
Abstract
Woodhouse-Sakati syndrome (WSS) is a rare eponymous disease described by Drs. Woodhouse and Sakati in 1983 as a syndrome of hypogonadism, alopecia, diabetes mellitus, intellectual disability, and ECG abnormalities. A couple of years later, a variant in the gene DCAF17 (DDB1 and CUL4-associated factor 17) was labeled as the founder mutation in most cases of WSS in the Arabian Peninsula and the Middle East. Reports around the world started to emerge on variable presentations of the syndrome, expanding its phenotypic spectrum. In addition, the discovery of new variants in the same gene grew our understanding of this multi-systemic syndrome. Genotype and phenotype expansion is increasing with the growing number of diagnosed cases owing to the availability and advances in clinical genetic testing. This review describes the current understanding of the DCAF17 gene with its molecular implication in WSS. We also provide an extensive analysis of the documented genetic changes associated with the syndrome, describing the geographical prevalence of these genetic variations. Additionally, we examine the disorder’s extensive manifestations and clinical presentations and describe a case of intra-familial phenotypic heterogeneity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
What is today known as Woodhouse-Sakati syndrome was first described as “A familial syndrome of deafness, alopecia and hypogonadism” in 1973 in three male siblings, and it was only 10 years later that Drs Woodhouse and Sakati described “A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities” that led to its current nomenclature [15, 46]. The syndrome follows an autosomal recessive mode of inheritance, and cases have been reported worldwide, starting in the Middle East and the Arabian Peninsula and spanning across Europe and East Asia [7, 11, 42, 48].
Most reported cases present to the pediatric clinic for amenorrhea or delayed secondary sexual characteristics for girls and boys respectively with evident temporal alopecia. The diagnosis is confirmed with a positive pathogenic or likely pathogenic variant in DCAF17 (OMIM 612515).
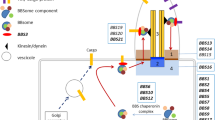
DCAF17 is known to be linked to E3 ligase function, and nucleolar functions, and is ubiquitously expressed in almost all human tissues [6, 27, 44, 45].
In this review, we aim to report on a family clinically followed for 20 years with intra-familial heterogeneity. Additionally, we will do a comprehensive analysis of the recognized genetic alterations associated with WSS, focusing on their geographical distribution, and examine the phenotypic variations and clinical presentations of the disease.
Clinical presentation
Case presentation
We present a family of eight siblings from a first-degree consanguineous marriage. Four out of the eight siblings are affected and presented for clinical evaluation (III-1, III-2, III-7, III-8). The two eldest sisters (III-1, III-2) were clinically diagnosed in 2003 with WSS and were described by our team [32]. These siblings are now at the age of 37 and 36 years, respectively. The four middle children of the family have not reported any complaints. The two youngest siblings (III-7, III-8) have been diagnosed with WSS during this current presentation. The seventh child is a 27-year-old female, and the eighth child is a 26-year-old male. The patients presented to the clinic for a follow-up of previous diagnoses and assessment of the two youngest siblings (Fig. 1). The four siblings, presented in this report, suffer from moderate intellectual disabilities, which lead to the termination of their education before high school level. They are also not independent in their daily activities. Their parents designated individual III-4 as their legal guardian.

Family pedigree with shaded shapes indicating affected individuals and Roman numerals I, II, and III indicating the generation number
The three female siblings (III-1, III-2, III-7) were complaining of underdeveloped secondary sexual characteristics and alopecia. The alopecia in the three sisters was more significant on the temporal sides and sparse eyebrows were noted. The skin of the patients was very distinctive and appeared very thin with evident wrinkles, giving it a progeroid appearance. An endocrinology-focused physical examination revealed delayed Tanner stages of development for all the sisters. From the oldest to the youngest, the stages were as follows: Stage II, Stage II, and Stage I. Neurologic assessment of the three sisters did not reveal significant findings except for mild intellectual disability for all three in addition and moderate dysarthria for the youngest sister.
The youngest brother (III-8) complained mostly of a similar appearance to his elder sisters and similar intellectual complaints. In addition, the patient complained of mild hearing difficulty. Upon physical examination, he also had temporal alopecia, sparse eyebrows, and progeroid skin with less pronounced features compared to his sisters (Fig. 2). When focusing on the endocrinological physical examination, we were surprised to find a normally developed gonadal and pubic development corresponding to Tanner’s stage V. Neurologic assessment focused on reported decreased peripheral sensation and motor function in his lower extremities. Physical examination revealed moderate dysarthria, poor saccadic eye movements, weak eye closure, and marked lower facial weakness. He was also found to have temporal and facial muscular wasting. Focusing on motor function, proximal strength, and reflexes were mostly normal. Peripheral motor strength was decreased in upper and lower extremities extensors. Atrophy of hand extensors was also noted. The patient also had a steppage gait, in addition to Parkinsonian-like tremors in the upper extremities are more pronounced on the right than the left. Hearing assessment in the clinic did not yield any significant results due to poor patient compliance; the patient was referred for formal hearing assessment.

Photograph showing the physical examination findings for the younger brother showing progeroid skin, temporal alopecia, and triangular facies
Laboratory and workup
The four affected siblings underwent a laboratory workup primarily for endocrinologic assessment including a complete blood count with differential, complete lipid profile, fasting blood glucose, HbA1c, insulin levels, vitamin D levels, anti-thyroperoxidase and anti-thyroglobulin serologies, thyroid function tests, and gender-dependent hormonal panel. The three sisters were found to have primary hypogonadism with estradiol levels at 6.59, 5.88, and 5 pg/ml respectively from the youngest to the oldest of the three sisters. The brother had normal levels of both gonadotropins and testosterone. The two eldest sisters and the youngest brother were all anemic. The two youngest sisters were shown to have low levels of free T4 with normal levels of free TSH. However, the youngest brother was shown to have high levels of TSH and a normal free T4 level. Finally, the second oldest affected sister (III-2) and her youngest brother (III-8) were shown to have low insulin levels with normal glycemic indices.
The three sisters underwent abdominopelvic ultrasounds with the following results. The eldest sister’s (III-1) ultrasound showed a small “inhomogeneous” uterus (20 × 19 × 11 mm) with the absence of both ovaries. The second oldest affected sister (III-2) was shown to have a more homogeneous but small-for-age uterus (23 × 18 × 25 mm), also with the absence of both ovaries. The ultrasound of the youngest affected sister (III-7) showed an “inhomogeneous” small uterus (25 × 14 × 22 mm) with the absence of both ovaries. No genital ultrasound was done for the brother because of the age-adequate development of gonads. However, semen analysis for the assessment of gonadal function was declined by the family due to personal religious beliefs.
Individual III-8 also underwent a formal hearing test and the audiogram showed a moderate sensorineural hearing deficit more prominent on the right side. He also had a brain MRI, which showed sub-centimetric circular white matter lesions at the white–gray matter junction bilaterally that were reported as non-significant during neurological assessment. Electromyography was deferred due to financial limitations by the patient and family.
Genetic testing
Blood samples were drawn from the second eldest sister (second child) for genetic testing. Whole exome sequencing (WES) showed a deletion in the fourth exon of DCAF17 gene c.436delC leading to a frameshift mutation and leading to early truncation of the protein. This deletion was labeled to be a pathogenic variant and has been previously reported in cases of WSS, which confirmed the diagnosis [36].
Case management and follow-up
After establishing the diagnosis in the family described above, we scheduled a clinical follow-up to discuss the implications and the management of the disease. Addressing the primary complaint of hypogonadism, mainly in the three sisters, we recommended hormone replacement therapy aiming to help the development of secondary sexual characteristics even though expectations for success were not very high due to the age at initiation of treatment. The symptomatic management was prescribed after explaining to the three sisters and the family that the infertility could not be treated because of absent and/or underdeveloped gonads.
The younger brother, after denying the sperm analysis, confirmed that he did not wish to procreate waiving all plans of treatment and management in that regard.
Levothyroxine supplementation was prescribed for the two youngest patients (III-7 and III-8) due to their hypothyroid state.
Neurological management for the youngest brother focused on follow-up assessments for surveillance of symptoms progression would warrant further workup and possible medical therapy.
It was agreed with the family to schedule a follow-up 6 months after the initial consult. Due to the deterioration in the familial socioeconomic status, the follow-up was done over the phone. The three sisters discontinued their hormone replacement therapy due to a lack of improvement in secondary sexual characteristics. The two youngest siblings were also not compliant with levothyroxine therapy for the same reason.
Upon further discussion during the second follow-up, the family reported deterioration in the neuro-psychiatric state of the youngest brother. They described episodes of emotional outbursts with harmful threats to his sisters and parents. He also had two episodes of described syncope with no defined triggers that were characterized by a post-ictal phase with low muscle tone and urine loss. He was not able to seek adequate medical attention and refused neurologist and psychiatry referrals. A second follow-up was scheduled for all the family for a physical exam and assessment.
One year later, only the brother presented to the follow-up. The visit focused on a neurological assessment by a neurologist specialized in neuromuscular and peripheral nervous system disease. The exam showed choreoathetosis movements in the right upper extremity, dystonic features in the right foot during ambulation, ataxic gait, and significant dysarthria that was increased compared to the initial presentation. Deep tendon reflexes were normal in both upper and lower extremities. Babinski reflex was negative bilaterally. The patient was prescribed Levodopa for symptomatic management and to halt the progression of neurological symptoms.
Discussion
The first cases of patients that were later labeled as WSS were reported in 1973 and 1979. The first cases were described in California as three brothers with progressive deafness, alopecia, and hypogonadism thought to be Björnstad syndrome (#262000), which is characterized as deafness and pili torti (fragile hair) [15]. However, this diagnosis was ruled out because of the hypogonadism in the three brothers which is not found in Björnstad syndrome. Later, in 1979, the first Middle Eastern cases were described in two siblings and a maternal cousin as a familial syndrome of hypogonadism and alopecia [38]. After that, six Saudi patients were described and the phenotype was established to add endocrinological manifestations namely diabetes mellitus, intellectual disabilities or mental retardation, and electrophysiologic cardiac findings [46]. This review describes in depth the spectrum of the phenotypic characterization of most WSS cases present in the literature, in addition to including a description of four siblings from a Lebanese family with intra-familial variability. This phenotypic variation has been previously described in case series or case reports in the literature [4, 11]. The phenotypic spectrum continues to expand which can make it difficult to have a unifying clinical presentation of the syndrome. However, this review showed consistencies in most patients diagnosed with WSS: hypogonadism, temporal alopecia, and prematurely aged skin.
All reported WSS patients share pathogenic or likely pathogenic variants in a DCAF17. The most common variant worldwide remains c.436delC, which causes premature transcript termination and interrupts the cell cycle and nucleolar function [4, 17, 23, 27]. Other variants in the same gene were also found in WSS patients most of which also cause early truncation of the protein or variable splicing due to splice-site modifications [5]. The repertoire of around 20 different loci in the DCAF17 linked to WSS cases confirms the importance of normal nucleolar localization of this protein for normal cell cycle and homeostasis [13, 17].
Reviewing the major complaints and the gaps in molecular pathophysiology, clinical work should focus on obtaining the correct diagnosis early. The quality of life is heavily impacted in this rare disease due to many factors namely the triangular face, progeroid skin appearance, and most importantly in female patients lack of secondary sexual characteristics. All of these can lead to social exclusion, especially in Arab and Middle Eastern countries in which consanguineous families commonly live in peripheral cities. In addition, these families can be reluctant to genetic counseling due to cultural and religious beliefs, which limits even further the possible intervention of the healthcare professionals after a late diagnosis in families [47]. Finally, intra-familial disparities in the presentation can sometimes also be a limitation in explaining the genetic aspect and importance of prenatal assessment. The review of such rare diseases is important to have integral descriptions of the disease and establish screening recommendations. Similar catalogues exist for the Middle East region and show that five author groups have described cases of Woodhouse-Sakati including the initial case by Drs. Woodhouse and Sakati [10].
Genetic review
Molecular mechanism
The first mutations causing WSS were discovered in 2008 by Alazami and his colleagues, approximately 25 years after the first clinical reports of the disease. The gene was initially named C2orf37 (Chromosome 2 Open Reading Frame 37) due to its location on chromosome 2q33.2 and its uncharacterized function [4]. It was later given the name DCAF17 denoting its function as DDB1 and CUL4-associated factor 17 [4, 6, 27]. The gene was found to play a significant role in various biological processes, including DNA repair, protein degradation, cell cycle regulation, and apoptosis [6, 27]. It is known to have two major transcripts in humans, the first one is composed of 520 amino acids, and the second of 453 amino acids [7, 16].
CUL4 and DDB1 proteins have been studied due to their high expression in pluripotent stem cells, more specifically in the nucleolus, and have been linked to the mechanism of differentiation and regulation of the cell cycle in these cells [8, 17]. In addition, DCAF17 knock-out models revealed its importance in spermatogenesis and cell cycle homeostasis while the exact mechanism is yet to be established [35].
The DCAF proteins belong to the WD40-containing protein family. This larger family of proteins is characterized by an amino-acid sequence of 40–60 units that are recognized by terminal dipeptides of tryptophan (W) and aspartate (D) [43]. This conserved region helps bind to damage DNA binding 1 (DDB1) protein which has been linked to multi-systemic diseases involving neurological, endocrinological, and overall oncologic systems [28, 35, 39]. However, DCAF17 and some other DCAF proteins, such as DCAF15 and DCAF16, do not appear to have this conserved interaction domain region but can still interact with DBB1 through other binding domains [23, 35]. DCAFs also create the proximity needed between E3 ligase and its substrate leading to the ubiquitination of the targeted protein [27].
Woodhouse-Sakati syndrome genotype
In 2008, Alazami and his colleagues identified the founder variant of WSS in 8 families from the Arabian Peninsula. This DCAF17 variant leads to disruption of the nucleotide transcription and ultimately loss of function of the gene [4]. The gene is composed of 17 exons.
Different loci are reported within the gene in patients with WSS, and some specific variants are linked to distinctive phenotypes [24]. The variants seem to be linked to ancestry and race. Most cases in the Arabian and Middle Eastern regions have the c.436delC variant [24]. This deletion in exon 4 causes a frameshift mutation that leads to early truncation of the native 520 amino acids [4, 5]. This deletion is described in 21 reports including Qatari, Kuwaiti, Bahraini, Saudi Arabian, Palestinian, Tunisian, and Lebanese families.
Another Middle Eastern splice-site variant was also reported by Alazami et al. in 2008 as a substitution of Thymine in position 1091 by Guanine leading to exon 10 skipping.
Pakistani cases have been described, and three different variants were reported in 2011, 2016, and 2020. One of which is a deletion of an Alanine in the 270th position of the transcript leading to the truncation of the sequence to 96 amino acids [6]. Another report found an intronic splice-site variant report (c.321 + 1 G > A) [21]. The most recently described patient has a novel variant affecting the initiation codon (c.1A > G) [42].
Two cases of compound heterozygotes have been described in 2018. A Saudi Arabian patient described by Zou et al. in 2018 has the following variants c.256 T > C, c.159 T > C. The second patient is the only WSS described in the USA with compound loss of function mutations in exon 5 (c.C535T) and in exon 9 (c.G906A) [20].
Four different variants in Turkish cases of WSS have been described: a splice-site mutation (c.127-3delTAGinsAA), two frameshift mutations (c.127-1G > C and c.1091 + 1G > A), and a duplication mutation c.270dup [5].
Five different variants have been reported in European cases in Italy, Slovenia, France, and Portugal. Italian cases have been described with nonsense mutations, the first one being a substitution of cytosine by adenine in position 341 and the second one also a substitution of guanine by adenine in position 906 [3]. The Slovene variant is a cytosine deletion at position 50 of the gene causing a frameshift [31]. French reports described gypsy patients with a unique nonsense mutation due to a substitution of the 387th nucleotide from guanine to adenine [3]. The most recent European variant reported in a Portuguese WSS patient, c.1091 + 2 T > C, is a splice-site mutation in intron 10 causing skipping of the tenth exon [29].
Several East Asian cases are present in the literature. Two Chinese cases have been described. The first, c.1488_1489delAG variant is present in exon 14, and the second, c.1111delA is present in exon 12 and both lead to early truncation of the protein [13, 48]. Two Japanese brothers with WSS have a substitution at position 796 from a guanine to a thymine [30].
Finally, three Indian variants, two of which are reported to be splice-site mutations, c.1422 + 5G > T and c.459-7_499del, and one frameshift c.1238delA exist in the literature [1, 4].
Phenotype review
The first clinical cases that are recognized in the literature as WSS were described in Italy by Crandall et al. in 1973 and in Lebanon by Salti and Salem in 1979 [15, 38]. Stemming from these described cases, WSS is today defined as an autosomal recessive disease with hypogonadism, deafness, alopecia, mental retardation, diabetes mellitus, and progressive extrapyramidal defects. The eponymous syndrome’s clinical features continue to expand. The two features that remain constant in all described patients are hypogonadism and dermatologic findings namely alopecia and/or progeroid skin [3, 5, 7, 24, 31]. The endocrinological features, namely diabetes mellitus and hypothyroidism, are reported in approximately 50% and 30% of the described patients in the literature, respectively [5, 24]. Intellectual disability is also reported in the majority of WSS patients and can be linked to the occurrence of sensorineural hearing loss.
Neurological involvement in WSS has been extensively described. The most common neurological finding involves extrapyramidal signs such as dystonia and dysarthria [11]. Dysarthria is believed to precede the occurrence of motor dystonia. The latter which happens in a descending manner, ultimately leads to paraplegia in most severe cases, leading to patients being wheelchair-bound [7]. In rare cases, other neurologic findings have been described such as tremors, ataxia, and seizures [11]. Neurological assessment across all the cases has not been consistent. Some patients have non-specific white matter lesions, while others have described leukodystrophic changes and most recently iron deposition. From the reported natural history of the disease, brain MRI is becoming an integral part of WSS workup after diagnosis [8, 19, 22].
The cardiac system can also be affected in WSS patients. The most common findings are electrophysiological changes such as ST-segment lengthening and T-wave flattening which were described in the initial cases by Drs. Woodhouse and Sakati and later in Indian and Middle Eastern cases [25, 40, 46]. One case has been described with a ventricular septal defect present at birth that has not been directly linked to the mutation in the DCAF17 gene [41].
Intra-familial heterogeneity
The diagnosis of Woodhouse-Sakati syndrome relies on the most common clinical manifestations such as alopecia, endocrinological irregularities, and developmental delay. However, specific phenotypic characterization can be found in individual patients. The family described in the report shows the importance of thorough assessment due to underrecognized phenotypes. In this family, the affected siblings comprise 3 females and 1 male. Even though all of them had typical characteristics of WSS, they also had unique traits (Table 1). Alopecia was consistent among all four reported patients; however, it affected different regions of the head with more severe temporal alopecia in the sisters compared to their brother. The premature aging of the skin followed the same pattern with the sisters having a slightly more pronounced phenotype. However, the neuro-psychiatric phenotype was almost absent in the three sisters, compared to their brother. He initially complained of mild tremors, peripheral weakness, and mild hearing difficulty. Over the years, his symptoms worsened to include foot drop and life-impairing tremors. More than that, he also developed a severe psychiatric phenotype with outbursts of violent and menacing behaviors against his siblings and other family members.
Differential diagnosis
The diagnosis of WSS can be confirmed with the clinical criteria or based on milder clinical presentation with pathogenic variants in the DCAF17 gene. Most patients diagnosed with WSS after genetic workup initially present for primary amenorrhea in their pubertal years with or without neurologic complaints [4]. Thus, in the workup of WSS, other possible diagnoses should be ruled out. The first disease to rule out for WSS-like presentation is isolated GnRH deficiency (OMIM#308700). This disorder may present as a spectrum, of which Kallmann syndrome is the most common and has a distinctive presentation with olfactory dysfunction. This can be ruled out simply by a normal GnRH level in the patients and by the absence of anosmia [18, 46]. Recently, WSS has been described in the spectrum of neurodegeneration with iron brain accumulation (NIBA) (OMIM #234200) disorders and should be kept on the differential for final diagnosis [12]. Iron accumulation in the globus pallidus can be present in WSS but is not specific to the disease and can be present in other types of NIBA such as pantothenate kinase-associated neurodegeneration (PKAN) (OMIM#606160) and PLA2G6-associated neurodegeneration (PLAN) (OMIM#610217) [40]. Even though the clinical features of each NIBA may differ slightly, the most definite way to diagnose a specific NIBA is genetic testing for known variants [19].
Alopecia is also a major characteristic of WSS, which can be the initial complaint of patients in the clinic; it is thus important to rule out other pathologies that could present with similar dermatological complaints. CARASIL (cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy, OMIM#600142) presents with gait disturbances around the age of 20 years with diffuse alopecia and neurological complaints [33]. In CARASIL, patients normally present primarily with focal neurological complaints and alopecia concentrated on the scalp area. Neurological complaints occur in a more acute presentation due to the pathophysiology of the disease. CARASIL patients also lack endocrinological complaints that are characteristic of WSS [26]. Gomez-Lopez-Hernandez syndrome (GLHS) is also an important diagnosis to have in the differential of patients presenting for cephalic alopecia. Patients with GLHS present between childhood and adulthood with neurological manifestations ranging from mild developmental delay to more prominent focal symptoms such as the typical trigeminal mononeuropathy or truncal ataxia [14]. Other characteristic findings in GLHS patients are typical facies such as turricephaly and brachycephaly in addition to neuroradiological findings, namely rhombencephalosynapsis with subsequent hydrocephalus [37]. The absence of typical imaging findings and lack of focal GLHS neurological signs such as trigeminal neuralgia and lack of GLHS facies help rule out this diagnosis for our patients.
Treatment
Establishing a treatment plan for patients diagnosed with Woodhouse-Sakati is complex due to the different patient-specific complaints. Amenorrhea and lack of secondary sexual characteristics are the major complaints in female and male patients respectively, and thus, endocrinological treatment is at the center of management. The early initiation of hormonal replacement therapy is fundamental to optimize results and avoid long-term complaints and stigma [2, 9]. Diabetes mellitus and hypothyroidism are also common endocrinological complaints, which are treated with insulin and thyroid replacement therapy respectively [2].
Considering neurological management, the treatment focuses on symptomatic relief. For dystonia, the patients are normally started on a trial of anticholinergic medications or benzodiazepines [11, 34]. If the patient’s symptoms are refractory to the initial treatments, botulinum toxin injections can be tried, and if symptoms are persistent, some patients are considered for deep-brain stimulation [34].
Follow-up
Initial assessment most of the time includes a complete endocrinological panel screening for diabetes mellitus, gonadotropic, gonadic, and thyroid function. An electrocardiogram is always recommended at the time of diagnosis due to some patients presenting with conduction defects. Finally, even if patients do not present initially with neurologic symptoms, a baseline assessment of hearing function and brain MRI is useful to establish a baseline and to keep on record if the patient later develops new neurological symptoms.
The evolution of the disease is patient-dependent, and we recommend yearly endocrinological assessment and neurological screening every 5 years unless the patient develops new symptoms that would indicate a neurological pathologic process.
Conclusion
Variants in DCAF17 have been linked to all cases of WSS. The advancement in diagnostic genetic testing helped reveal around 20 different mutations in the gene of interest. In addition, the expansion of the phenotypic syndrome is important for clinical consideration and management of diagnosed patients. The defining features of WSS remain hypogonadism, alopecia with dermatomal findings, intellectual disability, and neurological complaints ranging from Parkinsonian-like tremors to seizures. It is important for cases raising suspicion for WSS to have early genetic testing, to ameliorate the molecular understanding of this disease and ameliorate the clinical management and surveillance of patients.
Availability of data and materials
All data underlying the results are available as part of the article and no additional source data are required.
References
Abdulla MC, et al. Novel compound heterozygous frameshift mutations of C2orf37 in a familial Indian case of Woodhouse-Sakati syndrome. J Genet. 2015;94(3):489–92. https://doi.org/10.1007/s12041-015-0544-7.
Agopiantz M, et al. Endocrine disorders in Woodhouse-Sakati syndrome: a systematic review of the literature. J Endocrinol Invest. 2014;37(1):1–7. https://doi.org/10.1007/s40618-013-0001-5.
Alazami A, et al. C2orf37 mutational spectrum in Woodhouse-Sakati syndrome patients. Clin Genet. 2010;78(6):585–90. https://doi.org/10.1111/j.1399-0004.2010.01441.x.
Alazami AM, et al. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am J Hum Genet. 2008;83(6):684–91. https://doi.org/10.1016/j.ajhg.2008.10.018.
Ali R, et al. Expanding on the phenotypic spectrum of Woodhouse-Sakati syndrome due to founder pathogenic variant in DCAF17: report of 58 additional patients from Qatar and literature review. Am J Med Genet A. 2022. https://doi.org/10.1002/ajmg.a.62501.
Ali RH, et al. Exome sequencing revealed a novel biallelic deletion in the DCAF17 gene underlying Woodhouse Sakati syndrome. Clin Genet. 2016;90(3):263–9. https://doi.org/10.1111/cge.12700.
Almeqdadi M, et al. Phenotypic variability of c.436delC DCAF17 gene mutation in Woodhouse-Sakati syndrome. Am J Case Rep. 2018;19:347–53. https://doi.org/10.12659/AJCR.907395.
Arber CE, et al. Review: insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol Appl Neurobiol. 2016;42(3):220–41. https://doi.org/10.1111/nan.12242.
Bakhsh H, Alqntash N, Almajed E. The successful management of primary amenorrhea in Woodhouse-Sakati syndrome: a case report and a literature review. Life. 2023;13(10):2022. https://doi.org/10.3390/life13102022.
Bizzari S, et al. Catalogue for transmission genetics in Arabs (CTGA) database: analysing Lebanese data on genetic disorders. Genes. 2021;12(10):1518. https://doi.org/10.3390/genes12101518.
Bohlega S, et al. Patterns of neurological manifestations in Woodhouse-Sakati syndrome. Parkinsonism Relat Disord. 2019;69:99–103. https://doi.org/10.1016/j.parkreldis.2019.10.007.
Brugger F, et al. Neurodegeneration with brain iron accumulation (NBIA) syndromes presenting with late-onset craniocervical dystonia: an illustrative case series. Mov Disord Clin Pract. 2016;4(2):254–7. https://doi.org/10.1002/mdc3.12393.
Chen G, et al. Case report: a deletion variant in the DCAF17 gene underlying Woodhouse-Sakati syndrome in a Chinese consanguineous family. Front Genet. 2021;12:741323. https://doi.org/10.3389/fgene.2021.741323.
Choudhary N, Prabhakar A, Bhatia V, Gupta PC. Gomez-López-Hernandez syndrome: the triad of cerebello-trigemino-dermal dysplasia. BMJ Case Rep. 2021;14(10):e246189. https://doi.org/10.1136/bcr-2021-246189.
Crandall BF, et al. A familial syndrome of deafness, alopecia, and hypogonadism. J Pediatr. 1973;82(3):461–5. https://doi.org/10.1016/S0022-3476(73)80121-0.
Fozia F, et al. Novel splicing-site mutation in DCAF17 gene causing Woodhouse-Sakati syndrome in a large consanguineous family. J Clin Lab Anal. 2022;36(1):e24127. https://doi.org/10.1002/jcla.24127.
Gao J, et al. The CUL4-DDB1 ubiquitin ligase complex controls adult and embryonic stem cell differentiation and homeostasis. eLife. 2015;4:e07539. https://doi.org/10.7554/eLife.07539. Edited by A.J. Wagers.
Gonzalez-Latapi P, Sousa M, Lang AE. Movement disorders associated with hypogonadism. Mov Disord Clin Pract. 2021;8(7):997–1011. https://doi.org/10.1002/mdc3.13308.
Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep. 2011;11(3):254–61. https://doi.org/10.1007/s11910-011-0181-3.
Gurbuz F, et al. Novel inactivating mutations of the DCAF17 gene in American and Turkish families cause male infertility and female subfertility in the mouse model. Clin Genet. 2018;93(4):853–9. https://doi.org/10.1111/cge.13183.
Habib R, et al. A novel splice site mutation in gene C2orf37 underlying Woodhouse-Sakati syndrome (WSS) in a consanguineous family of Pakistani origin. Gene. 2011;490(1):26–31. https://doi.org/10.1016/j.gene.2011.09.002.
Hogarth P. Neurodegeneration with brain iron accumulation: diagnosis and management. J Mov Disord. 2015;8(1):1–13. https://doi.org/10.14802/jmd.14034.
Jin J, et al. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23(5):709–21. https://doi.org/10.1016/j.molcel.2006.08.010.
Kohil A, et al. Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review. Orphanet J Rare Dis. 2023;18(1):22. https://doi.org/10.1186/s13023-023-02614-8.
Koshy G, et al. Three siblings with Woodhouse-Sakati syndrome in an Indian family. Clin Dysmorphol. 2008;17(1):57–60. https://doi.org/10.1097/MCD.0b013e3282beb59e.
Kronlage C, Healy DG. Mystery Case: Bilateral alopecia as clue to diagnosis of Gomez-Lopez-Hernandez syndrome in a 38-year-old man. Neurology. 2019;93(9):408–10. https://doi.org/10.1212/WNL.0000000000008004.
Lee J, Zhou P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol Cell. 2007;26(6):775–80. https://doi.org/10.1016/j.molcel.2007.06.001.
Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci. 2001;58(14):2085–97. https://doi.org/10.1007/pl00000838.
Louro P, et al. Woodhouse–Sakati syndrome: first report of a Portuguese case. Am J Med Genet A. 2019;179(11):2237–40. https://doi.org/10.1002/ajmg.a.61303.
Matsuno A, et al. Japanese siblings with Woodhouse-Sakati syndrome: the first family in East Asia. J Neurol Sci. 2017;381:457. https://doi.org/10.1016/j.jns.2017.08.3498.
Medica I, Sepcić J, Peterlin B. Woodhouse-Sakati syndrome: case report and symptoms review. Genet Couns. 2007;18(2):227–31.
Mégarbané A, et al. Primary hypergonadotropic hypogonadism, partial alopecia, and müllerian hypoplasia: report of a second family with additional findings: second family with additional findings. Am J Med Genet A. 2003;119A(2):214–7. https://doi.org/10.1002/ajmg.a.20170.
Meschia JF, Worrall BB, Elahi FM, Ross OA, Wang MM, Goldstein ED, Rost NS, Majersik JJ, Gutierrez J. Management of inherited CNS small vessel diseases: the CADASIL example: a scientific statement from the American Heart Association. Stroke. 2023;54(10). https://doi.org/10.1161/STR.0000000000000444.
Messina C. Woodhouse-Sakati syndrome: a review. Rev Neurol (Paris). 2024:S0035-3787(24)100023-7. https://doi.org/10.1016/j.neurol.2023.11.008.
Mistry BV, et al. Expression profiling of WD40 family genes including DDB1- and CUL4- associated factor (DCAF) genes in mice and human suggests important regulatory roles in testicular development and spermatogenesis. BMC Genomics. 2020;21(1):602. https://doi.org/10.1186/s12864-020-07016-9.
Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. https://doi.org/10.1038/gim.2015.30.
Rush ET, Adam MP, Clark RD, Curry C, Hartmann JE, Dobyns WB, Olney AH. Four new patients with Gomez–Lopez‐Hernandez syndrome and proposed diagnostic criteria. Am J Med Gene Part A. 2013;161(2):320–6. https://doi.org/10.1002/ajmg.a.v161.2, https://doi.org/10.1002/ajmg.a.35817.
Salti IS, Salem Z. Familial hypogonadotropic hypogonadism with alopecia. Can Med Assoc J. 1979;121:428–34.
Schapira M, et al. WD-repeat domain proteins: a novel target class? Nat Rev Drug Discovery. 2017;16(11):773–86. https://doi.org/10.1038/nrd.2017.179.
Schneider SA, Bhatia KP. Excess iron harms the brain: the syndromes of neurodegeneration with brain iron accumulation (NBIA). J Neural Transm. 2013;120(4):695–703. https://doi.org/10.1007/s00702-012-0922-8.
Sendur SN, et al. A case of Woodhouse-Sakati syndrome with pituitary iron deposition, cardiac and intestinal anomalies, with a novel mutation in DCAF17. Eur J Med Genet. 2019;62(8):103687. https://doi.org/10.1016/j.ejmg.2019.103687.
Shah K, et al. Woodhouse–Sakati syndrome in a family is associated with a homozygous start loss mutation in the DCAF17 gene. Clin Exp Dermatol. 2020;45(2):159–64. https://doi.org/10.1111/ced.14046.
Suganuma T, Pattenden SG, Workman JL. Diverse functions of WD40 repeat proteins in histone recognition. Genes Dev. 2008;22(10):1265–8. https://doi.org/10.1101/gad.1676208.
The Human Protein Atlas. 2024. Available at: https://www.proteinatlas.org/. Accessed 31 Jan 2024.
Uhlén M, et al. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. https://doi.org/10.1126/science.1260419.
Woodhouse NJ, Sakati NA. A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J Med Genet. 1983. https://doi.org/10.1136/jmg.20.3.216.
Zayed H, Ouhtit A. Accredited genetic testing in the Arab Gulf region: reinventing the wheel. J Hum Genet. 2016;61(7):673–4. https://doi.org/10.1038/jhg.2016.22.
Zhou M, et al. Case report: a Chinese family of Woodhouse-Sakati syndrome with diabetes mellitus, with a novel biallelic deletion mutation of the DCAF17 gene. Front Endocrinol. 2021;12:770871. https://doi.org/10.3389/fendo.2021.770871.
Acknowledgements
Not applicable.
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Contributions
VW, CM, and EC designed the review and the case report. VW, GW, MD, AH, and AA completed the physical examination of the patients and elaborated the case report. VW, CM, EC, MJB, and GW wrote the article.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The current study and signed informed consent were approved by the Institutional Review Board (IRB) of the Lebanese American University (# IRB00006954 LAUIRB#1).
Consent for publication
Written consent was obtained from the legal guardian and oral ascent from the patients in this case report for participation in the study as well as publication of the images of the patients.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wakim, V., Dassouki, M.E., Azar, A. et al. Woodhouse-Sakati syndrome: genotype–phenotype review and case of intra-familial heterogeneity. J Rare Dis 3, 20 (2024). https://doi.org/10.1007/s44162-024-00045-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44162-024-00045-y