
Abstract
Purpose
Our previous study has shown that PTPRZ1-MET (ZM) fusion is a viable target for MET inhibitors in gliomas. However, the diversity and prevalence of somatic MET alterations in diffuse gliomas are still elusive and need to be extensively characterized for identifying novel therapeutic targets.
Methods
Totally, 1,350 glioma patients and 31 patient-derived cells were collected from the Chinese Glioma Genome Atlas (CGGA) and published data. All kinds of MET fusions and/or splicing variants (MET F/SVs) were identified by bioinformatical methods. Single-cell RNA sequencing (scRNA-seq) were used for validation. In vitro experiments of drug resistance were conducted for the possibility of MET-targeted treatment.
Results
MET F/SVs but not genomic amplification, were highly enriched in the secondary glioblastomas (sGBM) and marked worse prognosis. Further molecular and scRNA-seq analysis revealed that MET F/SVs were induced in the course of glioma evolution and highly associated with MET overexpression. Subsequent in vitro and the clinical study showed that cells and patients harboring MET F/SVs have better response to MET inhibitors.
Conclusion
Our findings expanded the percentage of gliomas with abnormal MET alterations and suggested that a subgroup of gliomas harboring MET F/SVs may benefit from MET-targeted therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Diffuse glioma is the most common, lethal, and highly heterogeneous type of primary malignant in adult brain [1,2,3]. Despite comprehensive treatment, including surgery, chemotherapy, fraction radiotherapy, and tumor treating fields, glioma patients still suffered a poor survival [4, 5]. Novel targets for individualized targeted strategies are urgently needed. Our previous studies revealed the poorly prognostic and targeted predictive role of PTPRZ1-MET (ZM) fusion and MET exon 14 skipping (METex14) in secondary GBM (sGBM) [6]. However, the diversity and prevalence of somatic MET alterations in diffuse gliomas are still elusive.
Aberrant MET overexpression and activation is widely observed in various neoplasms, triggering cell proliferation, migration and angiogenesis. Several MET alterations, including fusions and splicing variants (F/SVs), have been found and used as biomarkers for targeted therapy in melanoma, hepatoma, lung cancer, and so on [7,8,9].
Here, we comprehensively analyzed the MET F/SVs in a large number of diffuse gliomas from public and in-house data sets. Integrating the genomic and transcriptomic profiles of pan-gliomas, we found MET F/SVs marked MET overexpression and strongly associated with malignant biological phenotypes and late molecular events in diffuse gliomas. Functional studies of patient derived cell lines (PDCs) and clinical case demonstrated the feasibility of MET-targeted therapy for patients with MET F/SVs. Taken together, we revealed the predictive role of MET F/SVs in diffuse gliomas for MET-targeted therapy.
2 Methods
2.1 Sample acquisition
Glioma tissues, the corresponding genomic data, and patients’ follow-up information (histology, WHO grade, gender, age, extent of resection, and OS) were obtained from the Chinese Glioma Genome Atlas (CGGA, http://www.cgga.org.cn) [10], which includes patient samples collected from Beijing Tiantan Hospital, Sanbo Hospital in Beijing, Tianjin Medical University General Hospital, The First Affiliated Hospital of Nanjing Medical University, Harbin Medical University, and China Medical University. The glioma tissues were snap-frozen in liquid nitrogen immediately after surgical resection and preserved in liquid nitrogen.
2.2 Sample processing and sequencing
Genomic DNA from tumors and matched blood samples was extracted and confirmed DNA integrity by 1% agarose gel electrophoresis. The DNA was subsequently fragmented, and underwent quality control. Then, pair-end libraries were prepared. For whole-exome sequencing, Agilent SureSelect kit v6 was used for target capture. The sequencing was done using Illumina Hiseq platform with a 150-bp pair-end sequencing strategy.
Total RNA from each tumor sample was extracted, and then tRNA and rRNA removed. After quality control and quantification, mRNA was reverse-transcribed to cDNA. Next, library was prepared and the samples sequenced. The human reference genome hg19 and genome annotation file were downloaded from the UCSC genome browser. Clean RNA sequencing reads were mapped to the reference genome using STAR 2.5.2b. Gene expression, in FPKM, was calculated by RESM v1.2.31 [11].
2.3 Mapping and mutation calling
Valid DNA sequencing data were mapped to the reference human genome (UCSC hg19) using the Burrows-Wheeler Aligner (BWA v0.7.12, bwa mem) with default parameters [12]. Then, SAMtools v1.2 [13] and Picard v2.0.1 (Broad Institute) were used to sort the reads by coordinates and mark duplicates. Statistics such as sequencing depth and coverage were calculated using the resultant BAM files. SAVI2 was used to identify somatic mutations (including single-nucleotide variations and short insertion/deletions), as previously described [6, 14]. In this pipeline, SAMtools mpileup and bcftools v0.1.19 were used for variant calling. Then, the preliminary variant list was filtered to remove positions with no sufficient sequencing depth, positions with only low-quality reads, and positions that were biased toward either strand. Somatic mutations were identified and evaluated by using an Empirical Bayesian method. In particular, mutations with the mutation allele frequency in tumors significantly higher (fisher p value < 0.05) than that in normal control were selected.
2.4 Copy number alteration analysis
For whole-exome sequencing data, CNVkit v0.9.4[15] was used to detect copy number changes.
2.5 Detection of gene fusions in RNA-seq data
STAR-Fusion v1.2.0 (https://www.biorxiv.org/content/10.1101/120295v1) was used to detect gene fusion in RNA-seq data, with default parameters.
2.6 Detection of MET exon skipping in RNA-seq data
In the study, MET exon 10, 14 and 19 skipping were detected based on reads spanning the junction of exons 9 and 11, exons 13 and exon 15, and exon s18 and exon 20, respectively. Briefly, RNA-seq reads were aligned with the reference genome (hg19) using STAR v2.5.2b [16]. Then, the reads containing a jump of 4,429 bp from chr7:116,398,664, 3,226 bp from chr7:116,411,700, and 1,3666 bp from chr7:116,422,042 were counted to identify the candidate MET exon 10, 14, and 19 skipping events, respectively. The spanning reads were manually checked in Integrative Genomic Viewer (IGV) to remove false-positives, such as PCR artifacts and potential mapping errors. IGV was used to visualize the skipped reads.
2.7 Validation of MET exon skipping by PCR and Sanger sequencing
RNA extraction and reverse-transcription of the glioma specimens were performed as mentioned. MET exon skipping was detected by PCR. The following primers were used to detect exon 14 skipping: forward: 5′-AATCTTTTATTAGTGGTGGGAGCACAAT-3′; and reverse: 5′-GAATTAGGAAACTGATCTTTAATTTGC-3′. The reverse primer crosses exons 13 and 15 of MET. The following primers were used to detect MET exon 10 skipping: forward: 5′- CAAATCTTTTATTAGGCATGTCAACATC -3′; reverse: 5′- CTTCTGGAAAAGTAGCTCGGTAGTCT-3′. The forward primer crosses exons 9 and 11 of MET. The following primers were used to detect MET exon 19 skipping: forward: 5′-GATCAGTTTCCTAATTCATCTCAGAAC-3′; reverse: 5′- GCCAAAGGACCAATACAGTTTCTT-3′. The reverse primer crosses exons 18 and 20 of MET. In all reactions, DNA polymerase (GoTaq, Promega) was used to amplify a 626-bp fragment, with 30 cycles of 94 °C, 30 s; 58 °C, 30 s; 72 °C, 60 s; and a final extension at 72 °C for 10 min. The product bands were extracted from agarose gel (1%) after electrophoresis and verified by Sanger sequencing.
2.8 Sample and library preparation for scRNA-seq
Samples were collected from Beijing Tiantan Hospital, Capital Medical University from four glioblastoma patients. Fresh clinical samples were first trypsinized, followed by isolation of single cell and dead cell removal to obtain cell suspensions. Cell viability reached 80% with cell count reached 5 × 105 cells. cDNA libraries were prepared using the Chromium Single Cell 3’ Library and Gel Bead kit v2/3 (10 × Genomics) according to the manufacturer’s instructions. RNA-seq was performed using the Illumina HiSeq 2500 platform.
2.9 Single-cell data processing, quality control, and analysis
The Cell Ranger guideline (v3.0.2, 10 × Genomics) was followed to convert Illumina base-call files to FASTQ files, aligning FASTQ files with the hg19 reference for human samples, and producing a digital gene-cell counts matrix. Cells in gene-cell matrices with fewer than 200 transcripts, and genes with fewer than two counts in two cells were removed. The matrix was then normalized such that the number of UMIs in each cell was equal to the median UMI count across the dataset and log-transformed. The 10X Genomics datasets were integrated using the harmony R package. To pare-down the gene expression matrix to the most relevant features, principal components analysis (PCA) was used; this changes the dimensionality of the dataset. FindClusters function was performed to generate four clusters. To visualize data in 2D space, the PCA-reduced data were analyzed by t-distributed stochastic neighbor embedding (t-SNE), a non-linear dimensional reduction method. The differentially expressed genes between clusters were screened using the FindMarkers function and a comprehensive biological functional annotation of the gene list was performed in R package clusterProfiler. Cell subtypes (malignant cells) were identified by cell-specific markers. We use Seurat CellCycleScoring function to obtain cell cycle phases.
2.10 Cell culture
Patient-derived cells (PDC) were obtained from fresh surgical specimens of human primary GBM and cultured as tumor spheres in DMEM/F12 medium supplemented with B27 supplement (Life Technologies), bFGF and EGF (20 ng/ml each), at 37 °C in a humidified atmosphere of 5% CO2.
2.11 Determination of cell sensitivity to drugs
Cell drug sensitivity was evaluated by using the CCK-8 kit (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s protocol. Briefly, 3,000 PDCs were grown in each well of 96-well plate in normal medium for 24 h. The cells were then incubated in a medium containing specific concentrations of TMZ or crizotinib for 72 h, with six repeats for each concentration tested. Next, 10 μl of CCK-8 reagent was added to each well, and sample optical density at 450 nm and 630 nm was determined after 2 h incubation and 37 °C in a humidified atmosphere of 5% CO2. The number of living cells was calculated as OD450–OD630.
2.12 Immunohistochemistry
Immunohistochemical staining was performed as previously described [17, 18]. Briefly, tissue sections were deparaffinized and boiled in citrate antigen-retrieval buffer. Then, the primary antibodies (anti–c-MET, 1:1000, Abcam, Cambridge, UK; anti–p-MET, 1:200, CST, MA, USA; anti–Ki-67 1:100, ZSGB-Bio; anti-MYC, 1:150, Abcam; anti-CDK6, 1:150, Abcam; anti-CDC20,161 1:150, Abcam) were incubated with the sections, overnight at 4 °C. After incubation with ethanol containing 3% hydrogen peroxide for 10 min and washing with PBS buffer, the sections were incubated with the secondary antibodies (1:100, ZSGB-Bio, Beijing, China) at 25 °C for 1 h. The images were captured by using Axio Imager 2 (Zeiss) after 3,3′-diaminobenzidine and hematoxylin staining.
2.13 Statistical analysis
Statistical analyses were performed by fisher’s exact test. Survival curves were estimated with the Kaplan–Meier method. All statistical analyses were conducted and obtained using the R v4.0.3 software (https://www.r-project.org).
3 Results and discussion
3.1 MET alterations frequently occurred and enriched in sGBMs
To identify gliomas that will benefit from MET targeted therapy, we identified all MET alterations in 1,147 glioma cases and 31 patient-derived cells (PDC) from the Chinese Glioma Genome Atlas Network (CGGA, http://www.cgga.org.cn) and 203 samples from previously published data (Fig. S1a).
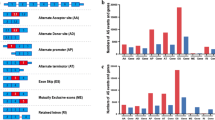
We figured out a variety of MET structural alterations, including MET fusions (34/1,104 cases), MET exon 10 skipping (METex10) (28/1,104 cases), MET exon 14 skipping (METex14) (23/1,104 cases), and MET exon 19 skipping (METex19) (19/1,104 cases) in gliomas and patient-derived cells (PDCs; 7/31 cell lines) from RNA-seq data. Of them, some have been reported in Pan-cancer, and some are newly discovered MET alterations (Fig. 1A-B and Supplementary Table 1, 2, 3). Particularly, MET fusions included C7orf55-LUC7L2-MET, CAPZA1-MET, CAPZA2-MET, ELAVL3-MET, KCND2-MET, LHFPL3-MET, PTPRZ1-MET (ZM), RSBN1L-MET, ST7-MET, ZMIZ2-MET and ZNF277-MET. Notably, two novel breakpoints of MET were identified to fuse to two different breakpoints of other genes in more than one sample (LHFPL3-MET and RSBN1L-MET, Fig. S1b). Importantly, two recurrent MET exon skipping format were newly observed in gliomas. The METex10 has been previously identified in lung cancer cell lines [19], but firstly identified in 28 glioma cases and 7 PDCs, while METex19 was a novel MET alteration identified in 19 glioma cases and 1 PDC (Fig. S1c-d). We then validated the presence of METex10 and METex19 in clinical glioma samples via gel-based PCR and Sanger sequencing (Fig. 1C-D and Fig. S1e-f). Unlike METex14, which induces MET signaling activation though skipping the JM domain of MET protein [19], METex10 and METex19 could result in truncated MET proteins (Fig. S2a-b).

MET alterations frequently occurred and enriched in sGBMs. A A summary of all reported MET RNA alterations in pan-cancer. Red indicated firstly identified MET F/SVs. Turquoise indicated MET F/SV that previously reported in other tumor but firstly identified in the brain tumor. Purple indicated previously identified MET F/SVs in brain tumor. Black indicated MET F/SVs which we didn’t identify in this study. B Frequencies of each MET RNA alterations in glioma subtypes and patient derived glioma cells in this study. C-D Sashimi plot of RNA-seq data from two METex10-positive sGBM (S0373 and S0179, C) and a METex19-positive sGBM (S0179, D). E The number of cases with multiple MET variants or single MET variant. F Frequencies of MET alterations in different subtypes of gliomas. G Ternary plot of mutation frequency in driver genes in primary LGG (pLGG), secondary GBM (sGBM), and IDH-wildtype primary GBM (pGBM), and the node size represents their overall frequency in glioma. H, Overall survival of sGBM patients (first-diagnosis) with and without IDH mutation and MET status (Median survival, 356 days, 254 days, 195 days for IDH Mutation & MET Wildtype, IDH Wildtype, IDH Mutation & MET Alteration, respectively). I The proportion of previously reported MET alterations and newly identified MET alterations in all cases
Furthermore, the different MET variants sometimes simultaneously presented in the same case (Fig. 1E), which was also observed in TCGA dataset (Fig. S2c and Supplementary Table 4). The MET fusions, METex10, METex14, and METex19 were all highly enriched in secondary GBM (sGBM, WHO grade 4) (Fig. 1F and Fig. S2d-g). Similarly, comparison of genomic alterations in primary LGGs (pLGG, WHO grade 2–3) and primary IDH-wildtype GBMs (pGBM, WHO grade 4), sGBMs revealed a high frequency of these MET fusions and splicing variants (Fig. 1G and Supplementary Table 5). We next checked whether all different MET variants were linked to sGBM patients’ survival. We defined the sGBM overall survival (OS) as the period from the first diagnosis of sGBM until patient death. Similar to METex14 and MET fusion, METex10 and METex19 separately stratified the OS of sGBM patients (Fig. S2h-k). Based on these, the different MET alterations may concurrent lead a consistent biological process. The OS of IDH-mutant sGBM patients with MET F/SVs was significantly poorer than that of IDH-wt sGBMs without MET F/SVs (P = 1.7 × 10–3, log-rank test) (Fig. 1H), underscoring the role of MET F/SVs as a prognostic marker in sGBM. Compared with our previous study6, these findings expanded the number of gliomas with MET F/SVs from 38 (involved with ZM and/or METex14) to 60 in our cohort (Fig. 1I).
3.2 MET F/SVs are later events
To investigate the stage of the identified MET F/SVs during glioma progression and evolution, we performed a novel bioinformatics tools, which we have recently reported for efficient prediction of the natural evolution stage of glioma based on their transcriptional features. Among well-known molecular events of glioma, we found that MET F/SVs were at a relative late stage of glioma natural evolution in 338 gliomas, including 100 sGBMs, for which both genetic and transcriptomic data were available (Fig. 2A, Fig. S3a and Supplementary Table 5). Indeed, gliomas with MET alterations were at a late stage of tumor evolution, consistent with the observation that MET alterations were enriched in sGBM.

MET alterations are later events. A Distribution of progression scores of key molecular events in gliomas (n = 338). B Progression scores of malignant cells from cases with or without MET F/SVs. C, Comparison of single-cell subpopulations of malignant cells from cases with (n = 2) or without (n = 2) MET F/SVs. D Single cell clusters of GBM samples with or without MET F/SVs (Left), the plot of t-SNE of pseudotime in single-cell subpopulations of malignant cells (Right). E The distribution of MET F/SVs expression in 8 patients with paired initial tumors and progressed sGBM available. F The mutational landscape of sGBM with or without MET F/SVs
We next performed single-cell RNA-seq (scRNA-seq) analysis of four GBM samples to study the progression status of malignant cells of tumors with or without MET F/SVs (Fig. S3b-d and Supplementary Table 6). The malignant cells of tumors with MET F/SVs showed significantly higher progression scores than that of tumors without MET F/SVs (Fig. 2B and Fig. S3e). Using a single-cell trajectory analysis, malignant cells of subpopulation C0, C2 and C3, whose proportion is most increased in cases with MET F/SVs, were in the latest stage of cancer development by pseudo-time prediction (Fig. 2C-D). Functional annotation also showed that hallmarks associated with advanced tumor stages, such as hallmarks of epithelial mesenchymal transition and cell cycle, were highly enriched in subpopulation C0, C2 and C3 (Fig. S3f and Supplementary Table 7). Beyond that, we analyzed longitudinal RNA-seq data of eight paired samples to explore features associated with occurrence of MET alterations in the secondary GBM (sGBM) patients. We observed that the MET F/SVs is elevated during glioma progression in six out of eight patients (six patients progressed from LGG to sGBM; and two patients progressed from sGBM to sGBM) (Fig. 2E and Supplementary Table 8). Importantly, MET F/SVs had been found presenting in the initial tumor diagnosed as LGG in three patients, indicating that MET F/SVs might be associated with glioma evolution at the stage of LGG. Analysis of longitudinal whole-exome or whole-genome sequencing data revealed that the occurrence of MET F/SVs were accompanied with the appearance of mutant genes associated with DNA damage repair in three paired cases with MET F/SVs (Fig. S3g). It has been reported that IDH-mutant non-co-deletion cases with CDKN2A/B homozygous deletion were associated with genomic instability [2, 20]. Here, we noticed that IDH1, TP53, ATRX, CDKN2A/B homozygous deletion, and RB1 alterations significantly co-occurred with MET F/SVs in sGBM (Fisher P < 0.05) (Fig. 2F). These findings suggested that the occurrence of MET alterations associated with genomic instability during glioma evolution. The CDKN2A/B homogenous deletion has been a prognostic and high-grade marker in IDH-mutant astrocytic gliomas and Barrett’s esophagus [21,22,23,24,25]. Here, we found that sGBMs with MET F/SVs and CDKN2A/B loss indicated the worst OS compared with those with MET wildtype, CDKN2A/B loss and MET wild-type, CDKN2A/B wildtype (Fig. S3h).
3.3 Transcriptomic features of glioma with and without MET F/SVs
The MET overexpression can activate cell proliferation, reduce apoptosis, and promote tumor migration [26, 27]. However, MET overexpression does not always accompany MET amplification or METex14 in lung cancer [28]. And the correlation between MET overexpression and MET F/SVs was also unclear in gliomas. Here, we investigated the association between MET overexpression and the MET F/SVs in sGBM. Interestingly, we observed that MET expression significantly increased with an increasing number of MET F/SVs events (Fig. 3A). The MET expression was also significantly increased in sGBM with each variant and all types of MET F/SVs compared with in the wild-type patients (Kruskal–Wallis P < 8.6 × 10–9) (Fig. S4a-e). To examine whether sGBM harboring MET F/SVs presents biological characteristics which could be triggered by MET signaling activation, we analyzed the biological function between bulk samples with and without MET F/SVs in CGGA dataset. As a result, 672 genes were found to be differentially expressed in MET F/SVs and over-expressed sGBMs. The CDK6 and CDC20 in cell-cycle progression pathway and CDKN2A in RB pathway showed elevated expression in both patients with MET F/SVs and overexpression. The fold change of the differentially expressed genes in samples with MET F/SVs and overexpression were highly correlated (Pearson’s correlation coefficient = 0.88, P < 2.2 × 10–16), indicating similar effects of MET F/SVs and MET overexpression (Fig. 3B).

Transcriptomic features of glioma with and without MET F/SVs. A The distribution of MET expression in glioma with and without MET F/SVs. B The differentially expressed genes in sGBM with MET F/SVs (compared to sGBMs without MET F/SVs) and sGBMs with MET overexpression (compared to sGBMs without MET overexpression). C MET expression in malignant cells of sGBM with or without MET F/SVs at single-cell level. D Hallmarks enriched in malignant cells of sGBM with or without MET F/SVs. E Cell cycle analysis of each single-cell transcriptome from four GBM samples. F The IHC of c-MET, p-MET, Ki-67, CDK6, and CDC20 and CDK4 in sGBM with and without MET F/SVs. Bar = 50 µm
In addition, scRNA-seq analysis showed that MET expression significantly increased in malignant tumor cells in cases with MET F/SVs (Fig. 3C). Functional analysis of differentially expressed genes between malignant tumor cells of cases with or without MET F/SVs showed that epithelial mesenchymal transition hallmarks enriched in cases with MET F/SVs (Fig. 3D). Cell cycle analysis of each single-cell transcriptome revealed increase of G2M or S phase cells in MET F/SVs patients, suggesting that MET F/SVs were associated with increased malignant features, such as cell proliferation (Fig. 3E).
We validated the differentially expressed genes in patients with and without MET F/SVs by IHC. Total MET (c-MET), phosphorylated MET (p-MET), Ki-67, CDK6, and CDC20 levels were elevated in sGBM with MET F/SVs, compared with sGBM without MET F/SVs (Fig. 3F). Collectively, these observations suggested that MET F/SVs are robust biomarkers to predict the MET overexpression and MET overexpression associated functions in sGBM. We subsequently observed that the proliferation rate of PDCs harboring MET F/SVs (PDC_25_1_S with METex10 and METex19, and PDC_28_1_S with METex10) was higher than that of PDCs without MET F/SVs (PDC_18_1_S and PDC_22_1_S, Fig. S4f). Taken together, the observations suggest that MET F/SVs could be indicative of the MET overexpression, which could enhance cell proliferation and transcriptional activities, in sGBM.
3.4 Preclinical testing and translation of a MET inhibitor treatment into a clinical setting
To delineate the characterization of MET alterations on drug response, we investigated drug sensitive test on response to TMZ and a MET inhibitor Crizotinib in PDCs. We observed that PDC_25_1_S and PDC_28_1_S harboring MET F/SVs exhibited less sensitive to TMZ than PDC_18_1_S and PDC_22_1_S (Fig. 4A), consistent with our previous report that gliomas with high MET expression are resistant to TMZ [29]. Meanwhile, PDC_25_1_S and PDC_28_1_S were more sensitivity to crizotinib, an FDA-approved multi-targets tyrosine kinase inhibitor, compared with MET wildtype PDCs (PDC_18_1_S and PDC_22_1_S) (Fig. 4B). The findings indicated that gliomas with MET F/SVs might be sensitive to MET-targeted therapy.

Preclinical testing and translation of a MET inhibitor treatment into a clinical setting. A-B The relative viability of cells (represented by the relative CCK8 value) after treatment of TMZ (A) and crizotinib (B). C Overall survival of sGBM patients (with MET F/SVs) with or without MET inhibitors (PLB1001). D Diagram of the glioma progression and treatment in patient S1451. E Diagram showing the presence of ST7-MET in primary glioma sample of this patient. F IHC of p-MET, Ki-67, CDK6, and CDC20 from primary glioma sample of this patient. Bar = 50 µm. G All MRI images represent axial T1-weighted scans with contrast enhancement of the patient at baseline and at indicated time points after initiation of treatment. Crizotinib (250 mg bid) were administrated orally at recurrence after 4 months from the diagnosis. Marked tumor shrinkage at the site of the main lesion was observed (arrows)
In our clinical trial (NCT02978261) studying of a c-MET inhibitor PLB1001 in glioma patients, all of six sGBM patients harboring MET F/SVs who had received MET-specific small-molecule inhibitor PLB-1001 have reached the end of follow-up. The OS of these six patients with MET inhibitor treatment significantly improved, compared with OS of other twelve sGBM patients with MET F/SVs without MET inhibitor treatment (Fig. 4C), suggesting patients with MET F/SVs conferred more sensitivity to MET-targeted therapy.
Encouragingly, we found that a recurrent glioma patient with MET fusion (ST7-MET) in her initial tumor well response to MET-targeted therapy after resistant to standard chemoradiotherapy (Fig. 4D-G). This patient was a 37-year-old female, diagnosed with astrocytoma with IDH mutation, WHO grade 2, which was resected followed by TMZ concurrent with radiotherapy (Fig. 4D). the ST7-MET fusion was detected in the specimen by RNA-sequencing (Fig. 4E). The IHC staining showed the initial tumor with characteristics that tended to evolve to sGBM with MET F/SVs (Fig. 4F). The patient subsequently underwent crizotinib treatment (250 mg bid) at recurrence after 4 months from the diagnosis. Tumor shrinkage and symptom relief were observed after 4 months of MET inhibitor treatment. The patient is still alive, with an over 2-year post-progression survival (Fig. 4G). This indicates that the MET inhibitor treatment is beneficial to not only sGBM patients who harbor ZM and/or METex14, but also to those with other MET F/SVs. Considering the similarity among various MET F/SVs identified in this study, MET inhibitor, might represent a promising therapeutic option for glioma patients with MET F/SVs.
4 Conclusion
In conclusion, we revealed the landscape of MET F/SVs in glioma and demonstrated that they were induced in the course of glioma evolution. In addition, MET F/SVs could predict the overexpression of MET and were strongly associated with proliferative biological phenotypes in glioma. Clinically, the combination of MET F/SVs expanded the proportion of glioma patients who would benefit from MET-targeted therapy.
Availability of data and material
The RNA sequencing and WES data used in this study is available on the CGGA website, which can be accessed freely.
References
Jiang T, et al. Clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. 2021;499:60–72.
Barthel FP, et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature. 2019;576:112–20.
Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2014–2018. Neuro Oncol. 2021;23:iii1–105.
Louis DN, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23:1231–51.
Chai R, et al. Predictive value of MGMT promoter methylation on the survival of TMZ treated IDH-mutant glioblastoma. Cancer Biol Med. 2021;18:272–82.
Hu H, et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell. 2018;175:1665–78.
El-Khoueiry AB, Hanna DL, Llovet J, Kelley RK. Cabozantinib: An evolving therapy for hepatocellular carcinoma. Cancer Treat Rev. 2021;98: 102221.
Camidge DR, et al. Crizotinib in Patients With MET-Amplified NSCLC. J Thorac Oncol. 2021;16:1017–29.
Yang X, Li Y, Duan Q, Zhang Q, He M. Identification of a Novel MET Exon 14 Skipping Variant in Lung Adenocarcinoma Sensitive to Crizotinib Treatment. J Thorac Oncol. 2021;16:e17–9.
Zhao Z, et al. Chinese Glioma Genome Atlas (CGGA): a comprehensive resource with functional genomic data from Chinese gliomas. Genomics Proteomics Bioinformatics. 2021;19(1):1–12.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Wang J, et al. Clonal evolution of glioblastoma under therapy. Nat Genet. 2016;48:768–76.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol. 2016;12: e1004873.
Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Chang YZ, et al. METTL3 enhances the stability of MALAT1 with the assistance of HuR via m6A modification and activates NF-kappaB to promote the malignant progression of IDH-wildtype glioma. Cancer Lett. 2021;511:36–46.
Chai RC, et al. The molecular characteristics of spinal cord gliomas with or without H3 K27M mutation. Acta neuropathol Commun. 2020;8:40.
Ma PC, et al. c-MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63:6272–81.
Giam M, Rancati G. Aneuploidy and chromosomal instability in cancer: a jackpot to chaos. Cell Div. 2015;10:3.
Bian YS, Osterheld MC, Fontolliet C, Bosman FT, Benhattar J. p16 inactivation by methylation of the CDKN2A promoter occurs early during neoplastic progression in Barrett’s esophagus. Gastroenterology. 2022;122:1113–21.
Aoki K, et al. Prognostic relevance of genetic alterations in diffuse lower-grade gliomas. Neuro Oncol. 2018;20:66–77.
Appay R, et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019;21:1519–28.
Shirahata M, et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018;136:153–66.
Yang RR, et al. IDH mutant lower grade (WHO Grades II/III) astrocytomas can be stratified for risk by CDKN2A, CDK4 and PDGFRA copy number alterations. Brain Pathol. 2020;30:541–53.
Day EK, et al. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020;30(3383–3396): e3387.
Paik PK, et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 2015;5:842–9.
Guo R, et al. MET IHC Is a Poor Screen for MET Amplification or MET Exon 14 mutations in lung adenocarcinomas: data from a Tri-institutional cohort of the lung cancer mutation consortium. J Thorac Oncol. 2019;14:1666–71.
Li MY, et al. Low c-Met expression levels are prognostic for and predict the benefits of temozolomide chemotherapy in malignant gliomas. Sci Rep. 2016;6:21141.
Acknowledgements
The authors thank the patients, study investigators, and staff who participated in this study.
Funding
This project was supported by grants from the Natural Science Foundation of China (NSFC)/Research Grants Council (RGC), Hong Kong, China Joint Research Scheme (81761168038); the National Natural Science Foundation of China (81802994, 81903078, 81972337, 81972816, 82002647, 82192894, 82103623, and 82002994), the Mainland-Hong Kong Joint Funding Scheme ITC grant MHP/004/19 and MOST grant 2019YFE0109400; the Beijing Natural Science Foundation (JQ20030); Sino-German Center for Research Promotion (M-0020); and the Beijing Nova Program (Z201100006820118).
Author information
Authors and Affiliations
Contributions
J.T., Z.Z. and Q.W. conceptualized the project. K.Z., Z.Z., Z.B., R.C. and L.W. analyzed and interpreted the sequencing data. J.C., R.C., K.Z., and H.L. performed the experiments. G.L., Y.C. and Q.W. prepared patient samples for sequencing and partially performed data pre-processing. R.T. carried out the clinical patients with MET inhibitor. Z.S. and Z.W. performed single cell analysis. Q.M., H.H., F.Z., Z.W., F.W., Y.Z. and Y.L. undertook the prospective patient enrolment, diagnosis, and clinical follow-up. J.W., W.Z. and C.K. and guided data analysis. U.D.K., K.Z., Z.B., R.C. and Z.Z. wrote the manuscript, which was further revised by D.H.N., T.J., J.W., and Q.W. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All studies performed were approved by the Institutional Review Board (IRB) of Beijing Tiantan Hospital, Capital Medical University. Written informed consent were obtained from all patients.
Consent for publication
The authors agreed to publish this manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Data summary and validation of MET alterations. a, The data composition of this study. b, schematic diagrams of novel fusion genes (LHFPL3-MET and RSBN1L-MET). c-d, Sashimi plot of RNA-seq data of all cases with METex10 (c) and METex19 (d). e-f, Sanger sequencing of PCR products of case S0179 with METex10 and METex19.
Additional file 2: Fig. S2.
Characteristic of METex10 and METex19. a, Diagram of the protein products of different MET exon skipping. b, The truncated protein band of METex10 (single star) and METex19 (double stars). c, The number of cases with multiple MET variants or single MET variant. d-g Frequencies of METex10, METex14, METex19, and MET fusions in pLGG, rLGG, pGBM, rGBM and sGBM. h-k, Overall survival of sGBM patients (first-diagnosis) stratified by the METex10 (The median survival is 150 days and 302 days for METex10 and non-METex10, respectively. h,), METex14 (The median survival is 224 days and 301 days for METex14 and non-METex14, respectively. i), METex19 (The median survival is 192 days and 299 days for METex19 and non-METex19, respectively. j), and MET fusions (The median survival is 195 days and 310 days for MET fusion and non-MET fusion, respectively. k).
Additional file 3: Fig. S3.
MET alterations are results of tumor evolution. a, Distributions of key molecular events in different gliomas. b-c, tSNE visualization of two GBM patients with MET F/SVs and two without MET F/SVs. d, Feature plot showing progression scores of malignant cells of sGBMs with or without MET F/SVs. e, The most enriched cancer hallmarks for applying the gene set enrichment analysis (GSEA) in different single-cell subpopulations of malignant cells. f, Evolutionary trees of three patients (two with novel MET alterations occurred in the recurrent samples, and one with inherit MET alterations from pre-recurrent samples) evaluated by whole-exome or whole-genome sequencing. Selected driver mutations are labeled in black. g, Survival of IDH-mutant sGBM with CDKN2A/B homozygous deletion and/or MET F/SVs.
Additional file 4 Fig. S4.
Characteristics of gliomas with MET alterations. a-d, The distribution of MET expression in sGBM with or without METex10 (a), METex14 (b), METex19 (c), and MET fusions (d). e, The distribution of MET expression in sGBM with different type of MET F/SVs. f, The proliferation rate of patient derived cells with or without MET F/SVs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, KN., Zhao, Z., Chen, J. et al. MET fusions and splicing variants convergently define a subgroup of glioma sensitive to MET inhibitors. Holist Integ Oncol 1, 18 (2022). https://doi.org/10.1007/s44178-022-00014-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44178-022-00014-9