, patients need to have biopsy-proven EGFRvIII expression before they can be enrolled. And it has implications for follow-up studies. When the expression of molecular targets (such as EGFRvIII) is unstable or easy to be lost, the effectiveness of monotherapy will be limited, and multi-target co-treatment should be considered.
4.2 Phase III clinical studies of personalized peptide vaccination (PPV)
Personalized peptide vaccination (PPV) is multi-targeted vaccine with individual differences. Phase III clinical trials are aimed at HLA-A24+ relapsed GBM patients, using PPV monotherapy, and the control group receives a placebo. There was no significant difference in OS and PFS between the experimental group and the placebo control [31]. The OS of the experimental group and the control group were 8.4 months and 8.0 months, respectively. The researchers found that the selection of SART2–93 polypeptide was a negative factor for clinical benefit. The mOS of patients who selected SART2–93 was 6.6 months, which was significantly shorter than that of the control group of 22.0 months. Patients with negative SART2–93 had longer relative OS. < 70 years old, ≤70 kg, PS 0–2 are also good prognostic factors. When patients have both SART2–93 negativity and one of the other prognostic factors, survival can be significantly prolonged. At present, the mechanism of SART2–93’s immune response and survival benefit of patients is not clear, and further research is needed.
5 The main failure reasons for peptide vaccines in GBM
-
1.
High intra-tumor heterogeneity can exacerbate immunotherapy resistance. Gliomas are highly heterogeneous, and targeting a single antigen often fails to cover all tumor cells. In addition, in 114 pairs of newly diagnosed-relapsed GBM patients, intense complex intratumoral evolution was observed, with 63% of patients relapsed with different tumor subtypes and accessible targets than newly diagnosed [47], exacerbating the difficulty in treating tumors after relapse. Even if treatment-sensitive clones are eliminated after immunotherapy, there may still be treatment-resistant clones within the tumor, which may even increase the difficulty of re-treatment.
-
2.
There are fewer tumor antigens and a low mutation load in GBM. Tumor-specific antigens and tumor neoantigens can be recognized by the immune system and elicit an immune response, but both are present at lower levels in GBM. In non-CNS tumors, tumor mutation burden (TMB) has been demonstrated as an independent predictor of immunotherapy efficacy. However, in GBM, there is no clear correlation between TMB and the efficacy of immunotherapy [48], and several studies have found that patients with lower TMB have longer survival [49]. Glioma de novo hypermutation is very rare, mostly due to mismatch repair (MMR) gene defect or POLE gene mutation, which may benefit from immunotherapy. More commonly, hypermutation detected at progression or recurrence after the TMZ regimen, is often subclonal with low immunogenicity and is not significantly associated with immunotherapy benefit [50]. Compared with calculating TMB, more important are the immune neoantigens of mutations, the clonality of neoantigens, the immune pressure of the tumor, and the selection of immunotherapy regimens [48].
-
3.
Systemic and local immunosuppression is strong in GBM. Both preclinical and clinical studies have shown that GBM could cause systemic and local immunosuppression [51]. Local immunosuppression is associated with multiple factors in the tumor microenvironment, including regulatory T cells [52], tumor-associated macrophages (TAM), tumor-killing T cell depletion, and high expression of inhibitory immune regulatory molecules. Systemic immunosuppression limits the access of peripheral immune cells to the local CNS tumor. Both TMZ and radiotherapy exacerbate systemic immune exhaustion, deplete the efficient generation of memory T cells and inhibit the efficacy of immunotherapies such as checkpoint inhibitors. Glucocorticoids also reduce peripheral CD4 + CD8+ T cells, inhibiting the efficacy of immunotherapy [53, 54].
-
4.
The depth of patient screening is insufficient. In almost all trials, investigators found that a subset of patients had a better/worse survival benefit. These various factors suggest the potential efficacy of this immunotherapy, and they are also important findings in phase III clinical trials, which can guide further research. To improve the efficiency of clinical studies and benefit more patients, it is recommended to use an adaptive trial design to fully detect the patient’s status, and if necessary, transfer patients between groups to achieve the best therapeutic effect.
-
5.
There are differences in imaging interpretation. According to the methods in published studies, most clinical trials utilize RANO criteria for imaging interpretation or do not specifically describe them. Clinical studies have confirmed that the RANO criteria itself may be insufficient in the evaluation of immunotherapy, such as the determination of pseudo-progression, the necessity of re-examination after progression, the significance of new lesions, and whether to continue treatment after imaging progression [55]. Using RANO criteria or simply using MRI before and after contrast to determine tumor progression may lead to premature discontinuation of immunotherapy and reduce the potential benefit to patients. After an independent, cross-border multidisciplinary expert symposium, the iRANO criteria are more applicable to the evaluation of the efficacy of immunotherapy [56]. Currently, the iRANO criteria are recommended instead of RANO for the imaging determination of the efficacy of glioma immunotherapy.
6 The challenges and future of peptide vaccines in GBM
The emerging field of immunotherapy offers great promise for the treatment of GBM. Studies on oncolytic viruses, peptide vaccines, DC vaccines, CAR-T therapy, and checkpoint inhibitors have provided new treatment ideas for adjuvant GBM therapy, which may improve patient prognosis and overall survival [3]. And more targets for peptide vaccines are worth exploring. In future research, it is necessary to analyze the factors that lead to the failure of immunotherapy from previous investigations, and further improve the trial design and treatment plan.
More appropriate treatment regimens, such as individualized multi-target vaccines, combined with multiple treatment methods and neoadjuvant therapy, can improve the therapeutic benefit [57]. Current clinical data show that individualized vaccines containing multiple antigenic peptides or using peptides from tumor cell self-lysate have initial benefits, which can significantly reduce immune escape and tumor growth pathway activation. Combination therapy has received increasing attention [58]. Immunotherapy in combination with standard care is one of the most studied combination treatments. Chemoradiation and chemotherapy can enhance tumor antigenicity and reduce immunosuppression in the tumor microenvironment, but standard therapy, especially temozolomide, can lead to severe lymphopenia, decreased CD8 T cell infiltration, and T cell failure. Studies have shown that booster doses of temozolomide do not lead to T cell failure, however, standard doses lead to up-regulation of markers of exhaustion [59, 60], so the dose of temozolomide needs to be weighed. There may be synergistic effects between different immunotherapies to improve immune response and prolong survival. In preclinical trials in GBM mice, it has been found that a combination of multiple immunotherapies is more effective in activating immunity in vivo and improving survival. Peptide vaccine combined with anti-PD-1 is better than single therapy [61]. Anti-LAG-3 combined with anti-PD The − 1 antibody also significantly improved mouse survival [62]. In addition to immune-led treatment, vaccines can also assist other treatment options, such as the use of peptide vaccines to alleviate resistance to radiotherapy and chemotherapy and vaccines combined with targeted therapy.
However, it is a remarkable fact that several studies have reported severe adverse effects of immune therapy including peptide vaccines in various solid tumors. Kitamura et al. have explained that long peptide vaccination could lead to lethality through CD4+ T cell-mediated cytokine storm [63]. Based on the previous study results, several adverse effects of peptide vaccines in GBM were also screened, of which the most common are injection site reactions, chiefly erythema and pruritus, which hardly exceed grade 2 toxicity [19, 21]. Other regimen-related toxicities include brain edema, myalgias, fatigue and flu-like syndrome [22, 24, 26, 46]. Thus, future monitoring of adverse events during regimens of peptide vaccines in GBM is still of great importance.
In addition, new clinical trial designs, such as molecular marker-based trial design, adaptive trial design, umbrella design and basket design may be used to group patients through more sophisticated methods of clinical trials, therefore improving data quality and research efficiency, and obtaining greater potential for patient benefits [64]. In data analysis, it is necessary to explore excellent molecular markers and screen for potentially beneficial patient subgroups to promote individualized and precise treatment [65, 66]. Although the effectiveness of the current phase III clinical trials is not satisfactory, future larger prospective studies may help to clarify the role of immunotherapy in glioma patients.
Availability of data and materials
Not applicable.
Abbreviations
- GBM:
-
Glioblastoma multiforme
- TMZ:
-
Temozolomide
- GAM:
-
Glioblastoma-associated macrophages
- M-CSF:
-
Macrophage colony-stimulating factor
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- MHC:
-
Major histocompatibility complex
- Treg:
-
T regular cell
- TCR:
-
T cell receptor
- EGFRvIII:
-
Epidermal growth factor receptor variant III
- CAR-T:
-
Chimeric antigen receptor T-cell immunotherapy
- HSPs:
-
Heat shock proteins
- HSPPCs:
-
HSP-peptide complexes
- APC:
-
Antigen presenting cell
- PFS:
-
Progression free survival
- OS:
-
Overall survival
- MGMT:
-
Methyl guanine methyl transferase
- MRD:
-
Minimal residual
- SRD:
-
Large residual
- PPV:
-
Personalized peptide vaccination
- TMB:
-
Tumor mutation burden
- MMR:
-
Mismatch repair
- TAM:
-
Tumor-associated macrophages
References
Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro-Oncology. 2020;22(12 Suppl 2):iv1–iv96.
Grochans S, Cybulska AM, Simińska D, et al. Epidemiology of glioblastoma Multiforme-literature review. Cancers (Basel). 2022;14(10):2412.
Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20(1):12–25.
Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the immune system in glioblastoma. Br J Cancer. 2018;119(10):1171–81.
Strepkos D, Markouli M, Klonou A, Piperi C, Papavassiliou AG. Insights in the immunobiology of glioblastoma. J Mol Med (Berl). 2020;98(1):1–10.
Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int J Mol Sci. 2018;19(2):436.
Gieryng A, Pszczolkowska D, Bocian K, et al. Immune microenvironment of experimental rat C6 gliomas resembles human glioblastomas. Sci Rep. 2017;7(1):17556.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9.
Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci U S A. 2008;105(51):20428–33.
Wainwright DA, Balyasnikova IV, Chang AL, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–21.
Wiendl H, Mitsdoerffer M, Hofmeister V, et al. A functional role of HLA-G expression in human gliomas: an alternative strategy of immune escape. J Immunol. 2002;168(9):4772–80.
Cuoco JA, Benko MJ, Busch CM, Rogers CM, Prickett JT, Marvin EA. Vaccine-based Immunotherapeutics for the treatment of glioblastoma: advances, challenges, and future perspectives. World Neurosurg. 2018;120:302–15.
Hirayama M, Nishimura Y. The present status and future prospects of peptide-based cancer vaccines. Int Immunol. 2016;28(7):319–28.
Nejo T, Yamamichi A, Almeida ND, Goretsky YE, Okada H. Tumor antigens in glioma. Semin Immunol. 2020;47:101385.
Andrews DW, Judy KD, Scott CB, et al. Phase Ib clinical trial of IGV-001 for patients with newly diagnosed glioblastoma. Clin Cancer Res. 2021;27(7):1912–22.
Pollack IF, Jakacki RI, Butterfield LH, et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J Clin Oncol. 2014;32(19):2050–8.
Schijns VE, Pretto C, Devillers L, et al. First clinical results of a personalized immunotherapeutic vaccine against recurrent, incompletely resected, treatment-resistant glioblastoma multiforme (GBM) tumors, based on combined Allo- and auto-immune tumor reactivity. Vaccine. 2015;33(23):2690–6.
Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–9.
Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-Oncology. 2011;13(3):324–33.
Schuster J, Lai RK, Recht LD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-Oncology. 2015;17(6):854–61.
Reardon DA, Desjardins A, Vredenburgh JJ, et al. Rindopepimut with bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): results of a double-blind randomized phase II trial. Clin Cancer Res. 2020;26(7):1586–94.
Weller M, Butowski N, Tran DD, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–85.
Fenstermaker RA, Ciesielski MJ, Qiu J, et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother. 2016;65(11):1339–52.
Ahluwalia MP, David P, Ciolfi M, Schilero C, Hobbs B, Ciesielski M, et al. Phase II study of pembrolizumab plus SurVaxM for glioblastoma at first recurrence. J Clin Oncol. 2020;38:TPS2581–1 10.1200.
Bloch O, Crane CA, Fuks Y, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro-Oncology. 2014;16(2):274–9.
Bloch O, Lim M, Sughrue ME, et al. Autologous heat shock protein peptide vaccination for newly diagnosed glioblastoma: impact of peripheral PD-L1 expression on response to therapy. Clin Cancer Res. 2017;23(14):3575–84.
Rampling R, Peoples S, Mulholland PJ, et al. A cancer research UK first time in human phase I trial of IMA950 (novel multipeptide therapeutic vaccine) in patients with newly diagnosed glioblastoma. Clin Cancer Res. 2016;22(19):4776–85.
Hilf N, Kuttruff-Coqui S, Frenzel K, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240–5.
Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–9.
Narita Y, Arakawa Y, Yamasaki F, et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro-Oncology. 2019;21(3):348–59.
Maire CL, Ligon KL. Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro-Oncology. 2014;16(Suppl 8):viii1–6.
Felsberg J, Hentschel B, Kaulich K, et al. Epidermal growth factor receptor variant III (EGFRvIII) positivity in EGFR-amplified glioblastomas: prognostic role and comparison between primary and recurrent tumors. Clin Cancer Res. 2017;23(22):6846–55.
Huang K, Liu X, Li Y, et al. Genome-wide CRISPR-Cas9 screening identifies NF-kappaB/E2F6 responsible for EGFRvIII-associated Temozolomide resistance in glioblastoma. Adv Sci (Weinh). 2019;6(17):1900782.
Johnson LA, Scholler J, Ohkuri T, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7(275):275ra222.
Uematsu M, Ohsawa I, Aokage T, et al. Prognostic significance of the immunohistochemical index of survivin in glioma: a comparative study with the MIB-1 index. J Neuro-Oncol. 2005;72(3):231–8.
Ampie L, Choy W, Lamano JB, Fakurnejad S, Bloch O, Parsa AT. Heat shock protein vaccines against glioblastoma: from bench to bedside. J Neuro-Oncol. 2015;123(3):441–8.
Pollack IF, Jakacki RI, Butterfield LH, et al. Immune responses and outcome after vaccination with glioma-associated antigen peptides and poly-ICLC in a pilot study for pediatric recurrent low-grade gliomas. Neuro-Oncology. 2016;18(8):1157–68.
Runcie K, Budman DR, John V, Seetharamu N. Bi-specific and tri-specific antibodies- the next big thing in solid tumor therapeutics. Mol Med. 2018;24(1):50.
Dutoit V, Herold-Mende C, Hilf N, et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain. 2012;135(Pt 4):1042–54.
Han MH, Kim CH. Current immunotherapeutic approaches for malignant gliomas. Brain Tumor Res Treat. 2022;10(1):1–11.
Dunn GP, Sherpa N, Manyanga J, Johanns TM. Considerations for personalized neoantigen vaccination in malignant glioma. Adv Drug Deliv Rev. 2022;186:114312.
Ciesielski MJ, Kozbor D, Castanaro CA, Barone TA, Fenstermaker RA. Therapeutic effect of a T helper cell supported CTL response induced by a survivin peptide vaccine against murine cerebral glioma. Cancer Immunol Immunother. 2008;57(12):1827–35.
Galbo PM Jr, Ciesielski MJ, Figel S, et al. Circulating CD9+/GFAP+/survivin+ exosomes in malignant glioma patients following survivin vaccination. Oncotarget. 2017;8(70):114722–35.
Srivastava PK, Callahan MK, Mauri MM. Treating human cancers with heat shock protein-peptide complexes: the road ahead. Expert Opin Biol Ther. 2009;9(2):179–86.
Migliorini D, Dutoit V, Allard M, et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro-Oncology. 2019;21(7):923–33.
Wang J, Cazzato E, Ladewig E, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48(7):768–76.
Merchant M, Ranjan A, Pang Y, et al. Tumor mutational burden and immunotherapy in gliomas. Trends Cancer. 2021;7(12):1054–8.
Brown MC, Ashley DM, Khasraw M. Low tumor mutational burden and immunotherapy in gliomas. Trends Cancer. 2022;8(5):345–6.
Gatto L, Franceschi E, Tosoni A, Nunno VD, Bartolini S, Brandes AA. Hypermutation as a potential predictive biomarker of immunotherapy efficacy in high-grade gliomas: a broken dream? Immunotherapy. 2022;4(10):799–813.
Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. 2019;20(9):1100–9.
Antonios JP, Soto H, Everson RG, et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro-Oncology. 2017;19(6):796–807.
Giles AJ, Hutchinson MND, Sonnemann HM, et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J Immunother Cancer. 2018;6(1):51.
Kelly, William J, and Mark R Gilbert. “Glucocorticoids and immune checkpoint inhibitors in glioblastoma.” J Neuro-Oncol. 2020;151(1):13–20. https://doi.org/10.1007/s11060-020-03439-2.
Wen PY, Chang SM, Van den Bent MJ, Vogelbaum MA, Macdonald DR, Lee EQ. Response assessment in neuro-oncology clinical trials. J Clin Oncol. 2017;35(21):2439–49.
Okada H, Weller M, Huang R, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. Lancet Oncol. 2015;16(15):e534–42.
Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422–42.
Barbari C, Fontaine T, Parajuli P, et al. Immunotherapies and combination strategies for Immuno-oncology. Int J Mol Sci. 2020;21(14):5009.
Karachi A, Yang C, Dastmalchi F, et al. Modulation of temozolomide dose differentially affects T-cell response to immune checkpoint inhibition. Neuro-Oncology. 2019;21(6):730–41.
Park J, Kim CG, Shim JK, et al. Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. Oncoimmunology. 2019;8(1):e1525243.
Liu CJ, Schaettler M, Blaha DT, et al. Treatment of an aggressive orthotopic murine glioblastoma model with combination checkpoint blockade and a multivalent neoantigen vaccine. Neuro-Oncology. 2020;22(9):1276–88.
Harris-Bookman S, Mathios D, Martin AM, et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int J Cancer. 2018;143(12):3201–8.
Kitamura H, Sedlik C, Jacquet A, et al. Long peptide vaccination can lead to lethality through CD4+ T cell-mediated cytokine storm. J Immunol. 2010;185(2):892–901.
Aldape K, Brindle KM, Chesler L, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol. 2019;16(8):509–20.
Hung AL, Garzon-Muvdi T, Lim M. Biomarkers and immunotherapeutic targets in glioblastoma. World Neurosurg. 2017;102:494–506.
Lynes JP, Nwankwo AK, Sur HP, et al. Biomarkers for immunotherapy for treatment of glioblastoma. J Immunother Cancer. 2020;8(1):e000348.
Acknowledgements
Not applicable.
Funding
This research was funded by Beijing Municipal Natural Science Foundation [grant number: 7202150].
Author information
Authors and Affiliations
Contributions
All authors designed and conducted this review. Yu Wang had primary responsibility for the final content. Tianrui Yang and Yixin Shi wrote the original draft. Wenbin Ma and Yu Wang supervised the writing of the manuscript. Tingyu Liang and Hao Xing assisted on critical revision of the article for important intellectual content. Tianrui Yang and Yixin Shi equally share the first authorship. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, T., Shi, Y., Liang, T. et al. Peptide vaccine against glioblastoma: from bench to bedside. Holist Integ Oncol 1, 21 (2022). https://doi.org/10.1007/s44178-022-00021-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44178-022-00021-w