
Abstract
The search for new antimicrobial agents due to the development of antimicrobial resistance is one of the greatest points of concern among medicinal chemists. It has been observed that the infections due to the antimicrobial resistance pathogens are one of the reasons for the mortality. Therefore, it is really important to find out some new low molecular weight antimicrobial drugs with different mode of action. Despite of being a better antibiotic, ciprofloxacin represented resistance against many pathogens. Herein, this study reported the synthesis of novel analogues of ciprofloxacin (1) including 1,3,4-oxadiazole, thiazolidine-4-one, 1,3,4-oxazoline, 1,2,4-triazole, Schiff's base, hydrazide, and 1,3,4-thiadiazole (2–8), following their computational assessment. All analogues were then synthesized and characterized using FT-IR, NMR, Mass spectroscopy, etc. The prepared analogs were then assessed for antimicrobial properties against bacterial pathogens and fungal isolates using disc diffusion and serial dilution methodology. To observe the toxicity of the prepared analogs the MTT assay was performed against HepG2 cells. The receptor glucosamine-6-phosphatase (GlcN-6P) and lanosterol 14-alpha-demethylase (CYP51) were used for the molecular docking assay using Auto Dock Tools-1.5.6 to evaluate the degree of hydrogen bonding and binding affinities. It was observed that analogue (2) established strong hydrogen bonds with the both receptors, with great binding affinities.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Presently, one of the most serious health issues in the world is the spontaneous increase in antibiotic resistance. Infections caused by antimicrobial-resistant bacteria are responsible for more than 21,000 annual deaths in the United States, according to the literature [1]. Studies claimed that the misuse and overuse of current medications may be a major contributor to genetic mutations in all pathogenic and nonpathogenic microorganisms. These factors were listed as the causes of the growth of antimicrobial resistance by pathogens, along with excessive or incomplete antibiotic use [2]. Ciprofloxacin, a second-generation fluoroquinolone, portrayed the significant antimicrobial chemotherapeutic effects but with some side effects [3]. The compounds or derivatives containing ciprofloxacin moiety portrayed various chemotherapeutic potentials such as antimicrobial [4], anticancer and topoisomerase I and II inhibitory [5, 6], antiplasmodial [7], anti-tubercular [8], anti-HIV-1 integrase [9], antimalarial [10], anti-ischemic [11]. Sultana et al. reported an identical strategy aimed at the synthesis of a carboxamide analogue of ciprofloxacin, which they tested for antimicrobial activity against gram-positive and gram-negative pathogenic microbes, and found that some of the analogues displayed superior antimicrobial activity compared to ciprofloxacin [12]. Similarly, Sharma P.C., et al. recently reported the synthesis and antibacterial assessment of the analogues of fluoroquinolones annulated with 6-substituted-2-aminobenzothiazoles and found that the analogues were more potent than the ciprofloxacin [13]. More recently one study reported by Rabbani M. G. and Islam, M. R., aimed the synthesis of some NH-analogues of ciprofloxacin and tested them for antibacterial, antifungal, and cytotoxic activities. The authors concluded that the biological activity was enhanced for the synthesized analogues [14]. Additionally, the derivatives with carbohydrazide functional group, Schiff's base, 1,3,4-thiadiazole, 1, 3, 4-oxadiazole,1,2,4-triazole-3-thiones, 1,3,4 oxadiazoline, 1,3,4-thiazolidin-4-one have been reported to exhibit versatile pharmacological applications [15,16,17,18]. The versatile pharmacological potential of these functional moieties as well as the reported enhancement of the biological activities of ciprofloxacin analogues prompted us to design a new class of analogues of ciprofloxacin fused with these functional moieties, with an aim that it will be a better approach to our continuous search for new antimicrobial chemotherapeutic agents.
2 Experimental
The probable structures and the schematic diagrams were created through ChemDraw. The computational studies such as the drug-likeness score and physicochemical characteristics were estimated using software (Molinspiration chemoinformatics). This investigation employed solvents and reagents purchased from Sigma Aldrich and Merck. Additionally, they were of analytical quality and were used as given. The reactions were conducted in round-bottom flasks and refluxed using magnetic hot plate stirrers to achieve homogeneous mixing. Utilizing a Heraeus Vario EL III analyzer, the melting points of several compounds were determined. Using a Bruker Avance 300 MHz spectrometer for NMR spectra, a Micromass Quattro II triple quadrupole mass spectrometer for Mass spectra, and a Perkin-Elmer FT-IR spectrometer, the structures of the analogues were determined. HIMEDIA provided the chemicals, reagents, and media required for the biological studies. For antimicrobial research, a laminar flow cabinet was used, and an ELISA test was utilized to quantify MTT. For docking studies, Pymol, AutoDock-Tools 1.5.6, and Autodock vina were used.
2.1 Computational analysis
The computational analyses of ciprofloxacin and its analogues (1–8) were carried out using molinspiration chemoinformatics software, as described before [19].
2.2 Chemistry
General procedure for the synthesis of analog (2): Ciprofloxacin (10 mmol), methanol (40 ml) and conc. H2SO4 was poured into a 100 mL flask and refluxed for 6 h. After 6 h, the hydrazine hydarate (10 mmol) was injected drop by drop to the flask and refluxed again for 7 h. The final output was formed as a precipitate that was dried, filtered, and recrystallized [20].
General procedure for the synthesis of analog (3): Equal amounts of ciprofloxacin hydrazide (2) (10 mmol) and of 2-aminothiazole-5-carboxaldehyde (10 mmol) were dissolved in ethanol (60 mL), and then refluxed in the mixture for 14 h [21]. As a catalyst, glacial acetic acid (1 mL) was added to the reaction mixture to produce a precipitate, which was then filtered, dried, and re-crystallized.
General procedure for the synthesis of analog (4): A suspension of the hydrazide analogue of ciprofloxacin (2) (10 mmol), and thiosemicarbazide (10 mmol) in POCl3 (30 mL) was heated under reflux for 7 h. The mixture was permitted to cool, after which the distilled water was added drop by drop and the refluxing process was repeated for 7 h [22]. When the reaction was completed, a precipitate of the product was generated, which was observable after being neutralized with potassium hydroxide, and filtered.
General procedure for the synthesis of analog (5): The potassium hydroxide (15 ml) was dissolved in absolute ethanol and refluxed for 3 h. The hydrazide analogue of ciprofloxacin (2) (10 mmol) and carbon disulfide (13 mmol) was also added and stirred for 6 h. Diethyl ether (40 ml) was used to dilute the mixture, following which hydrazine hydrate (30 ml) dissolved in distilled water (30 ml) was added and the mixture was refluxed for 6 h [23]. The finished product underwent filtration, drying, and recrystallization.
General procedure for the synthesis of analog (6): The Schiff’s base analogue of ciprofloxacin (3) was placed in a flask with acetic anhydride (60 ml) and refluxed for 20 h. The output precipitated when the reaction mixture was added to ice-cooled water, filtered, dried, and recrystallized [24].
General procedure for the synthesis of analog (7): The Schiff's base analogue of ciprofloxacin (3), (20 mmol), and thioglycolic acid, (20 mmol), were mixed in dimethyl formamide (60 mL) and refluxed for 22 h in a round-bottom flask [22]. During the reaction anhydrous zinc chloride was also introduced in it. To produce the desired product the mixture was poured into cold water, and the precipitate was produced which was filtered, dried, and recrystallized.
General procedure for the synthesis of analog (8): For 15 h, a mixture of Schiff's base analogue of ciprofloxacin (3) (20 mmol) and POCl3 (60 ml) was heated under reflux. After completion, the solution was added to ice-cold water to get the precipitate, which was vacuum-filtered and dried then e-crystallized [24].
2.3 Characterization
1-Cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carbohydrazide (2): Yield: 92%; m.p. 179–181 °C: Creamy white crystals; FT-IR (cm−1): 3321, 3188, 2932, 1774, 1726, 1045, and 1013; 1H-NMR (DMSO-d6, Figure S1) δ (ppm): 1.101–1.1.405 (m, 5H, -cyclopropyl), 2.310 (s, 4H, –N–CH2–CH2–N), 3.759 (s, 4H, CH2–CH2-Piperazine), 7.208 (s, 1H, Ar–H), 7.513 (s, 1H, Ar–H), 7.903 (s, 1H, Ar–H), 9.065 (s, 1H, NH), 9.539 (s, 1H, NH), 9.892 (s, 2H, NH2); 13C-NMR (DMSO-d6, Figure S2) δ (ppm): 7.9 (CH2-Cyclopropyl), 7.9 (CH2-Cyclopropyl), 35.2 (CH-Cyclopropyl), 46.1, 50.8, 103.2, 110.8, 112.8, 121.3, 134.0, 145.5, 147.0 (C-N), 152.2 (C-F), 167.4 (CONH), 176.7 (CO); Anal. calc. for C17H20FN5O2: C 59.12, H 5.84, N 20.28, found: C 59.20, H 5.82, N 20.31; MS (m/z): [M+ + 1] 345.16 (345.19).
N'-[(Z)-(2-amino-2,5-dihydro-1,3-thiazol-5-yl)methylidene]-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (3): Yield: 88%; m.p. 185–188 °C: Grey crystals; FT-IR (cm−1): 3381, 3106, 3077, 2895, 1784, 1718, 1625, 1052 and 1021; 1H-NMR (DMSO-d6, Figure S3) δ (ppm): 1.089–1.400 (m, 5H, -cyclopropyl), 2.303 (s, 4H, –N–CH2–CH2–N), 3.777 (s, 4H, CH2-CH2-Piperazine), 7.156 (s, 1H, Ar–H), 7.499 (s, 1H, Ar–H), 7.925 (s, 1H, Ar–H), 8.216 (s, 1H, HC=N), 8.362 (s, 1H, Ar–H), 9.077 and 9.577 (s, 2H, 2NH), 10.026 (s, 2H, NH2); 13C NMR (DMSO-d6, Figure S4) δ (ppm): 7.6 (CH2-Cyclopropyl), 7.6 (CH2-Cyclopropyl), 32.6, (CH), 35.5 (CH-Cyclopropyl), 45.6 (2CH2), 52.8 (2CH2), 104.6 (CH), 113.7 (CH), 118.2, 121.8, 136.4, 145.5 (C-N), 148.3, 155.2 (CN), 152.5 (C-F), 163.8 (C=N), 169.5 (CONH), 177.5 (C=O); Anal. calc. for C21H24FN7O2S: C 55.13, H 5.29, N 21.43, found C 59.20, H 5.82, N 20.31; MS (m/z): [M+ + 1] 457.52 (457.54).
3-(5-Amino-1,3,4-thiadiazol-2-yl)-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)quinolin-4(1H)-one (4): Yield: 86%; m.p. 181–183 °C: Grey crystals; FT-IR (cm−1): 3246, 3019, 2914, 1727, 1663, 1628, 1059, and 1025; 1H-NMR (DMSO-d6, Figure S5) δ (ppm): 1.113–1.408 (m, 5H, -cyclopropyl), 2.431 (s, 4H, –N–CH2–CH2–N), 3.807 (s, 4H, CH2-CH2-Piperazine), 7.590 (s, 1H, Ar–H), 7.972 (s, 1H, Ar–H), 8.219 (s, 1H, Ar–H), 9.611 (s, 1H, NH), 9.904 (s, 2H, NH2); 13C-NMR (DMSO-d6, Figure S6) δ (ppm): 7.7 (CH2-Cyclopropyl), 7.7 (CH2-Cyclopropyl), 36.5 (CH-Cyclopropyl), 46.6 (2CH2), 52.2 (2CH2), 102.9, 113.2, 119.1, 116.9, 135.8, 141.8, 148.7, 152.9, 158.3, 162.5, 175.5; Anal. calc. for C18H19FN6OS: C 55.94, H 4.96, N 21.75, found C 55. 94, H 4.96, N 21.73; MS (m/z): [M+ + 1] 386.13 (386.14).
3-(4-Amino-5-sulfanyl-4H-1,2,4-triazol-3-yl)-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl) quinolin-4(1H)-one (5): Yield: 85%; m.p. 187–189 °C: pale grey crystals; FT-IR (cm−1): 3337, 3180, 2932, 2534, 1781, 1733, 1670, 1633, 1041,and 1020; 1H-NMR (DMSO-d6, Figure S7) δ (ppm): 1.003–1.420 (m, 5H, –cyclopropyl), 2.372 (s, 4H, –N–CH2–CH2–N), 3.332 (s, 1H, SH), 3.661 (s, 4H, CH2-CH2-Piperazine), 7.692 (s, 1H, CH-Ar), 7.992 (s, 1H, CH-Ar), 8.316 (s, 1H, CH-Ar), 9.544 (s, 1H, NH), 9.935 (s, 2H, NH2); 13C-NMR (DMSO-d6, Figure S8) δ (ppm): 7.5 (CH2-Cyclopropyl), 7.5 (CH2-Cyclopropyl), 36.2 (CH-Cyclopropyl), 46.4 (2CH2), 51.01 (2CH2), 102.8, 111.6, 112.5, 118.4, 134.7, 141.7, 143.8, 152.8, 162.7, 167.9, 176.1; Anal. calc. for C18H20FN7OS: C 53.85, H 5.02, N 24.42, found C 53.90, H 5.05, N 24.44; MS (m/z): [M+ + 1] 402.14 (402.16).
3-[4-Acetyl-5-(2-amino-2,5-dihydro-1,3-thiazol-5-yl)-4,5-dihydro-1,3,4-oxadiazol-2-yl]-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)quinolin-4(1H)-one (6): Yield: 88%; m.p. 184–186 °C: Grey crystals; FT-IR (cm−1): 3321, 3064, 2932, 1758, 1730, 1689, 1636, 1058, and 1027; 1H-NMR (DMSO-d6, Figure S9) δ (ppm): 1.131–1.418 (m, 5H, -cyclopropyl), 2.106 (s, 3H, COCH3), 2.405 (s, 4H, -N-CH2-CH2-N), 3.797 (s, 4H, CH2-CH2-Piperazine), 7.675 (s, 1H, Ar–H), 7.867 (s, 1H, Ar–H), 8.323 (s, 1H, Ar–H), 8.452 (s, 1H, Ar–H), 9.732 (s, 1H, NH), 9.922 (s, 2H, NH2); 13C-NMR (DMSO-d6, Figure S10) δ (ppm): 7.1 (CH2-Cyclopropyl), 7.1 (CH2-Cyclopropyl), 23.4 (CH3), 35.9 (CH-Cyclopropyl), 41.7 (CH), 46.1 (2CH2), 51.5 (2CH2), 78.7 (S-CH), 81.2 (CH-NH2), 103.5, 110.7, 112.4, 121.0, 135.4, 139.1, 152.8, 165.7, 166.8, 168.8,177.1; Anal. calc. for C23H26FN7O3S: C 55.30, H 5.25, N 19.63, found C 55.31, H 5.23, N 19.65; MS (m/z): [M+ + 1] 500.18 (500.16).
N-[5-(2-Amino-2,5-dihydro-1,3-thiazol-5-yl)-2-oxo-1,3-thiazolidin-3-yl]-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (7): Yield: 90%; m.p. 186–188 °C: Grey crystals; FT-IR (cm−1): 3355, 3208, 3018, 2964, 1794, 1753, 1722, 1645, 1077, 1039, and 1011; 1H-NMR (DMSO-d6, Figure S11) δ (ppm): 1.006–1.406 (m, 5H, -cyclopropyl), 2.310 (s, 1H, S-CH2), 2.388 (s, 4H, -N-CH2-CH2-N), 3.811 (s, 4H, CH2-CH2-Piperazine), 7.590 (s, 1H, Ar–H), 7.970 (s, 1H, Ar–H), 8.215 (s, 1H, Ar–H), 9.788, and 9.902 (s, 1H, NH), 10.104 (s, 2H, NH2); 13C-NMR (DMSO-d6 Figure S12) δ (ppm): 7.4 (CH2-Cyclopropyl), 7.4 (CH2-Cyclopropyl), 35.3 (CH2-S), 36.1 (CH-Cyclopropyl), 46.4 (2CH2), 52.6 (2CH2), 61.6, 77.8, 102.5, 111.9, 112.8, 120.2, 135.4, 147.5, 144.3, 152.0, 163.7, 167.2, 169.6, 176.8; Anal. calc. for C23H26FN7O3S2: C 51.96, H 4.93, N 18.44, found C 51.98, H 4.91, N 18.47; MS (m/z): [M+ + 1] 532.16 (532.19).
3-[5-(2-Amino-2,5-dihydro-1,3-thiazol-5-yl)-1,3,4-oxadiazol-2-yl]-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)lquinolin-4(1H)-one (8): Yield: 83%; m.p. 192–194 °C: Grey crystals; FT-IR (cm−1): 3360, 3007, 2954, 1732, 1688, 1640, 1618, 1035, and 1026; 1H-NMR (DMSO-d6, Figure S13) δ (ppm): 1.129–1.530 (m, 5H, -cyclopropyl), 2.430 (s, 4H, -N-CH2-CH2-N), 3.877 (s, 4H, CH2-CH2-Piperazine), 7.488 (s, 1H, Ar–H), 7.675 (s, 1H, Ar–H), 8.196 (s, 1H, Ar–H), 8.405 (s, 1H, Ar–H), 9.550 (s, 1H, NH), 10.142 (s, 2H, NH2); 13C-NMR (DMSO-d6, Figure S14) δ (ppm): 7.3 (CH2-Cyclopropyl), 7.3 (CH2-Cyclopropyl), 36.5 (CH-Cyclopropyl), 39.4, 46.2 (2CH2), 52.7 (2CH2), 77.9 (S-CH-NH2), 102.5, 113.2, 116.9, 118.8, 135.4, 147.1, 141.3, 152.9, 160.3, 163.1, 164.2, 176.8; Anal. calc. for C21H22FN7O2S: C 55.37, H 4.87, N 21.52, found C 55.34, H 4.92, N 21.60; MS (m/z): [M+ + 1] 456.15 (456.17).
3 Pharmacology/biology
3.1 In-vitro antibacterial evaluation
The antibacterial activity of ciprofloxacin and its analogues (1–8) was tested using disc diffusion and sequential different concentrations [22]. Bacterial pathogens were examined for antibacterial resistance (Staphylococcus aureus, Staphylococcus epidermidis, Streptococcus pyogenes, Micrococcus luteus, Klebsiella pneumoniae, Pseudomonas aeruginosa, Escherichia coli, and Proteus mirabilis). To quantify the zone of inhibition, six-millimeter paper discs were made, dipped into the stock solutions, and put under laminar flow on the agar plate that had been previously prepared. Each stock solution for ciprofloxacin and its analogues (1–8) was made by dissolving 1 mg of each analogue in 100 mL of DMSO. The serial dilutions were created using a variety of concentrations, including 400, 200, 100, 50, 12.5, 6.25, and 3.125 g/ml, in order to estimate the MIC, or minimum inhibitory concentration, which is the concentration of a drug required to prevent the growth of pathogens.
3.2 In-vitro antifungal evaluation
The antifungal evaluation of ciprofloxacin and its analogues (1–8) in vitro was performed by serial dilution and agar disc diffusion. The investigation used many fungal infections, including (Candida albicans, Candida krusei, Aspergillus flavus, Aspergillus fumigates, Trichophyton interdigitale, and Penicillium marneffei). After cultivation on Sabourd Dextrose Plates, each fungal isolate was suspended by adding it to a 3 cubic centimetre saline solution. 1 mg of each analogue was mixed in 100 mL of DMSO to make stock solutions for ciprofloxacin and its analogues (1–8). The inhibition zone (in millimetres) and the lowest inhibitory concentration were used to determine the antifungal activity of the sample (MIC). Concentrations of 400, 200, 100, 50, 12.5, 6.25, and 3.125 g/mL were employed to make serial dilutions [22, 25].
3.3 MTT assay
To determine the cytotoxicity, or percent viability of cells against HepG2 cells (human hepatocellular carcinoma), the MTT test was used with ciprofloxacin and its analogues (1–8) [26, 27]. Figure 4 depicts the use of ELISA to test the viability of a sub-confluent population of HepG2 cells cultured in DMEM for ciprofloxacin and its analogues (1–8).
3.4 Molecular docking
The molecular docking was performed to evaluate the extent of hydrogen bonds and the binding affinities between the amino acid regions of the receptors GlcN-6P (PDB: 2VF5); lanosterol 14-alpha-demethylase (CYP51), and ciprofloxacin and its derivatives (1–8). Aautodock-tools 1.5.6 and Autodock-vina were used to accomplish the inquiry, and Pymol software was used to create the docking photos in accordance with the technique described in the literature [28, 29] ChemDrawUltra-12.0, which also provided as the platform for transferring the smiles format used to generate PDB files, was used to produce the initial structures of (1–8). These PDB files were then converted to PDBQT format with the help of Aautodock-tools 1.5.6, and the docked file was retrieved with the help of Autodock-vina. The files were used to generate the docked images using Pymol.
4 Results and discussion
4.1 Computational assessment
The bioactivity score and physicochemical properties were estimated using molinspiration, ChemDraw ultra 12.0 was used to construct the framework of ciprofloxacin and its analogues (1–8) and smiles formats were created. The smiles were employed to estimate the computational characteristics, and the results indicated that the analogues (2, 4, 6, and 8) as well as ciprofloxacin (1) were lying under the zone for active drug molecules as enzyme inhibitors. On the other hand, the physicochemical results indicated that only analogue (7) violated the Lipinski's rule of five in terms of molecular weight, while all other analogues fully adhered to the rule, as shown in Table 1, detailed calculations.
5 Chemistry
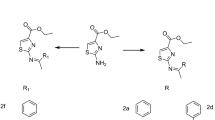
The schematic representation of the process used for synthesis of ciprofloxacin analogs (2–8), has been described in Fig. 1. The synthesis of 1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carbohydrazide (2), was performed by refluxing ciprofloxacin (1) with methanol and concentrated sulfuric acid, then an equimolar quantity of hydrazine hydrate was added. N'-[(Z)-(2-amino-2,5-dihydro-1,3-thiazol-5-yl)methylidene]-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (3) was prepared by the condensation of 1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carbohydrazide (2) and 2-aminothiazole-5-carboxaldehyde. Also, the synthesis of 3-(5 amino-1,3,4-thiadiazol-2-yl)-1-cyclopropyl-6-fluoro-7 (piperazin-1-yl)quinolin-4(1H)-one (4) was produced by the reaction of equimolar of ciprofloxacin derivative (1), and thiosemicarbazide in the presence of POCl3. In addition, the treatment of 1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carbohydrazide (2) with equal quantities of carbon disulfide and hydrazine hydrate led to the formation of the corresponding derivative (5). On refluxing the derivative of ciprofloxacin (3) with acetic anhydride yielded the corresponding thiazolyl analogue (6). Also, the N-[5-(2-amino-2,5-dihydro-1,3-thiazol-5-yl)-2-oxo-1,3-thiazolidin-3-yl]-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3 carboxamide (7), was formed by the reaction of equivalent amounts of the N'-[(Z)-(2-amino-2,5-dihydro-1,3-thiazol-5-yl)methylidene]-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carbohydrazide (3) and thioglycolic acid. Finally, ciprofloxacin carbohydrazide derivative (3) was refluxed with phosphorous oxychloride to yield the corresponding 3-[5-(2-amino-2,5-dihydro-1,3-thiazol-5-yl)-1,3,4-oxadiazol-2-yl]-1-cyclopropyl-6-fluoro-7-(piperazin-1-yl)lquinolin-4(1H)-one (8) in a good yield.

Representing the route and the probable structures of the ciprofloxacin and its analogue
6 Characterization
The structures of ciprofloxacin analogues (2–8) were supported by spectroscopic methods such as FT-IR, 1H-NMR, 13C-NMR, etc., and the detailed findings are reported above in experimental section. The synthesis of the hydrazide analogue of the ciprofloxacin (2), was supported by the appearance of the characteristic FTIR bands due to the new NH and NH2 at 3188 cm−1 and 3321 cm−1 which confirmed the conversion of the COOH group to the CONHNH2. FT-IR spectrum of the compound (3) showed the appearance of additional bands at 1625 cm−1 owing to C=N, as well as a variation in the FT-IR band due to NH2 at 3381 cm−1, which further verified the condensation reaction between aldehyde and amine and the generation of derivative (3). Also, the disappearance of the NH group band at 3188 cm−1, in the structure of analogue (2) as well as the simultaneous appearance of bands characteristic to the functional groups –C=N, C=N, NH, and NH2 respectively at 1628 cm−1, 1663 cm−1, 3019 cm−1, and 3246 cm−1, confirmed the cyclization of the hydrazide analogue of ciprofloxacin (2) into the 1,3,4-thiadiazole analogue (4). The presence of the FT-IR bands at 2534 cm-1, in the FT-IR spectra of (5) owing to the SH functional group, in addition to the appearance of bands at 3361 cm-1 for NH2 which confirmed the cyclization reaction of the hydrazide analogue of ciprofloxacin into the 1,2,4-triazole analogue (5). Additionally, the presence of absorption bands at 1638 and 1669 cm−1 was characteristic of the C=N groups. Furthermore, that is additional evidence for supporting the above-mentioned cyclization. The analogue (6) was derived from the analogue (3), therefore on comparing the FT-IR spectra of both compounds, it was observed that there was the absence of an FT-IR band at 3188 cm−1 due to the NH and the appearance of the FT-IR band at 1689 cm−1 due to C=N functional moiety. The FT-IR spectra of the thiazolidinedione analogue of ciprofloxacin (7), represented the presence of the FT-IR band at 1794 cm−1 attributed to an additional CO group in the structure of compound (7). Some additional bands were also observed at 1011 cm−1, 1039 cm−1, 1079 cm−1, and 1645 Attributed to C–N and C=N groups respectively. FT-IR spectrum of derivative (8) presented the absence of absorption bands at 3188 cm−1 attributed to the NH group and band at 1784 cm−1 Attributed to the CO group, as well as the simultaneous appearance of the absorption bands at 1026, 1035, 1618, 1640, 1688, and 1732 cm−1, attributed to C–N and C=N groups respectively, which strongly supported the cyclization process of compound (3) to afford derivative (8). The structures of the analogues of ciprofloxacin (2–8), were further supported by the 1H-NMR spectra, the 1H-NMR spectra of the hydrazide analogue of ciprofloxacin (2), represented the presence of singlet signals at δ 9.065, 9.539, and 9.892 ppm attributed to NH and NH2 groups which strongly proposed that conversion of COOH group to CONHNH2 group as well as the synthesis of the ciprofloxacin hydrazide analogue (2). Similarly, the 1H-NMR spectra of the Schiff base analogue ciprofloxacin (3) showed a new singlet at δ 8.216 ppm attributed to the characteristic HC=N group that strongly supported the condensation of hydrazide to ciprofloxacin Schiff base analogue (3). Also, the disappearance of the singlet signal at δ 9.539 ppm was attributed to the NH group and the simultaneous appearance of the singlet signal at δ 9.904 ppm was attributed to the amino group which recommended the cyclization of the hydrazide analogue of ciprofloxacin (2) to the 1,3,4-thiadiazole analogue of the ciprofloxacin (4). In the 1H-NMR spectra of (5), the absence of a singlet signal at δ 9.539 and 9.892 ppm was attributed to NH and NH2 groups respectively and the existence of a singlet signal at δ 3.332 ppm characteristic of the SH group and at δ 9.935 ppm was attributed to the NH2 group strongly assisting the formation of the compound (5). The absence of a singlet signal was attributed to NH in the 1H-NMR spectrum of (3), and the appearance of the singlet signal at δ 2.106 ppm is characteristic of COCH3 protons in the 1H-NMR spectrum of (6). The presence of the characteristic singlet signal at δ 2.310 ppm due to S-CH2 proton in the 1H-NMR spectrum of (7), recommended the formation of ciprofloxacin thiazolidinedione analogue (7). The structure of the analogue (8) was confirmed by comparing the 1H-NMR spectra of compounds (3) and (8). the 1H-NMR spectrum of (8) revealed the absence of a singlet signal at δ 9.077 ppm attributed to the CO–NH group while it appeared in the 1H-NMR spectrum of (3), hence, confirming the synthesis of (8). The 13C-NMR spectra of (2–8), exhibited some common peaks due to the presence of central skeleton in all the analogues around δ 7.1 ppm to 7.9 ppm, 35.2 ppm to 36.5 ppm, 50.8 ppm to 52.8 ppm, and 45.6 ppm to 46.6 ppm attributed to the CH2, two cyclopropyl, CH2 of piperazine moieties groups respectively. 13C-NMR spectra of all prepared analogues showed some additional signals such as the presence of signals at δ 167.4 ppm characteristic of the CONHNH2 group for the ciprofloxacin hydrazide analogue (2) strongly supported its synthesis. On the other hand, the 13C-NMR spectrum of (3), displayed the signal at δ 163.8 ppm attributed to CN, which recommended the condensation of compound (2) with an aldehyde. The 13C-NMR spectrum of (4), depicted the absence of signal at 167.4 ppm attributed to the CONHNH2 group and the presence of signals at δ 158.3 and 162.5 ppm attributed to the two C=N carbons, similarly, the 13C-NMR spectra of (5), the peaks at δ 162.7 and 167.9 ppm attributed to C=N carbons confirmed the corresponding cyclization. The characteristic signals in the 13C-NMR spectrum of derivative (6), represented were at δ 23.4 ppm due to CH3 carbon of acetyl group, δ 78.7 ppm attributed to S-CH carbon, 81.2 ppm for CH-NH2 carbon. Similarly, the peaks in the 13C-NMR spectrum of (7), at δ 35.3 ppm due to S-CH2 carbon, 163.7 for C=N carbon and 167.2 attributed to additional C=O carbon and the in the 13C-NMR spectrum of (8), at δ 77.9 ppm for S-CH-NH2, 160.3 ppm, 163.1 ppm and 164.2 attributed to three C=N groups recommended the synthesis of (7–8).
7 Pharmacology/biology
7.1 Antimicrobial activity
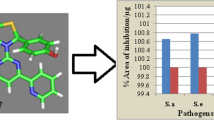
The antimicrobial activity of all ciprofloxacin and its analogues (1–8) was performed by disc diffusion and sequential concentration. The antimicrobial potential was established on the bacterial pathogens [Gram-positive bacteria: S. aureus, S. epidermidis, S. pyogenes, and M. luteus], [Gram-negative bacteria: K. pneumoniae, P. aeruginosa, E. coli, and P. mirabilis], and fungal pathogens [Fungi: C. albicans, C. krusei, A.flavus, A. fumigates, T. interdigitale, and P. marneffei). The antimicrobial activities were quantified in terms of zone of inhibition (mm) and MIC (minimum inhibitory concentration) in (µg/mL), and the results are shown in Tables 2a, b, and 3. To compare the antibacterial and antifungal findings with the reference drugs, we also calculated the per cent area of inhibition for all the analogues and the results are presented in Figs. 2, and 3. The results of antibacterial screening exhibited that the analogues (2–8) were found to represent significant antibacterial effects. The results also reported that the analogue (2), (4), and (8), represented better antibacterial potential than ciprofloxacin approximately against all the studied pathogens. The antifungal potential of analogues (2–8) revealed considerable antifungal effects from all the analogues; it was observed that only analogue (2) exhibited better antifungal potential than Amphotericin B, against all the fungal isolates. On the other hand, the analogue (4) and (8) also exhibited better antifungal effects than amphotericin B against the fungal strains (A.flavus) and (P. marneffei).

Representing the per cent area of inhibition/µg of ciprofloxacin and its derivatives (1–8)

Representing the per cent area of inhibition/µg of ciprofloxacin and its derivatives (1–8)
8 MTT assay
Using HepG2 cells, the MTT test was employed to determine the percentage of viable cells for ciprofloxacin and its derivatives (1–8). According to the scientific literature, the MTT experiment is based on the conversion of the MTT complex to the formazan dye by an enzyme located in the mitochondria of living cells; hence, only alive cells can convert the MTT complex to the formazan dye. Based on prior research conducted on the percent viability of cells, it was obvious that it was inversely related to concentration. The MTT test findings demonstrated that the analogs were less hazardous to HepG2 cells, with 71–79% viability of the cells at 100 M, as shown in Fig. 4.

The percent of HepG2 cells those are resistant to ciprofloxacin and its derivatives (1–8)
9 Molecular docking
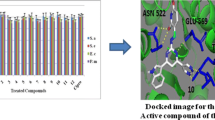
Molecular docking was performed using the receptor GlcN-6P and lanosterol 14-alpha-demethylase (CYP51) using AutoDock Tools-1.5.6 to estimate the level of hydrogen bond formation and binding affinities. Molecular docking was detected in agreement with antibacterial experiment results, Figs. 5 and 6. In addition, it was observed that analog 2 displayed extremely strong H-bonding with the amino acid residues of GlcN-6P (2VF5) and lanosterol 14-alpha-demethylase (CYP51), with binding affinities ranging from – 6.3 to – 7.1 kcal/mol, and – 5.8 to – 9.0 kcal/mol, respectively, Table 4.

The molecular docking images of most active analogues 2, 4, 8 with the receptor GlcN-6P

The molecular docking images of most active analogues 2, 4, 8 with the receptor lanosterol 14-alpha-demethylase (CYP51)
10 Conclusion
In this study a novel series of ciprofloxacin analogues (2–8), was designed and screened for drug-likeness. It was observed that the bioactivity score of most of the analogues was present under the zone of the active drug molecule. The analogues (2–8), were synthesised and characterized by various analytical methods. The antimicrobial potential was assessed by disc diffusion and serial dilutions method, and the outcomes revealed that analogues (2, 4, and 8), exhibited better antimicrobial effects than ciprofloxacin (1). It was also observed that some of the analogues have even better antimicrobial activity than ciprofloxacin. The MTT assay confirmed that the analogues (2–8) were found to be less hazardous towards HepG2 cells based on the percent viability of cells. The molecular docking was performed and it was observed that the analogue (2) established strong hydrogen bonds with the residues, GlcN-6P (2VF5) and lanosterol 14-alpha-demethylase (CYP51), with binding affinities ranging from -6.3 to -7.1 kcal/mol, and -5.8 to -9.0 kcal/mol. Over all on the basis of these promising findings, further studies can be performed using these moieties aiming the in vivo analysis, for the development of novel chemo-core to treat antimicrobial infections.
Data availability
The data will be available on request.
References
Dighe SN, Collet A (2020) Recent advances in DNA gyrase-targeted antimicrobial agents. Eur J Med Chem 199:112326. https://doi.org/10.1016/j.ejmech.2020.112326.
Zanni R, Galvez-Llompart M, Machuca J, Garcia-Domenech R, Recacha E, Pascual A, Rodriguez-Martinez JM, Galvez J (2017) Molecular topology: A new strategy for antimicrobial resistance control. Eur J Med Chem 137:233–246. https://doi.org/10.1016/j.ejmech.2017.05.055
Verderosa AD, de la Fuente-Núnez C, Mansour SC, Cao J, Lu TK, Hancock REW, Fairfull-Smith KE (2017) Ciprofloxacin-nitroxide hybrids with potential for biofilm control. Eur J Med Chem 138:590–601
Wang R, Yin X, Zhang Y, Yan W (2018) Design, synthesis and antimicrobial evaluation of propylene-tethered ciprofloxacin-isatin hybrids. Eur J Med Chem 156:580–586. https://doi.org/10.1016/j.ejmech.2018.07.025
Chrzanowska A, Roszkowski P, Bielenica, A, Olejarz W, Stępień, K, Struga M, (2020) Anticancer and antimicrobial effects of novel ciprofloxacin fatty acids conjugates. Eur J Med Chem 185:111810, https://doi.org/10.1016/j.ejmech.2019.111810.
Abdel-Aziz M, Park SE, El-Din G, Abuo-Rahma AA, Sayed MA, Kwon Y (2013) Novel N-4-piperazinyl-ciprofloxacin-chalcone hybrids: Synthesis, physicochemical properties, anticancer and topoisomerase I and II inhibitory activity. Eur J Med Chem 69:427–438. https://doi.org/10.1016/j.ejmech.2013.08.040
Dixit SK, Mishra N, Sharma M, Singh S, Agarwal A, Awasthi SK, Bhasin VK (2012) Synthesis and in vitro antiplasmodial activities of fluoroquinolone analogs. Eur J Med Chem 51:52–59. https://doi.org/10.1016/j.ejmech.2012.02.006
Hu YQ, Xu Z, Zhang S, Wu X, Ding JW, Lv ZS, Feng LS (2017) Recent developments of coumarin-containing derivatives and their anti-tubercular activity. Eur J Med Chem 136:122–130. https://doi.org/10.1016/j.ejmech.2017.05.004
Sriram D, Yogeeswari P, Senchani G, Banerjee D (2007) Newer tetracycline derivatives: synthesis, anti-HIV, antimycobacterial activities and inhibition of HIV-1 integrase. Bioorg Med Chem Lett 17:2372–2375. https://doi.org/10.1016/j.bmcl.2006.11.055
Hu YQ, Gao C, Zhang S, Xu L, Xu Z, Feng LS, Wu X, Zhao F (2017) Quinoline hybrids and their antiplasmodial and antimalarial activities. Eur J Med Chem 139:22–47
Park CH, Lee J, Jung HY, Kim MJ, Lim SH, Yeo HT, Choi EC, Yoon EJ, Kim KW, Cha JH, Kim SH, Chang DJ, Kwon DY, Li F, Suh YG (2007) Identification, biological activity, and mechanism of the anti-ischemic quinolone analog. Bioorg Med Chem 15:6517–6526. https://doi.org/10.1016/j.bmc.2007.07.009
Sultana N, Saeed AM, Bushra SRS, Haroon U (2011) Synthesis, Characterization and Biological Evaluations of Ciprofloxacin Carboxamide Analogues. Bull Korean Chem Soc 32:483. https://doi.org/10.5012/bkcs.2011.32.2.483
Sharma PC, Jain A, Shahar Yar M, Pahwa R, Singh J, Goel S (2015) Synthesis and antibacterial evaluation of novel analogs of fluoroquinolones annulated with 6-substituted-2-aminobenzothiazoles. Arab J Chem 8:671–677. https://doi.org/10.1016/j.arabjc.2011.04.008
Rabbani MG, Islam MR (2020) Synthesis and Characterization of Some NH-Analogues of Ciprofloxacin on Antibacterial, Antifungal, and Cytotoxic Activities. J Sci Res 12:349–362. https://doi.org/10.3329/jsr.v12i3.42804
Zulfiqar A, Ahmed D, Fatima R, Yousuf S (2020) Green synthesis, urease inhibitory activity and antioxidant potential of 4-bromo -2-(((2′-chloro-4′-nitrophenyl)imino)methyl)phenol Schiff base. J Mol Structure 1202:127263, https://doi.org/10.1016/j.molstruc.2019.127263.
Shashikant VB, Kailash G, Mayuresh KR, Ajit AP, Aniket PS, Vinod JM (2008) Design, Synthesis and Evaluation of Antiinflammatory, Analgesic and Ulcerogenicity studies of Novel S-Substituted phenacyl-1,3,4-oxadiazole-2-thiol and Schiff bases of Diclofenac acid as Nonulcerogenic Derivatives. Bioorg Med Chem 16:1822–1831. https://doi.org/10.1016/j.bmc.2007.11.014
Zhang J, Wang X, Yang J, Guo L, Wang X, Song B, Dong W, Wang W (2020) Novel diosgenin derivatives containing 1,3,4-oxadiazole/thiadiazole moieties as potential antitumor agents: Design, synthesis and cytotoxic evaluation. Eur J Med Chem 186:111897. https://doi.org/10.1016/j.ejmech.2019.111897.
Maddila S, Nagaraju K, Jonnalagadda SB (2020) Synthesis and antimicrobial evaluation of novel pyrano[2,3-d]-pyrimidine bearing 1,2,3-triazoles. Chemical Data Collections. 28:100486, https://doi.org/10.1016/j.cdc.2020.100486.
Ningegowda R, Chandrashekharappa S, Singh V, Mohanlall V, Venugopala KN (2020) Chemical Data Collections 28:100431. https://doi.org/10.1016/j.cdc.2020.100431
Alodeani EA, Arshad M, Izhari MA (2015) Anti-uropathogenic activity, drug likeness, physicochemical and molecular docking assessment of (E-)-N’-(substitutedbenzylidene)-2-(quinolin-8-yloxy) acetohydrazide. Asian Pac J Trop Biomed 5:676–683. https://doi.org/10.1016/j.apjtb.2015.04.010
Arshad M, Bhat AR, Pokharel S, Kim JE, Lee EJ, Athar F, Choi I (2014) Synthesis, characterization and anticancer screening of some novel piperonyl–tetrazole derivatives. Eur J Med Chem 71:229–236. https://doi.org/10.1016/j.ejmech.2013.11.008
Arshad M (2020) Design, Drug-likeness, Synthesis, Characterization, Antimicrobial, Molecular Docking and MTT Assessment of 1,3-thiazolidin-4-one Bearing Piperonal and Pyrimidine Moieties. Russ J Bioorg Chem 46:599–611. https://doi.org/10.1134/S1068162020040056
Arshad M (2014) An insight to the synthetically obtained triazole possessing numerous biological activities. Int J Pharm Pharm Sci 6(9):16–23
Arshad M (2014)1, 3, 4-Oxadiazole nucleus with versatile pharmacological applications: A Review, International Journal of Pharmaceutical Sciences and Research. 5 (2014) 1000–1013. https://doi.org/10.13040/IJPSR.0975-8232.5(4).1000-13
Clinical and Laboratory Standards Institute, Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically Approved Standard, Seventh ed. CLSI document M7-A7. Wayne, Pennsylvania, USA, 2006, pp. 1–49. https://clsi.org/media/1928/m07ed11_sample.pdf
Gupta MK, Neelakantan TV, Sanghamitra M, Tyagi RK, Dinda A, Maulik S, Mukhopadhyay CK (2006) Goswami SK (2006) An assessment of the role of reactive oxygen species and redox signaling in norepinephrine-induced apoptosis and hypertrophy of H9c2 cardiac myoblasts. Antioxid Redox Signal 8:1081–1093. https://doi.org/10.1089/ars.2006.8.1081
Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods, 1983, vol. 65, pp. 55. https://doi.org/10.1016/0022-1759(83)90303-4
Mouilleron S, Badet-Denisot MA, Golinelli-Pimpaneau B (2008) Ordering of C-terminal loop and glutaminase domains of glucosamine-6-phosphate synthase promotes sugar ring opening and formation of the ammonia channel. J Mol Biol 377(4):1174–1185. https://doi.org/10.1016/j.abb.2010.08.008
Trott, O., Olson A.J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem., 2010, vol. 31, pp. 455–461. https://doi.org/10.1002/2Fjcc.21334
Funding
This study was supported by University Grant Commission, The Government of India.
Author information
Authors and Affiliations
Contributions
This perspective paper was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
43994_2023_61_MOESM1_ESM.pdf
Details about NMR figures of the synthesized compounds in this study, Molecular docking images of the rest compounds 1 (PDF 4235 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Asghar, B.H., Arshad, M. Ciprofloxacin analogues: drug likeness, biological and molecular docking studies. J.Umm Al-Qura Univ. Appll. Sci. 9, 508–520 (2023). https://doi.org/10.1007/s43994-023-00061-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43994-023-00061-6