
Abstract
Demethoxylation was kinetically and spectroscopically studied over three catalysts with different Ru0/Ruδ+ ratios. In-situ spectroscopic tests demonstrated that the synergy between Ru0 and Ruδ+ was crucial, and Ru0 was in charge of H2 activation and adsorption of aromatic ring while Ruδ+ adsorbed with O in methoxyl. A Langmuir–Hinshelwood kinetic model was proposed, and ratio of Ru0/Ruδ+ was the key in deciding the rate-determining step (RDS): i) desorption of toluene was RDS over catalyst with high Ru0 ratio; ii) dissociation of H2 was RDS over Ruδ+ enriched catalyst; iii) demethoxylation was rate-determined by CO water–gas shift (WGS) when Ru0/Ruδ+ approached ~ 1. The best performance was obtained over Ru/NiAl2O4-200, which effectively enabled both C-O bond activation and rapid recovery of adsorption sites for aromatic rings. Finally, in-situ DRIFT studies on methoxy decomposition and CO-WGS unraveled that the electronic composition of Ru was more stable in Ru/NiAl2O4-200 which contributes to its excellence.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The design of catalysts for the upgrading of lignin-derived compounds which are generated from catalytic pyrolysis and reductive fractionation of biomass feedstocks has been a pressing issue in biomass conversion that would bring valuable chemicals and fuels in a sustainable way [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15]. Lignin-derived compounds containing plenty of oxygen species like phenolic and methoxy groups, usually have low energy density and poor stability [16,17,18]. As a common strategy, hydrodeoxygenation (HDO) is widely used to remove oxygen through breaking Caromatic-O bonds and obtain target products over metal-based catalyst.
Ru-based catalysts have been generally demonstrated to exhibit excellent performance on the HDO of lignin-derived compounds to valuable chemicals, especially for its reservation of aromatic rings during C-O bonds cleavage [19,20,21,22,23,24,25,26,27]. Selective hydrogenolysis of C-O bond, demethoxylation for instance, enables the removal of methoxys while preserving the phenolic hydroxys, which is widely used in the production of bio-based phenols [9, 28,29,30,31,32,33]. Recently, our group found that Ru exhibited better performance than Pt even in the self-reforming-driven hydrogenolysis of lignin and lignin oil to 4-alkylphenols that enables the full utilization of structural hydrogen in the substrate without the addition of external hydrogen sources, in which Ru also has a lower activation energy in the rate-determining step (RDS), i.e., the hydrogenolysis of methoxys among Pt, Pd, Ru/NiAl2O4 catalysts [34, 35]. Based on a fundamental study on C-O bond activation, Vlachos and co-workers have confirmed that the oxygen vacancies account for the dissociative adsorption of C-O bonds over RuO2 that follows a reverse Mars-van Krevelen mechanism [24]. The distinctive ability of RuO2 was reported and unraveled in their work, however, the contribution from metallic Ru species were not taken into consideration, and the synergy of both metallic Ru and oxidized Ru species were not elucidated for the HDO over supported and reduced Ru catalysts.
Herein, we report a detailed investigation on how the electronic states of Ru influence the hydrogenolysis of methoxy in anisole, namely demethoxylation, over Ru-based catalysts. According to our previous research [34,35,36], the spinel structure of NiAl2O4 possesses a large amount of oxygen vacancies, which have two important functions: (i) promoting the reduction of the loading metal that makes it closer to a metallic state, which is beneficial to dehydrogenation, and (ii) activating water with high efficiency [37], which accelerates the water − gas shift reaction to generate H2. In-situ Raman experiments helped determine the nature of the active sites, Ruδ+ species, that were responsible for the dissociative adsorption of the C-O bonds in anisole. TPSR-MS and in-situ DRIFT studies on anisole were carried out to capture the reaction intermediates and compare the activation of the C-O bonds over different Ru-based catalysts under the reaction conditions. It was found that the C-O bond could not be effectively activated when the catalyst possessed higher concentrations of either Ru0 or Ruδ+ species, which further unraveled the critical synergy between Ru0 and Ruδ+ species when dealing with the demethoxylation, i.e., demethoxylation is an electron-sensitive reaction. Furthermore, a simplified dual-site Langmuir–Hinshelwood kinetic model was proposed to decide the surface RDS in demethoxylation over three model catalysts with different Ru electronic compositions. We found that the water–gas shift (WGS) of CO after methoxy decomposition was the RDS over Ru/NiAl2O4-200 (Ru/NAO-200), which was the catalyst with best demethoxylation performance among all the tested samples. Lastly, in-situ DRIFT studies on methoxy decomposition and DRIFT-MS studies on CO-WGS were performed over both Ru/NAO-200 and Ru/NAO-400, showing that methoxys were both rapidly decomposed into CO, and were later turned into CO2 via WGS following a redox mechanism over these two catalysts. It was found that the reduction of Ruδ+ species was more difficult in Ru/NAO-200, leading to a longer residence time of CO species over catalyst surface during WGS. More importantly, we also found that the electronic states of Ru particles in Ru/NAO-200 showed no obvious differences before and after WGS, which also contributed to its excellent performance in demethoxylation.
2 Experimental methods
2.1 Preparation of catalyst
NiAl2O4 was prepared via a co-precipitation method with the Ni/Al mole ratio at 1:2 [38]. In a typical procedure, 40 mmol nickel nitrate hexahydrate (Ni(NO3)2·6H2O, Adamas-beta) and 80 mmol aluminum nitrate nonahydrate (Al(NO3)3·9H2O, General Reagent) were dissolved in 200 mL ultra-pure water (18.2 mΩ) under vigorous stirring. Ammonium hydroxide (NH3·H2O, General Reagent) was used as the precipitant until the pH value reached 9. After stirring for another one hour, the suspension was settled overnight for aging. Then the powder was separated by vacuum filtering and washed with deionized water until the pH value of the filtrate was around 7, followed by the overnight drying in a 100°C oven. Finally, the powder was calcined at 800 °C under N2 atmosphere for 8 h, 5°C·min−1and used as support. γ-Al2O3 support was prepared via the calcination of commercial boehmite, which was purchased from Aluminum Corporation of China, at 500°C for 4 h with a heating rate of 5°C·min−1.
Ru/NiAl2O4 (denoted as Ru/NAO-x, where x represents the reduction temperature) and Ru/γ-Al2O3 (denoted as Ru/AO) catalysts were synthesized by incipient wetness impregnation method using 10wt% RuCl3 aqueous solution as the precursor. Before each activity test, the catalysts were all activated at different temperatures (100°C, 200°C, 300°C and 400°C) in 10% H2/Ar for 3 h.
2.2 Catalyst characterization
X-ray power diffraction (XRD) patterns were recorded on a D8 Focus Diffractometer (CuKα1 radiation, k = 1.5406 Å) at 40 kV and 40 mA, with the scattering angles between 10–80 o. High angle annular dark field (HAADF)-STEM images were collected on a FEI Talos F200X G2 electron microscopy at 200 kV and recorded with a convergence semi angle of 11 mrad, and inner- and outer collection angles of 59 and 200 mrad, respectively. X-ray photoelectron spectra (XPS) were collected on a Thermofisher Scientific Escalab 250 Xi equipment with monochromatic Al Kα radiation, and all the results were calibrated using C 1 s at 284.8 eV. XAS measurements were conducted at the Canadian Light Source for the Ru L2-edge on the SXRMB/CLS beamline and Ru foil was used for energy calibration. X-ray absorption near-edge structure (XANES) data analysis was performed using Athena software.
CO chemisorption tests were operated on Huasi DAS-7200 automatic chemisorption instrument. Typically, 0.1 g catalyst was injected to the quartz sample tube. Before each run, the catalyst sample was in-situ reduced under 10% H2/Ar atmosphere (30 mL·min−1) for 1 h, and cooled down to ambient temperature with continuous He sweeping (30 mL·min−1). The CO chemisorption results were calculated based on the peak areas of CO pulse injection (200 μL per pulse, filled with 50% CO/He), which were monitored by a TCD detector (45 °C, 80 mA).
2.3 TPSR-MS andin-situ DRIFT evaluation of catalyst
The temperature-programmed surface reaction (TPSR) and in-situ diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy were both collected in a Nicolet iS50 FTIR (Thermofisher) equipped with a reaction chamber (Harreck) that was connected to a Mass Spectroscopy instrument. Before each test, the catalyst samples (50–70 mg) were all in-situ reduced under 5% H2/Ar for 1 h, swept in pure Ar for another 1 h, and finally cooled down to 30 °C. Then the substrate was introduced into the chamber by Ar bubbles (60 mL·min−1 as flow rate) with a quartz bubbler maintained at 150 °C in an oil bath. The temperature of the tubes, which was connected the bubbler to the reaction chamber, was constantly maintained at 90 °C via a heater. Before the collection of DRIFT spectra and fragment trends in MS, the physisorbed molecules were removed by pure Ar with a flow rate of 30 mL·min−1. For TPSR runs, the reaction chamber was heated from 30 °C to 440 °C, with a rate of 5 oC·min−1 and a 5%H2/Ar flow rate of 10 mL·min−1.
2.4 In-situ Raman experiments
Raman spectra were collected on a HORIBA LabRAM HR system exposed under the excitation line of 514 nm. The tests of the catalyst were divided into two parts, i.e., in-situ reduction from ambient conditions to the desired temperature, and surface HDO reaction using anisole as the probe molecule. First, the catalyst sample was dehydrated in testing cell under Ar flow at 100 °C for 10 min, then switched to 5%H2/Ar (10 mL·min−1), and the reduction started from 100 °C to 400 °C and kept for another 30 min. After reduction, the reaction cell was cooled down to room temperature again under the protection of Ar flow. Anisole was introduced into the cell at room temperature and swept by Ar (10 mL·min−1) for 20 min to remove the physiosorbed molecules. The Raman spectra were collected from 100 °C to 250 °C with a step of 50 °C. When approaching 200 °C, the reaction atmosphere was switched from Ar to H2/Ar (10 mL·min−1) for demethoxylation reaction.
2.5 Catalytic performance test and kinetic measurements
The catalyst performance was evaluated in a 50-mL stainless batch reactor with a magnetic stirrer. For each run, the reactor was loaded according to the expected dosage of the substrate, catalyst and solvent. After that, the reactor was sealed with graphite ring, purged with pure N2 for 3 times to expel the residue air, followed by the injection of 0.1 MPa pure H2, and kept stabilized by pure N2 at 2 MPa as the initial pressure. After the end of each reaction, the batch reactor was quickly removed from the heater and quenched in ice water. The liquid product was carefully extracted using 5 g ethyl-acetate from the aqueous reaction residue. The qualitative analysis of the extraction was carried out in a GC–MS system equipped with a HP-5 column, Agilent 7890A-5975C, and quantitatively analyzed by Agilent 7890B equipped with a HP-5 column and an FID detector, using tridecane as the internal standard.
Kinetic measurements were performed using 4-methylanisole as the reactant. Before testing, the diffusion and thermodynamic limitations were excluded by changing the stirring rate and by altering the catalyst-to-reactant ratio, and all the kinetic results were collected by keeping the conversion below 10%. The reaction rate was calculated according to the generation rate of deoxygenated product, toluene. The kinetic models were established according to the Langmuir–Hinshelwood model, which was illustrated in supporting information in detail.
3 Results and discussions
3.1 Characterization of catalysts
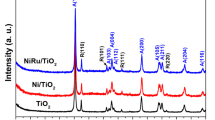
XRD patterns of the catalyst samples are presented in Fig. 1a, whereas no obvious diffraction peaks corresponding to Ru species can be observed over all these three catalysts, indicating the well-dispersion of Ru nanoparticles on NiAl2O4 and γ-Al2O3 supports. The diffraction peaks at 19.1°, 31.4 o, 37.0 o, 44.9 o, 59.7 o, and 65.5 o of Ru/NAO-200 (400) catalysts are assigned to (111), (220), (311), (400), (511), and (440) facets of NiAl2O4 spinel (JCPDS #10–0339). In contrast, only three diffraction peaks were observed for 2%Ru/AO-400 catalyst, which are at 37.4 o, 42.8 o, and 67.3 o, relating to (311), (321), and (522) facets of γ-Al2O3 (JCPDS #04–0880).

a XRD patterns and (b) Ru L2-edge XANES and (c) XPS spectra of Ru/NAO-400, Ru/NAO-200 and Ru/AO-400; HAADF-STEM images and Ru size-distributions on (d) Ru/NAO-400, (e) Ru/NAO-200 and (f) Ru/AO-400
Ru L2-edge XANES spectra for three reduced catalysts as well as Ru foil and RuO2 are present in Fig. 1b. In general, a large ‘white line’ refers to the more oxidized Ru, whereas a small one indicates the reduced-state of Ru species [36]. The reduction of the whiteline intensity indicates that the chemical state of Ru in Ru/NAO-400 catalysts was more electron-enriched than those in Ru/NAO-200 and Ru/AO [36, 39]. In Fig. 1c, two Ru species were obtained in the XPS spectra of Ru 3d5/2, in which the fitting peaks at around 280.2 eV and 281.0 eV were assigned to the metallic Ru0 and RuOx species, respectively [40,41,42]. According to the XPS analysis and fitting results in Fig. 1c and Table 1, we calculated the Ru0 proportions in three catalysts, which were 80.9%, 54.5% and 38.3% in Ru/NAO-400, Ru/NAO-200 and Ru/AO-400, respectively.
HAADF-STEM was carried out to count the Ru particle size distributions in three catalysts. As is illustrated in Fig. 1d-f, the average sizes of Ru particles are 1.2 nm, 1.2 nm and 1.8 nm in Ru/NAO-400, Ru/NAO-200 and Ru/AO-400, respectively. Figure S1 displays the EDS mapping images of Ru/NAO-400 sample, in which it can be seen Ru was well dispersed. Besides, these three catalysts also exhibited similar dispersion of Ru sites according to the CO pulse titration results (Table 1).
Table 1 summarized some other physical and chemical properties of these three catalysts and their corresponding catalytic activities in demethoxylation of 4-methylanisole. According to the demethoxylation rates over three different catalysts in Table 1, it can be preliminarily concluded that the Ru0/Ruδ+ ratio was crucial to the demethoxylation performance of the catalyst.
3.2 In-situ Raman observations on anisole demethoxylation
Figure 2 shows the in-situ Raman spectra of anisole demethoxylation over Ru/NAO-400 and NiAl2O4 support, which were performed to investigate the real sites responsible for the activation of C-O bonds during reaction. Under ambient conditions, we first focused on two intense peaks at 314 cm−1 and 475 cm−1 (Figure S2a) over Ru/NAO-400 corresponding to the Ni–O species and Ru–O species, respectively [43,44,45,46,47]. These peaks can also be observed in the in-situ Raman spectra of NiAl2O4 spinel, NiO/Al2O3 hybrid and RuO2 (shown in Figure S3). After dehydration at 100 °C for 10 min, all intensities reduced significantly. Further reduction at 400 °C in 5%H2/Ar atmosphere led to the disappearance of the signal at 475 cm−1, together with the appearance of a new peak at 356 cm−1. Compared with the in-situ Raman spectrum of RuO2 (Figure S3d), we can propose that the signal at 356 cm−1 can be ascribed to the partially reduced RuOx species. Interestingly, we also found the pair peaks, related to the RuOx species, at 375 cm−1 and 471 cm−1 over Ru/AO-400 (Figure S2b), in which the peak shifts could be attributed to the different electronic states of Ru over different supports [48]. In addition, a new band appeared at around 553 cm−1 over Ru/NAO-400, which could be assigned to the NiO phase with nickel incorporated into the subsurface of the alumina support [45, 49], emerged and kept its existence even after reduction.

In-situ Raman spectra of anisole hydrodeoxygenation over (a) Ru/NAO-400, and (b) pure NiAl2O4 spinel
In the Raman experiments, anisole was injected into the reaction cell at ambient temperature, and was further swept in Ar for 10 min. Before the adsorption of anisole, both samples were reduced and then cooled down to room temperature, whereas the procedures were in-situ operated in the testing cell. When the temperature raised to 100 °C, the peak at 470 cm−1 recovered simultaneously with the disappearance of the band at 356 cm−1, and kept steady even when the temperature reached 150 °C (Fig. 2a). This result indicates that the oxygen in anisole was adsorbed by RuOx species, and the interaction between Ru and adsorbed oxygen species contributed to the recovery of the Ru–O bending at 470 cm−1. Right after the introduction of hydrogen at 200 °C, the intensity of the newly formed Ru–O bond went down again with the increase of the temperature. Simultaneously, the reappearance of the peak at 356 cm−1 after reaction further indicates that the demethoxylation of anisole underwent the redox pathway over active RuOx species in Ru/NAO-400. In contrast, only two bands at 320 cm−1 and 555 cm−1 without obvious changes were observed during the reaction conditions over pure NiAl2O4 support was shown in Fig. 2b. According to the in-situ Raman spectra, we can conclude that RuOx species worked as the active sites that interacted with the O atom in methoxy during demethoxylation.
3.3 TPSR-MS andin-situ DRIFT studies on anisole demethoxylation
TPSR-MS of anisole was further carried out to investigate the ad/de-sorption behavior of the reactant, intermediate species and products, which could help unravel the possible reaction pathway. The starting points of demethoxylation over three catalysts could also be obtained from the uplifting temperatures of the generation of methanol and benzene. To be noted, the starting points of demethoxylation hereby were all determined by the uplift trend of methanol (m/z = 31), due to the fragment overlap between anisole and benzene at m/z = 78 (Figure S5).
Figure 3a and b show that the generation temperatures of methanol were quite close over two NiAl2O4-based catalysts, which were 103 °C and 107 °C over Ru/NAO-400 and Ru/NAO-200, respectively, suggesting they might possess similar activation ability for C-O bond cleavage. This speculation could also be confirmed via the in-situ DRIFT spectra of anisole adsorption. The bands corresponded to methoxy C-O bond at 1240 cm−1 and 1238 cm−1 in Fig. 3d were both red-shifted compared with gaseous anisole, indicating the existence of strong interaction between the substrate and the catalyst surface. As a contrast in Fig. 3c, the demethoxylation over Ru/AO-400 initiated from 115 oC, whereas only a slight redshift of 5 cm−1 in DRIFT was observed, indicating that the activation of C-O bond was less effective over Ru/AO-400. More interestingly, the adsorption of anisole on Ru/AO-400 was weak since the desorption peak of anisole was at 98 °C, right before the starting point of demethoxylation. Besides Ru/AO-400, the desorption peaks of anisole also vary between the other two catalysts, which were 210 oC and 168 oC over Ru/NAO-400 and Ru/NAO-200, respectively, which could be ascribed to the fact that the chemical compositions of Ru, namely the proportion of Ru0 species, play an important role in the adsorption of the aromatic ring in anisole, which was also mentioned by the previous work [50]. However, a higher concentration of Ru0 species can enable the stronger adsorption of the reactant, but may also make it more difficult for the desorption of produced aromatics, which will hinder the recovery of Ru0 species. These influences can be seen from the second desorption peaks of fragment at m/z = 78 at, 327 °C, 267 °C and 232 °C over Ru/NAO-400、Ru/NAO-200和Ru/AO-400, respectively. Hence, combined with the Raman results, an appropriate Ru0/Ruδ+ ratio played vital role in demethoxylation, and Ru/NAO-200 catalyst likely possessed a good ratio of Ru0/Ruδ+. This analysis is in good agreement with catalytic performance summarized in Fig. 2. In addition, we also observed the formation of H2 (m/z = 2) and formaldehyde (m/z = 30) when we raised the temperature, suggesting the decomposition of generated methanol, or methoxy, that later initiated the WGS according to the uplift of CO2 (m/z = 44) [34, 38, 51]. Interestingly, the generation temperature of CO2 was at 300 °C over Ru/NAO-200 in Fig. 3b, higher than 154°C and 118°C over Ru/NAO-400 and Ru/AO-400, respectively, which could be ascribed to the low WGS rate over Ru/NAO-200. The difference between Ru/AO-400 and Ru/NAO-400 was also thought to be related to the acidity of the support, and the more acidic Ru/AO-400 had lower desorption temperature. Combining the activity evaluation, in-situ Raman, DRIFT and TPSR-MS, we can conclude that the synergy between Ru0 and Ruδ+ is crucial for the demethoxylation reaction, i.e., Ru0 is in charge of the dissociation of H2 and suitable adsorption of the aromatics, while Ruδ+ is responsible for the activation of C-O bond.

TPSR-MS profiles of anisole demethoxylation over (a) Ru/NAO-400, (b) Ru/NAO-200 and (c) Ru/AO-400, operated in 5%H2/Ar atmosphere; (d) in-situ DRIFT spectra of anisole adsorption experiments
3.4 Kinetic measurements for anisole demethoxylation over different catalysts
According to the TPSR experiments, we found that the hydrogenolysis of methoxy underwent complicated reaction pathways, including both demethoxylation and methoxy decomposition, followed by the CO-WGS. To further investigate the influence of Ru electronic states in these reactions, we carried out the kinetic studies over these three catalysts, Ru/NAO-400, Ru/NAO-200 and Ru/AO-400. H2 and 4-methylanisole were selected as reactants in this reaction. Based on the in-situ Raman results, RuOx species, namely Ruδ+, were responsible for the activation of C-O bond, while the adsorption of aromatic ring and the dissociation of hydrogen were generally believed to occur on metallic M0 sites [50, 52]. Hence, the kinetic modeling was established on a dual active sites model, i.e., Ru0 for activation of hydrogen, and the synergy between Ru0 and Ruδ+ to dissociate the C-O bond in 4-methylanisole. As is described in SI, the demethoxylation of 4-methylanisole was divided into 9 elementary steps, which were i) dissociative adsorption of H2; ii) dissociative adsorption of 4-methylanisole; iii) surface reaction between adsorbed H and aromatic species; iv) desorption of generated toluene; v) decomposition of adsorbed methoxy into formaldehyde; vi) decomposition of adsorbed formaldehyde into CO; vii) adsorption of H2O molecules; viii) surface reaction of CO-WGS; and ix) desorption of generated CO2. The detailed derivation of the kinetic modeling is attached in the supporting information.
Table 2 shows the finest fitted rate expressions and regression forms with respect to hydrogen partial pressure (PH2) or 4-methylanisole concentration (CAn). As is illustrated in Table 2 and Figures S5-S7, the best fitness results indicate that the RDS would be the desorption of produced toluene (Step iv) over Ru/NAO-400, which had more Ru0 species; while the surface reaction of CO-WGS (Step viii) was the RDS over Ru/NAO-200 which possessed relatively more Ruδ+ species than Ru/NAO-400 (Table 1). This phenomenon was also in accordance with the TPSR-MS results on Ru/NAO-200 (Fig. 4b) that the temperature of CO2 generation was much higher over Ru/NAO-200 (after 300°C) than that over both Ru/NAO-400 and Ru/AO-400, which were 154 °C and 118 °C, respectively. Interestingly, the dissociation of H2 (Step i) is the rate-determining step for Ru/AO-400 catalyst as a result of its high ratio of RuOx species, with its lowest Ru0 concentration limiting the activation of H2 molecules. This is also suggested by the H2-TPD profiles of these three catalysts in Figure S8 that the adsorption amount of H2 was much lower over Ru/AO-400 than those over the other two catalysts.

a Steady-state kinetic studies and measurements for the demethoxylation of p-methylanisole to toluene over different Ru/NAO catalysts pretreated at different temperatures. b The figure show toluene production rates as a function inverse temperature. Reaction conditions (Con. < 10%): 250°C, 0.2 g 4-methylanisole in 10 mL H2O, 2 MPa (0.2 MPa H2 for measurement of rates, 0.1 MPa for measurement of activation energies, and balanced by 2 MPa N2), 10 mg catalyst, with a stirring rate of 800 rpm
Moreover, the demethoxylation rates over Ru/NAO catalysts with different reduction temperatures followed a volcanic distribution (Fig. 4a), in which Ru/NAO-200 exhibited the highest demethoxylation rate, approaching 20.6 mmol·h−1·gcat−1. In contrast, the rate was extremely low over NAO support, also suggesting that Ru was essential for this reaction. Thus, we can conclude that demethoxylation of 4-methylanisole was an electronic structure sensitive reaction, in which the synergy of different Ru species is sensitive in influencing the catalytic performances. The apparent activation energies for demethoxylation were subsequently measured over three catalysts, which was 39 ± 3 kJ·mol−1, 20 ± 1 kJ·mol−1 and 59 ± 6 kJ·mol−1 over Ru/NAO-400, Ru/NAO-200 and Ru/AO-400, respectively (Fig. 4b), further manifesting that an appropriate Ru0/Ruδ+ ratio could lower the energy barrier, thus promoting the demethoxylation reaction over Ru/NAO-200 catalyst.
3.5 In-situ DRIFT studies on methoxy decomposition and DRIFT-MS studies on WGS
Since the surface reaction of CO-WGS after methanol (or methoxy) decomposition was the RDS over Ru/NAO-200, which was the catalyst with the best demethoxylation activity. In-situ DRIFT study of methanol were carried out to investigate the decomposition of methoxy species over different catalysts. We chose Ru/NAO-400 and Ru/NAO-200 as two models that allowed the comparisons on the electronic effect of Ru on the methoxy decomposition. Figures 5a and b show the in-situ DRIFT spectra of methanol over Ru/NAO-400 and Ru/NAO-200. The peak sets of ~ 2950 cm−1 νas(C-H), ~ 2840 cm−1 (νs(C-H)), 1450–1350 cm−1 (δ(C-H)) and ~ 1030 cm−1 (ν(C-O)) represent a bridged methoxy over the catalyst surface while the band intensity of ν(C-O) decreased simultaneously with the emerging of the band at ~ 1100 cm−1 after adsorption, indicating the transformation of bridged mode to on-top adsorbed methoxys over both Ru/NAO-400 and Ru/NAO-200 catalysts [53, 54].

In-situ DRIFT spectra of methanol adsorption over (a) Ru/NAO-400 and (b) Ru/NAO-200, collected at 30°C in Ar sweep
Methoxy species immediately decomposed under the ambient conditions over both catalysts, as we also observed the bands of carbonyl species, which were 1649 cm−1 over Ru/NAO-400, and 1654 cm−1 over Ru/NAO-200, in which the band strengths were probably due to the difference in the interaction between the carbonyl species and Ru species with different electronic states [55]. More interestingly, adsorbed CO molecule (bands in the range of 2050 ~ 2070 cm−1) was even detected in the collected spectra, suggesting a deep decomposition of methoxy to CO. However, the residence time of CO was longer over Ru/NAO-200, and CO molecules kept its existence even after 20 min of Ar sweeping (Fig. 5b). In contrast, the peak intensity of CO was relatively weak at the initial stage, then disappeared with the prolonging sweeping time over Ru/NAO-400 (Fig. 5a). This can be ascribed to the possibility that CO was rapidly consumed via water–gas shift (WGS), once it was generated on the surface [56], in accordance with the anisole-TPSR results in Fig. 3a and b that the generation of CO2 was easier over Ru/NAO-400 than over Ru/NAO-200.
To verify the capability of CO conversion over these two catalysts, we further performed in-situ DRIFT-MS studies on WGS. In Fig. 6a and b, we can observe three bands of CO species on Ru catalysts at ~ 2135 cm−1, ~ 2070 cm−1 and ~ 2005 cm−1 which are assigned to multi-carbonyl species on Ruδ+ (Ruδ+-COn), linearly adsorbed CO on Ruδ+ (Ruδ+-CO), and linearly adsorbed CO on Ru0 (Ru0-CO), respectively [57, 58]. The bands corresponding to Ruδ+-CO and Ru0-CO were located at relatively higher frequencies over Ru/NAO-200, which can be ascribed to its positively charged Ru particles [59]. With the simultaneous increase in temperature in a continuous 10%CO/He flow with water, the intensity ratios of Ru0-CO and Ruδ+-CO first decreased (Figs. 6c and d), indicating that the linearly adsorbed CO molecules were transferred from Ru0 to Ruδ+ species. After 250 °C, the ratio curve of Ru0/Ruδ+ shot up steeply with the production of CO2 gas (m/z = 44), suggesting the reduction of some Ruδ+ species. In addition, the production of H2 (m/z = 2) over both catalysts existed at higher temperatures than those of CO2. All these phenomena indicate that CO molecules helped remove the active oxygen species on these two catalysts, leading to the formation of active sites for the dissociation of water. Thus, H2 was generated via water splitting at higher temperature ranges (both after 310 oC) following the redox mechanism [38, 60]. Moreover, the starting point of CO2 production was much higher over Ru/NAO-200, 196°C in Fig. 6d, compared to 139 °C over Ru/NAO-400 (Fig. 6c), suggesting that it was much more difficult to convert CO over Ru/NAO-200 during the WGS. More interestingly, we also observe that there was no obvious difference (Δ) in Ru0/Ruδ+ ratios over Ru/NAO-200 catalyst, comparing their initial and final states. However, some Ruδ+ species were completely reduced over Ru/NAO-400 after the test, in which the difference reached ~ 0.3 in Fig. 6c. All these phenomena suggested that the reduction of surface Ruδ+ species was much more difficult in Ru/NAO-200, contributing to a relatively longer residence of CO on the catalyst surface. As a result, the surface reaction of WGS is the rate-determining step over Ru/NAO-200 catalyst, which is in accordance with the conclusions from the kinetic measurement.

In-situ DRIFT-MS studies on CO-WGS over (a, c) Ru/NAO-400 and (b, d) Ru/NAO-200, collected from 50°C to 350°C, H2O was bubbled into the chamber with continuous 10%CO/He flow, 30 mL·min.−1. Ratios of band intensities were calculated as I~2005 cm-1/I~2070 cm-1
4 Conclusion
In summary, we studied the demethoxylation of lignin derived model compound over Ru/NAO-400, Ru/NAO-200 and Ru/AO-400 catalysts with different ratios of Ru0/Ruδ+. In-situ Raman experiments illustrated that surface Ruδ+ species is crucial to demethoxylation, whereas it worked as the active sites for the dissociative adsorption of the C-O bonds. TPSR-MS and in-situ DRIFT studies further demonstrated that the concentration of Ru0 species also had impact on the competitive adsorption of aromatic rings both in the substrate and the aromatic products, which would influence the subsequent catalytic cycle. The kinetic studies on demethoxylation of 4-methylanisole revealed that the RDS is sensitive to Ru states, which are the desorption of produced toluene over Ru0 enriched Ru/NAO-400, the surface reaction of CO-WGS over Ru/NAO-200 with both Ru0 and Ruδ+ are needed, and the dissociation of H2 over Ruδ+ enriched Ru/AO-400, respectively. We also figured out that the methoxys were rapidly decomposed to CO over the catalysts, and finally to CO2 by WGS, following a redox mechanism, in which the oxidation of CO to CO2 by Ruδ+ species was much more difficult over Ru/NAO-200 than that over Ru/NAO-400. Finally, the excellent demethoxylation performance of Ru/NAO-200 can be ascribed to the appropriate Ru0/Ruδ+ ratio, which resulted in i) the effective activation of H2 and C-O bond, ii) the rapid desorption of produced aromatics, and iii) a stable electronic state of Ru particles. This work helps reveal the real active sites for hydrodeoxygenation reaction, and also sheds lights on the designing of relating catalysts with higher catalytic performance.
Abbreviations
- RDS:
-
Rate-determining step
- WGS:
-
Water–gas shift
- HDO:
-
Hydrodeoxygenation
- Ru/NAO-x:
-
Ru/NiAl2O4-reduction temperature
- Ru/AO-x:
-
Ru/γ-Al2O3- reduction temperature
- Con.:
-
Conversion
References
Huber GW, Iborra S, Corma A (2006) Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering. Chem Rev 106:4044–4098
Alonso DM, Bond JQ, Dumesic JA (2010) Catalytic conversion of biomass to biofuels. Green Chem 12:1493–1513
Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM (2010) The catalytic valorization of lignin for the production of renewable chemicals. Chem Rev 110:3552–3599
Tuck CO, Pérez E, Horváth IT, Sheldon RA, Poliakoff M (2012) Valorization of biomass: deriving more value from waste. Science 337:695–699
Li C, Zhao X, Wang A, Huber GW, Zhang T (2015) Catalytic transformation of lignin for the production of chemicals and fuels. Chem Rev 115:11559–11624
Schutyser W, Renders AT, Van den Bosch S, Koelewijn SF, Beckham GT, Sels BF (2018) Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem Soc Rev 47:852
Jing YX, Guo Y, Xia QN, Liu XH, Wang YQ (2019) Catalytic production of value-added chemicals and liquid fuels from lignocellulosic biomass. Chem 5:1–27
Sun Z, Fridrich B, de Santi A, Elangovan S, Barta K (2018) Bright side of lignin depolymerization: toward new platform chemicals. Chem Rev 118:614–678
Liao Y, Koelewijn SF, Van den Bossche G, Van Aelst J, Van den Bosch S, Renders T, Navare K, Nicolai T, Van Aelst K, Maesen M, Matsushima H, Thevelein JM, Van Acker K, Lagrain B, Verboekend D, Sels BF (2020) A sustainable wood biorefinery for low-carbon footprint chemicals production. Science 367:1385–1390
Zhang C, Wang F (2020) Catalytic lignin depolymerization to aromatic chemicals. Acc Chem Res 53:470–484
Wong SS, Shu R, Zhang J, Liu H, Yan N (2020) Downstream processing of lignin derived feedstock into end products. Chem Soc Rev 49:5510–5560
Toledano A, Serrano L, Pineda A, Romero AA, Luque R, Labidi J (2014) Microwave-assisted depolymerisation of organosolv lignin via mild hydrogen-free hydrogenolysis: Catalyst screening. Appl Catal B Environ 145:43–55
Dou Z, Zhang Z, Wang M (2022) Self-hydrogen transfer hydrogenolysis of native lignin over Pd-PdO/TiO2. Appl Catal B Environ 301:120767
Yang J, Li S, Zhang L, Liu X, Wang J, Pan X, Ning L, Wang A, Yu C, Wang X (2017) Hydrodeoxygenation of furans over Pd-FeOx/SiO2 catalyst under atmospheric pressure. Appl Catal B Environ 201:266–277
Azadi P, Khan S, Strobel F, Azadi F, Farnood R (2012) Hydrogen production from cellulose, lignin, bark and model carbohydrates in supercritical water using nickel and ruthenium catalysts. Appl Catal B Environ 117:330–338
Meng J, Moore A, Tilotta DC, Kelley SS, Adhikari S, Park S (2015) Thermal and storage stability of bio-oil from pyrolysis of torrefied wood. Energy Fuels 29:5117–5126
Walker TW, Motagamwala AH, Dumesic JA, Huber GW (2019) Fundamental catalytic challenges to design improved biomass conversion technologies. J Catal 369:518–525
Romero Y, Richard F, Brunet S (2010) Hydrodeoxygenation of 2-ethylphenol as a model compound of bio-crude over sulfided Mo-based catalysts: Promoting effect and reaction mechanism. Appl Catal B Environ 98:213–223
Shao Y, Xia Q, Dong L, Liu X, Han X, Parker SF, Cheng Y, Daemen LL, Ramirez-Cuesta AJ, Yang S, Wang Y (2017) Selective production of arenes via direct lignin upgrading over a niobium-based catalyst. Nat Commun 8:16104
Li L, Dong L, Liu X, Guo Y, Wang Y (2020) Selective production of ethylbenzene from lignin oil over FeOx modified Ru/Nb2O5 catalyst. Appl Catal B Environ 260:118143
Van den Bosch S, Schutyser W, Koelewijn SF, Renders T, Courtin CM, Sels BF (2015) Tuning the lignin oil OH-content with Ru and Pd catalysts during lignin hydrogenolysis on birch wood. Chem Commun 51:13158–13161
Luo Z, Zheng Z, Wang Y, Sun G, Jiang H, Zhao C (2016) Hydrothermally stable Ru/HZSM-5-catalyzed selective hydrogenolysis of lignin-derived substituted phenols to bio-arenes in water. Green Chem 18:5845–5858
Xin Y, Dong L, Guo Y, Liu X, Hu Y, Wang Y (2019) Correlation of the catalytic performance with Nb2O5 surface properties in the hydrodeoxygenation of lignin model compound. J Catal 375:202–212
Goulas KA, Mironenko AV, Jenness GR, Mazal T, Vlachos DG (2019) Fundamentals of C-O bond activation on metal oxide catalysts. Nat Catal 2:269–276
Guo T, Xia Q, Shao Y, Liu X, Wang Y (2017) Direct deoxygenation of lignin model compounds into aromatic hydrocarbons through hydrogen transfer reaction. Appl Catal A Gen 547:30–36
Luo Z, Zhao C (2016) Mechanistic insights into selective hydrodeoxygenation of lignin-derived β-O-4 linkage to aromatic hydrocarbons in water. Catal Sci Tech 6:3476–3484
Luo Z, Zheng Z, Li L, Cui Y-T, Zhao C (2017) Bimetallic Ru–Ni catalyzed aqueous-phase guaiacol hydrogenolysis at Low H2 pressures. ACS Catal 7:8304–8313
Song S, Zhang J, Gözaydın G, Yan N (2019) Production of terephthalic acid from corn stover lignin. Angew Chem Int Ed 58:4934–4937
Liu X, Wang C, Zhang Y, Qiao Y, Pan Y, Ma L (2019) Selective preparation of 4-alkylphenol from lignin-derived phenols and raw biomass over magnetic Co–Fe@N-Doped carbon catalysts. Chemsuschem 12:4791–4798
Huang X, Ludenhoff JM, Dirks M, Ouyang X, Boot MD, Hensen EJM (2018) Selective production of biobased phenol from lignocellulose-derived Alkylmethoxyphenols. ACS Catal 8:11184–11190
Dong L, Xin Y, Liu X, Guo Y, Pao C-W, Chen J-L, Wang Y (2019) Selective hydrodeoxygenation of lignin oil to valuable phenolics over Au/Nb2O5 in water. Green Chem 21:3081–3090
Zhou H, Wang H, Sadow AD, Slowing II (2020) Toward hydrogen economy: Selective guaiacol hydrogenolysis under ambient hydrogen pressure. Appl Catal B Environ 270:118890
Ishikawa M, Tamura M, Nakagawa Y, Tomishige K (2016) Demethoxylation of guaiacol and methoxybenzenes over carbon-supported Ru-Mn catalyst. Appl Catal B Environ 182:193–203
Li L, Dong L, Li D, Guo Y, Liu X, Wang Y (2020) Hydrogen-free production of 4-Alkylphenols from lignin via self-reforming-driven depolymerization and hydrogenolysis. ACS Catal 10:15197–15206
Li L, Zhang T, Guo Z, Liu X, Guo Y, Huang Y, Wang Y (2021) Unraveling the role of metal in M/NiAl2O4 (M = Pt, Pd, Ru) catalyst for the self-reforming-driven hydrogenolysis of lignin. Ind Eng Chem Res 60:11699–11706
Sham TK, Ohta T, Yokoyama T, Takata Y, Kitajima Y, Funabashi M, Kuroda H (1991) Ru L-edge x-ray absorption studies of the formation of Ru–Cu bimetallic aggregates on Cu(100). J Chem Phys 95:8725–8731
Xu M, Yao S, Rao D, Niu Y, Liu N, Peng M, Zhai P, Man Y, Zheng L, Wang B, Zhang B, Ma D, Wei M (2018) Insights into interfacial synergistic catalysis over Ni@TiO(2–x) Catalyst toward water-gas shift reaction. J Am Chem Soc 140:11241–11251
Li D, Li Y, Liu X, Guo Y, Pao C-W, Chen J-L, Hu Y, Wang Y (2019) NiAl2O4 spinel supported pt catalyst: high performance and origin in aqueous-phase reforming of methanol. ACS Catal 9:9671–9682
Sham TK, Ohta T, Yokoyama J, Takada Y, Kitajima Y, Funabashi M, Kuroda H (1990) Ru L-edge X-ray absorption near edge structures (XANES) from Ru LM4,5M4,5 and M4,5N4,5N4,5 auger yields. J Electron Spectrosc 53:177–184
Okal J, Zawadzki M (2009) Catalytic combustion of butane on Ru/gamma-Al2O3 catalysts. Appl Catal B Environ 89:22–32
Adamska K, Okal J, Tylus W (2019) Stable bimetallic Ru-Mo/Al2O3 catalysts for the light alkane combustion: Effect of the Mo addition. Appl Catal B Environ 246:180–194
R. Insyani, A.F. Barus, R. Gunawan, J. Park, G.T. Jaya, H.S. Cahyadi, M.G. Sibi, S.K. Kwak, D. Verma, J. Kim, RuO2-Ru/H beta zeolite catalyst for high-yield direct conversion of xylose to tetrahydrofurfuryl alcohol, Appl. Catal. B Environ., 291 (2021).
Li W-Q, Zhou R-Y, Wang X-T, Hu L-Y, Chen X, Guan P-C, Zhang X-G, Zhang H, Dong J-C, Tian Z-Q, Li J-F (2021) Identification of the molecular pathways of RuO2 electroreduction by in-situ electrochemical surface-enhanced Raman spectroscopy. J Catal 400:367–371
Vedharathinam V, Botte GG (2013) Direct evidence of the mechanism for the electro-oxidation of urea on Ni(OH)2 catalyst in alkaline medium. Electrochim Acta 108:660–665
Vuurman MA, Stufkens DJ, Oskam A, Deo G, Wachs IE (1996) Combined Raman and IR study of MOx–V2O5/Al2O3(MOx= MoO3, WO3, NiO, CoO) catalysts under dehydrated conditions. J Chem Soc Faraday Trans 92:3259–3265
Bala N, Singh HK, Verma S, Rath S (2020) Magnetic-order induced effects in nanocrystalline NiO probed by Raman spectroscopy. Phys Rev B 102:024423
Mironovaulmane N, Kuzmin A, Steins I, Grabis J, Sildos I, Pärs M (2007) Raman scattering in nanosized nickel oxide NiO. J Phys Conf Ser 93:012039
Jo HC, Kim KM, Cheong H, Lee S-H, Deb SK (2005) In situ raman Spectroscopy of RuO2⋅xH2O. Electrochem Solid St Let 8:E39
Chan SS, Wachs IE (1987) In situ laser Raman spectroscopy of nickel oxide supported on γ-Al2O3. J Catal 103:224–227
Huš M, Bjelić A, Grilc M, Likozar B (2018) First-principles mechanistic study of ring hydrogenation and deoxygenation reactions of eugenol over Ru(0001) catalysts. J Catal 358:8–18
Mudiyanselage K, Al-Shankiti I, Foulis A, Llorca J, Idriss H (2016) Reactions of ethanol over CeO2 and Ru/CeO2 catalysts. Appl Catal B Environ 197:198–205
Dong L, Shao Y, Han X, Liu X, Xia Q, Parker SF, Cheng Y, Daemen LL, Ramirez-Cuesta AJ, Wang Y, Yang S (2018) Comparison of two multifunctional catalysts M/Nb2O5 (M = Pd, Pt) for one-pot hydrodeoxygenation of lignin, Catal. Sci Technol 8:6129–6136
Rousseau S, Marie O, Bazin P, Daturi M, Verdier S, Harlé V (2010) investigation of methanol oxidation over au/catalysts using operando ir spectroscopy: determination of the active sites, intermediate/spectator species, and reaction mechanism. J Am Chem Soc 132:10832–10841
Qi Z, Chen L, Zhang S, Su J, Somorjai GA (2021) Mechanism of methanol decomposition over single-site Pt1/CeO2 Catalyst: A DRIFTS Study. J Am Chem Soc 143:60–64
Guo W, Tong T, Liu X, Guo Y, Wang Y (2019) Morphology-tuned activity of Ru/Nb2O5 catalysts for ketone reductive amination. ChemCatChem 11:4130–4138
Han YF, Kahlich MJ, Kinne M, Behm RJ (2004) CO removal from realistic methanol reformate via preferential oxidation - performance of a Rh/MgO catalyst and comparison to Ru/γ-Al2O3, and Pt/γ-Al2O3. Appl Catal B Environ 50:209–218
Vignatti CI, Avila MS, Apesteguía CR, Garetto TF (2011) Study of the water-gas shift reaction over Pt supported on CeO2–ZrO2 mixed oxides. Catal Today 171:297–303
Chin SY, Williams CT, Amiridis MD (2006) FTIR studies of CO adsorption on Al2O3- and SiO2-supported Ru catalysts. J Phys Chem B 110:871
Komanoya T, Kinemura T, Kita Y, Kamata K, Hara M (2017) Electronic effect of ruthenium nanoparticles on efficient reductive amination of carbonyl compounds. J Am Chem Soc 139:11493–11499
Kalamaras CM, Americanou S, Efstathiou AM (2011) “Redox” vs “associative formate with –OH group regeneration” WGS reaction mechanism on Pt/CeO2: effect of platinum particle size. J Catal 279:287–300
Acknowledgements
The authors thank Prof. Minghui Zhu from East China University of Science & Technology for his help in Raman characterization. The authors also thank The Canadian Light Source for the help in XAS characterization.
Funding
This work was supported financially by the National Key Research and Development Program of China (No. 2022YFA1504900), the NSFC of China (No. 22072041, 21832002) and the Science and Technology Commission of Shanghai Municipality (10dz2220500).
Author information
Authors and Affiliations
Contributions
L.X. L. and Z.R.G.: preparation and characterization of catalysts, and performing the catalytic reactions; X.H.L.: supervision and discussion; M.S. and Y.F.H.: collection and analysis of XAS; Y.G. and Y.Q.W.: overall direction of the project and supervision; L.X.L. and Y.G.: wrote the manuscript with the help from all authors.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, L., Guo, Z., Liu, X. et al. Structure–activity relationships over Ru/NiAl2O4 catalysts in anisole demethoxylation: spectroscopic and kinetic studies. Carb Neutrality 3, 5 (2024). https://doi.org/10.1007/s43979-024-00080-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43979-024-00080-0