
Abstract
The reaction of methane with oxygen from La0.8Sr0.2FeO3-δ (LSF) and several LSF-Fe2O3 configurations was studied in chemical looping mode. Shell (LSF) and core (Fe2O3) (four catalysts called CS-3, CS-4, CS-5 and CS-6, over a range of coverage), LSF mixed uniformly with Fe2O3 (UM) and Fe2O3 (front) followed by LSF (PIS) were packed in a tubular reactor. The reaction was conducted at 900 °C and weight hourly space velocity (g methane/g catalyst/h) of 3 h−1 in 20 min reduction (10 mol% methane in nitrogen) and 20 min oxidation (10 mol% oxygen in nitrogen) cycles. LSF, CS, UM and PIS configurations yielded a significantly different performance (methane conversion, CO, CO2 and H2 selectivity and coke formation) measured in 10 reproducible cycles. The reaction and XRD data indicate that CO2 and steam formed by combustion of methane on Fe2O3 modify the phase composition of LSF, inhibits the initial LSF activity and improves the performance. Feeding a mixture containing 0.4 mol% CO2, 10 mol% methane in nitrogen to LSF confirms the positive effect of CO2 on the performance of LSF.
Similar content being viewed by others

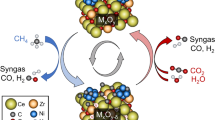

1 Introduction
Syngas is produced commercially from natural gas that contains mainly methane. A recent review [1] describes the processes developed to convert methane to syngas: steam methane reforming (SMR), auto-thermal reforming of methane (ATR), dry methane reforming (DMR), bi-reforming of methane, tri-reforming of methane and partial oxidation of methane (POM). One criterion for selection of the most suitable process is the hydrogen to carbon monoxide ratio that varies from > 3 in SMR to ≤ 1 in ATR or DMR. POM produces syngas with a ratio of 2, suitable as feedstock for many processes. The main reaction in the POM process (partial oxidation to syngas) is mildly exothermic. However, other reactions [2] that take place in parallel and in series, depending on the catalyst and operating conditions, are highly exothermic (combustion) or highly endothermic (steam reforming).
Feeding a mixture of methane and oxygen requires separation of oxygen from air, could create a safety issue and may affect the selectivity to syngas. In chemical looping, methane and oxygen are fed separately in two steps: methane reduction of solid oxides (metal oxides, perovskites) by reaction with active oxygen from the solid to produce syngas and regeneration of the active oxygen in the solid by oxidation with oxygen, carbon dioxide or steam to complete the cycle [4]. A book [3] and review papers [4, 5] published recently analyze the development of solid oxides and the progress of chemical looping in general and chemical looping of POM in particular. The thorough recent review [4] stresses the significant advantages of chemical looping processes, POM among them, based on the thermodynamic second law analysis and the reactive separation of reactants. Extensive research has been conducted in the recent years to develop solid oxides that display improved performance in terms of activity, selectivity, oxygen storage capacity and reduced coke formation [6,7,8,9,10,11]. The design of oxygen carrier solid is challenging and should be based on fundamental principles. The authors of the recent review [4] outline two basic areas that determine the suitability of the solid oxides to partial oxidation. The first is the thermodynamic need to provide the proper equilibrium partial pressure of oxygen to produce the desired products. The other is to eliminate non-selective, electrophilic oxygen species so that only lattice oxygen reacts. This requires tuning the oxygen flux from the bulk to the solid surface that can be accomplished by modifying the solid properties. Fundamental studies and application of promoters lead to improved catalytic properties of the redox catalysts [12,13,14,15,16,17].
Various configurations that combine a primary solid oxide as oxygen reservoir and a redox catalyst have been studied, specifically Fe2O3 and LSF [18,19,20]. The two solid components were combined in a shell (LSF) and core (Fe2O3) configuration. This structure enables optimal utilization of LSF and Fe2O3 properties by using the iron oxide as oxygen-carrying core for lattice oxygen storage and LSF as shell, which facilitates O2− and electron conduction inhibits primary oxide sintering and enhances activity and selectivity for POM [18,19,20]. LSF displayed a higher selectivity to H2 and CO than the core–shell catalyst while the LSF activity was lower. The dynamic nature of reactions between methane and core–shell Fe2O3@LSF materials was described by four stages: (1) combustion of CH4 to CO2, (2) competing combustion to CO2 and partial oxidation to CO, (3)partial oxidation of CH4 to CO, and 4. CH4 decomposition. These regions display the dynamic behavior of redox catalyst as a function of reduction time and the concentration and type of active oxygens that include [23] lattice oxygen O2− (Lo), defect-affected O2−VM, activated surface oxygen (Dao), activated surface oxygen resulting from oxygen adsorption: O2(g) → O−2ads → O2−2ads → O−ads → O2−surf (So), oxygen in carbonates and organic residual O = C (CO) and hydroxyl groups OH (OHO). The dynamic behavior of pure LSF was not investigated in these studies, and the information is limited. Therefore, the contribution of core–shell structure to LSF performance needs to be determined. Another study conducted on fully oxidized LaFeO3 [14] confirmed stages 1, 3 and 4, omitting stage 2. It infers that stage 1 occurs because of the low amount of vacancies. Controlling the oxidation step so that the oxygen deficiency was increased to > 0.08 shifted the selectivity to partial oxidation of methane to syngas.
Most studies used diluted oxygen or air to oxidize the solid oxide. Two recent studies employed diluted [12] and undiluted [13] CO2 to produce CO in the oxidation step, thus decreasing the H2/CO ratio in the process. Various configurations [12] of the chemical looping process were proposed to modify the H2/CO ratio and heat effect (endothermic or exothermic).
The packed-bed configuration used in the chemical looping experiments may create a gradient in the oxygen vacancies content of the solid oxides, especially at high methane conversion. A fluidized-bed reactor employed in a recent study [21] performs in a close to well-mixed mode. Experiments were conducted with La0.85Sr0.15FeO3-α at 800–950 °C and a range of space velocities of methane. The active oxygen conversion increased from 0.12 to 0.91 as the temperature increased from 806 to 927 °C during the 15 min reduction step. The methane (8.6% in N2) conversion increased from 6 to 74% and the H2/CO increased from 1.45 to 1.84, implying that the reverse water gas shift took place in parallel. The oxidation was conducted in two stage, first CO2 (7.1% in N2) for 10 min then O2 (18% in N2) for 5 min. Both CO2 and oxygen conversions were complete.
Addition of a primary solid oxide as oxygen reservoir to a redox catalyst, specifically adding Fe2O3 to LSF, may improve performance. The core–shell configuration was proposed in previous studies [17,18,19]. The purpose of our study is to examine and understand the effect of various potential configurations (core–shell catalyst with different levels of coverage and different reactor configurations of pure LSF and Fe2O3—uniformly mixed (UM) and Fe2O3 (front) followed by LSF (PIS) packed in a tubular reactor) on the performance of the combined system by comparing their dynamic behavior with pure LSF.
2 Experimental
2.1 Materials preparation
LSF perovskite was synthesized by the sol–gel method [22], as described in [23]. The perovskite precursors were dissolved separately in an aqueous solution (in molar ratio) and heated to 50 °C on a hot plate. 3.5 g iron nitrate nonahydrate (Fe(NO3)3·9H2O, Fluka) was dissolved in 6 ml water, 2.9 g lanthanum nitrate pentahydrate (La(NO3)3·5H2O, Sigma- Aldrich) was dissolved in 6 ml water, and 0.4 g strontium nitrate (Sr(NO3)2, Fluka) was dissolved in 2 ml water. Once fully dissolved, the perovskite precursors were mixed, and the complexants: 8.3 g citric acid (Sigma-Aldrich) and 4.9 g glycine (Sigma- Aldrich) were added to the solution (the ratio of moles complexants to total moles of perovskite precursors was 6.3). Then the mixed solution was heated to 80 °C on a hot plate until the gel formed. The material was dried overnight at 110 °C (5 °C/min), calcined in air at 200 °C (10 °C/min) for 2 h and then at 600 °C (5 °C/min) for 2 h.
Hematite Fe2O3 used as a core in preparing core–shell materials was purchased from NANOCAT®Superfine iron oxide (SFIO), MACH I manufacturer. It consists of porous aggregates of 3–10 nm Fe2O3 nanocrystals with a surface area of 229 m2/g. Before testing as pure catalyst in PIS configuration and mixing with LSF getting combined material of UM configuration it was calcined at 700 °C. After calcination it represented well crystallized pure Fe2O3 (hematite) phase with average crystal size of 50 nm and surface area of 8 m2/g.
60%wt LSF/40% Fe2O3 CS (core–shell) materials was synthesized by the infiltration method [23,24,25],as described in [23]. The infiltration-gelation was carried out at 60 °C while keeping the iron oxide powder on a hot plate. The infiltration was carried out in two steps due to the solubility limits of complexants. The ratio between the total moles of complexants (glycine, citric acid) and perovskite precursors (Fe, La, Sr) was 6.3. In the first step, aqueous solutions containing 6.6 mL water, perovskite precursors (0.6 g strontium nitrate, 4.3 g lanthanum nitrate pentahydrate, and 5.3 g iron nitrate nonahydrate) and complexants (6.5 g citric acid with 3.8 g glycine in preparation of CS-3; 5.3 g glycine for CS-4 and 7.8 g citric acid for CS-5) were infiltrated into 2.0 g iron oxide. Then, the material was dried overnight at 110 °C (5 °C/min). In the second step, considering the mass loss during drying, the materials obtained at the first step were infiltrated with aqueous solutions containing complexants (2.0 g citric acid with 1.2 g glycine dissolved in 13.9 mL water in preparation of CS-3; 3.0 g glycine dissolved within 14.0 mL water for CS-4 and 14.3 g citric acid dissolved in 24.6 mL water for CS-5). The materials were dried overnight at 110 °C (5 °C/min) and calcined at 200 °C (10 °C/min) for 2 h and then at 600 °C (5 °C/min) for 2 h. Further calcination was conducted at 700 °C (5 °C/min) for 2 h in the case of CS-3 and CS-5 and for 10 h for CS-4 to complete crystallization of the perovskite phase. After drying and calcination at 700 °C, the perovskite phase was formed and distributed at the outer surface of aggregates of hematite nanocrystals and in their pores.
45%wt LSF/55% Fe2O3 CS (CS-6) material was prepared as CS-3, the amounts of perovskite precursor and citric acid-glycine complexants were reduced to get 45wt% loading of LSF-perovskite component.
2.2 Materials characterization
The surface area, pore size, and pore volume of the catalysts were calculated from N2 adsorption–desorption isotherms using conventional BET (Brunauer–Emmett–Teller) and BJH (Barrett–Joyner–Halenda) methods, with NOVA 3200e Quantachrome adsorption analyzer. Before analysis, the samples were outgassed under vacuum at 250 °C for 2 h. Conventional wide-angle XRD patterns were measured with a Panalytical Empyrean Powder Diffractometer equipped with a position-sensitive detector X’Celerator fitted with a graphite monochromator, at 40 kV and 30 mA. The phases were analyzed with reference to the International Center for Diffraction Data (ICDD) database in HighScore software. The phase content in catalytic materials was calculated using Rietveld refinement.
High-resolution transmission electron microscopy (HRTEM) imaging was carried out using a JEOL JEM-2100F analytical TEM operated at 200 keV equipped with a GATAN 894 US1000 camera. Energy-filtered TEM (EFTEM) experiments were performed using a Gatan image filter. The lanthanum M-edge (832 eV) and iron L-edge (708 eV) were used for elemental mapping. The samples for HRTEM were prepared by depositing a drop of ethanol suspension of the solid catalyst on a silica-coated grid.
2.3 Experimental setup and analytical procedures
Chemical-looping experiments were conducted in the rig depicted in Fig. 1. It consisted of a quartz fixed bed reactor (1), 55 cm long and 1.3 cm ID, heated by a horizontal tube furnace (4) (Nabertherm P-330) and equipped with gas mass flow controllers (2) (Brooks Delta smart II SLA5850S) for nitrogen, methane, oxygen and carbon dioxide. Distilled water was fed from vessel (6) by a pump (7) (Jasco PU-1580) through a controlled valve to a nitrogen stream. The steam-nitrogen stream was heated in a tubular preheater (Carbolite MTF 10/25/130), combined with the other gas stream and fed to the reactor. Automatic valves (3) (Baccara GEM-T) were programmed to control the feed of methane, oxygen, water and carbon dioxide. The reactor was packed in three different configurations. In the first, LSF, CS and Fe2O3, 180–350 µm in size, diluted with quartz at a 1:2 ratio, were packed in the isothermal zone of the furnace. In the second, Fe2O3 (first bed) and LSF were packed in series (called PIS) and the third consisted of a uniform mixture of Fe2O3 and LSF (called UM). The gas composition (H2, CO, CO2, CH4 and O2) was analyzed continuously during the run by an IR device (5) (ABB AO2020). Periodic calibration of the gases was done. The measurements were reproducible with an estimated error of ± 5%.

Experimental chemical looping rig
A typical experiment consisted of four steps: reduction with methane diluted in nitrogen (10–30 min) followed by flushing with nitrogen (10 min), then oxidation with oxygen diluted in nitrogen (20 min) and finally purging with nitrogen (10 min). The automatic valves were turned on and off to set the start and end of methane and oxygen flow in the reduction and oxidation step, respectively.
Weight hourly space velocity (WHSV) is defined as mass of methane per mass of catalyst per hour.
The methane conversion was calculated as follows:
where x1t is the methane conversion, F10 and F1t are the methane flowrate at reactor inlet and outlet (at time t), respectively.
The selectivity of the partial oxidation, combustion and pyrolysis products and the hydrogen to carbon monoxide ratio were expressed as:
where S2t, S3t, S4t and Sctr are selectivity of hydrogen, carbon monoxide, carbon dioxide and coke respectively, r is the hydrogen to carbon monoxide, whereas F2t, F3t and F4t are the outlet hydrogen, carbon monoxide and carbon dioxide flowrates, respectively.
The oxygen conversion was calculated as follows:
where x5t is the oxygen conversion, F50 and F5t are the oxygen flowrate at reactor inlet and outlet (at time t), respectively.
The coke conversion by combustion to carbon monoxide \({S}_{c3}\) and dioxide \({S}_{c4}\) was expressed as:
where \({F}_{c3}\) and \({F}_{c4}\) are the carbon monoxide and dioxide outlet flowrates during combustion.
3 Results and discussion
3.1 Materials characterization
The phase composition and the texture characteristics of as-prepared materials after calcination are listed in Table 1. The crystal size of LSF phase in the four core–shell materials is like the pure LSF. γ-Fe2O3 maghemite phase was measured in CS-4 (prepared with glycine) and especially in CS-5 (prepared with citric acid) while CS-3 and CS-6 prepared with combined citric acid and glycine contained only original α-Fe2O3 hematite. The texture characteristics of three core–shell materials (CS-3, CS-4 and CS-6) are very similar. CS-5, prepared with citric acid, displayed higher surface area, pore volume and average pore diameter.
While the preparation method (complexants) has an effect on the phase composition and texture of the core–shell materials, its main effect is on the surface coverage of iron oxide core by LSF shell [23], calculated based on the surface metals concentration measured by XPS [23]. The coverage varied significantly for the four core–shell materials with the same phase composition from 71% for CS-4 to 98% for CS-5, in good agreement with estimations of relative core coverage according to HRTEM-EELS images in [23]. As expected, CS-6 prepared by the same method as CS-3 but with a lower LSF content (45% vs. 60%), yielded a lower (61% vs. 77%) surface coverage in agreement with the HRTEM-EELS image in [23] (CS-3) and in SI (Additional file 1: Figure S1- CS-6).
The phase composition of the LSF, Fe2O3 and CS materials measured by XRD, after the oxidation and the reduction steps in 10-cycles chemical looping of methane partial oxidation is listed in Table 2. Their composition after the oxidation step was the same as the fresh materials listed in Table 1. In contrast, XRD data measured after the methane reduction step indicate significant changes of the phase composition of the various materials tested.
85 wt% of the LSF was converted to other phases after the methane reduction: metallic iron, SrLaFeO4 (mixed oxide containing tetragonal Fe3+, ICDD card # 29-1305) with a structure of K2NiF4, and La(OH)3 (ICDD card # 83-2034). The LSF phase was converted from cubic perovskite to tetragonal SrLaFeO4 phase with significant change of the content of metal cations, from Sr0.2La0.8FeO3-δ to Sr1.0La1.0FeO4 [26]. This reductive transformation of LSF perovskite to trimetallic mixed oxide SrLaFeO4 enriched with Sr and La yielded monometallic Feo and La hydroxide phases. Most of pure Fe2O3 (83%) was converted mainly to Fe3O4, FeO and partially to Fe0. The reduction of Fe2O3 was reported to proceed in three stages with increasing temperature[27,28,29]: Fe2O3 Fe3O4 FeO Fe0. Reduction of Fe2O3 with methane is thermodynamically limited to form mainly Fe3O4 [5].
A new phase, lanthanum oxide carbonate La2O2CO3 (ICDD card # 84–1963), was measured in CS materials after methane reduction. The formation of this phase is observed at high temperatures in presence of CO2 where La2O3 or mixed oxides containing La react with carbon dioxide forming thermally stable La2O2CO3 [30,31,32]. Its content increased with decreasing LSF coverage from 8 wt% at 98% coverage to 17 wt% at 61% coverage. A significant variation in the amount of iron oxides (Fe2O3, Fe3O4, FeO) after methane reduction was found between CS-5 (6 wt%) and the partially covered CS-3, CS-4 and CS-6 (20–23 wt%). That is ultimately reflected in the amount of LSF after reduction, which increased from 8 to 20 wt% with increasing LSF coverage. Furthermore, the amount of SrLaFeO4 was similar in CS-3, CS-4 and CS-6, increasing to 18 wt% for CS-5.
The separate phase composition of the shell (S) and core (C) in the reduced state, estimated by accounting for the wt% of each component (i.e., CS-3 consists of 60 wt% shell and 40 wt% core), considering the relevant phases found in each component and normalizing them to 100% is listed in Table 3. The apparent composition of the shell (S-5) and the core (C-5) of CS-5 is different than CS-3 and CS-4. The SrLaFeO4 content (24 wt%) in C-5 is higher than that of C-3 (13 wt%) and C-4 (15 wt%) and the La2O2CO3 content (11 wt%) is much lower, implying the much less CO2 was formed in the nearly fully covered core of CS-5. Furthermore, the Fe0 content in C-5 (26 wt%) is by far higher than in the other materials (4–11 wt%). CS-6 prepared by the same method as CS-3 but with a lower LSF content resulting in a relatively lower core coverage, displayed a much higher content of La2O2CO3 (33 wt%) in C-6 than in C-3 (19 wt%) and a much lower LSF content (15 wt% vs. 26 wt%).
3.2 Performance of LSF and core–shell materials
10 mol% methane (reduction step) or 10 mol% oxygen (oxidation step) mixed with nitrogen were fed to a quartz fixed bed reactor. The runs were performed at WHSVLSF = 3 h−1 and 900 °C. In all experiments, 1 g LSF was packed in the reactor as pure phase or in CS and uniform mixture of LSF and Fe2O3. It means that in the case of core–shell catalysts, such as CS-3 (60 wt% LSF) and CS-6 (45% LSF), 1.67 g and 2.22 g of catalyst were packed, respectively. Normally, 10 cycles were conducted with each catalyst. Performance of cycles 2–10 was identical. 50 cycles performed in several runs displayed no changes in performance. The amount of coke, calculated by carbon balance in the reduction and oxidation steps was in agreement within 5%. Carbon, hydrogen and oxygen balances of the reduction and oxidation steps deviated by < 5%.
Time periods of 10, 20 and 30 min in the reduction step were tested for LSF and CS-3 to determine the proper time needed to capture the main features of catalysts performance. The time of the oxidation step was 20 min. The methane conversion, CO, CO2 and H2 selectivity, H2/CO molar ratio and coke selectivity, depicted in Fig. 2, were found to be independent of the time of the reduction step. The reduction time was selected to be 20 min in all experiments to capture its main features.

Performance of LSF, CS-3 and CS-6 in the reduction step measured at 900 °C and WHSV = 3 h−1. a methane conversion; b H2 selectivity and H2/CO molar ratio; c CO and CO2 selectivity; d coke combustion selectivity
LSF displayed a dramatically different performance than core–shell CS-3 and CS-6 (containing 60% and 45% LSF, respectively). Methane conversion measured with LSF, shown in Fig. 2a, increased from 20 to 50% after 4 min, then decreased gradually to 30% for the next 10 min, remaining constant for the rest of the reduction period. In contrast, CS-3 and CS-6 started at 15 and 12% methane conversion, increasing to maximum of 43% and 30% after 21 min and 15 min and decreasing to 30% and 22% at the end of the 30 min and 20 min cycle, respectively. CO selectivity depicted in Fig. 2c was even more dramatically different. On LSF, it increased slightly from 78 to 82% over the first 3 min, then decreased steeply to 30% over the next 11 min, indicating significant coking activity, shown in Fig. 2d. In the contrary, on CS-3 and CS-6, it started at 40% CO selectivity increasing to 70% and 80%, respectively, after 4 min. It increased further on CS-3 to 80% while decreasing on CS-6 to 60% after 12 min. Ultimately, it decreased to 46% on both materials after 30 min and 20 min cycle, respectively. The related coke formation after about 5 min in the reduction cycle is significantly higher on LSF than on the CS catalysts (Fig. 2d). This is a result of higher oxygen storage capacity of CS materials compared with LSF (23). The time of the reduction cycle to avoid consumption of reactive oxygen, that lead to coke formation, depends on the oxygen storage capacity.
LSF displayed negligible combustion to CO2 (Fig. 2c) and water. A relatively high combustion activity of CS-3 and CS-6 (40% CO2 selectivity in Fig. 2c) was observed at the start, decreasing to 10–12% CO2 between 4 to 10 min. The hydrogen selectivity on LSF, depicted in Fig. 2b, was 100% after 4 min, remaining constant mainly due to the coking activity (70% coke selectivity in Fig. 2d). It increased gradually on CS-3 and CS-6 to 100% after 15–20 min, forming much less coke then LSF. A different performance is also reflected in the H2/CO molar ratio that increased sharply from about 2 (partial oxidation) to 8 (coking) on LSF and remained about 2 for the first 15 min and 7 min on CS-3 and CS-6, respectively. Although the performance of CS-3 and CS-6 was somewhat different during the reduction step, the trends are very similar. The combustion activity of both core–shell catalysts that produced CO2 and water is the reason for the change to different phases found after the reduction step. This change may be the reason for the very different performance of the core–shell catalysts compared to LSF.
XRD measurements confirmed that the phases formed during the reduction step were oxidized to LSF and Fe2O3 original phases and the coke was completely oxidized to CO and CO2. LSF also displayed a different behavior than the two core–shell catalysts during the oxidation step, as displayed in Fig. 3. The oxygen conversion was complete for the first 4–5 min on all three catalysts. LSF was oxidized and its coke oxidized completely after 7 to 8 min, depending on the reduction step time that formed different coke amounts. Over 90% of the coke was oxidized to CO (Fig. 3b) and the rest to CO2. Oxidation and coke oxidation of CS-3 and CS-6 lasted for the entire 20 min cycle. 10% and 20% of the coke was oxidized relatively fast to CO2 on CS-6 and CS-3, respectively, while the rest was combusted to CO.

Performance of LSF, CS-3 and CS-6 in the oxidation step measured at 900 °C and WHSV = 3 h−1. a oxygen conversion; b coke conversion
The different behavior of LSF compared with CS-3 and CS-6 is the result of the direct access of methane to Fe2O3 in the core–shell catalysts. Therefore, two other configurations combining LSF and Fe2O3 were studied to uncover the interaction of the two components.
3.3 Performance of systems combining LSF and Fe2O3
Reduction of Fe2O3 in H2-TPR experiments [23] found that the amount of reacted oxygen was much higher than in the reduction of LSF, despite a smaller surface area. This is probably due to significant reduction to metallic state at relatively lower temperature of 560–870 °C, while LSF remains mostly in a mixed oxide form. Reduction of Fe2O3 with 10 mol% methane in nitrogen was conducted at 900 °C and WHSV of 1.25, 2.75 and 30 h−1. The combustion of methane to CO2 and water was completed in < 0.5 min and 2 min at WHSV of 1.25 h−1 and 30 h−1, respectively, as shown in Fig. 4. The amount of oxygen consumed during combustion of methane corresponds to the thermodynamic limits of Fe2O3 with methane. Fe2O3 mainly formed Fe3O4, as listed in Table 2 and reported in [5]. The carbon, hydrogen and oxygen mass balances during combustion and oxidation steps deviated by < 5%.

Methane combustion on Fe2O3 in chemical looping mode at 900 °C a methane conversion; b products composition (CO2 and H2O)
The methane conversion on CS-3 and CS-6 (Fig. 2) was low at the start of the reduction step and increased gradually, probably inhibited by the combustion products, CO2 and steam [33, 34], formed by the reaction of methane with Fe2O3. Actually, methane has access to the core in all CS catalysts, regardless of the shell coverage, due to the porous shell as observed in HRTEM images presented in [23]. It was suggested [33] that steam reacted with the active site of the PdO surface to form Pd(OH)2 phase, effectively blocking access of methane to the active PdO phase. La(OH)3 (ICDD card # 83-2034) and La2O2CO3 (ICDD card # 84-1963) phases measured after reduction at 900 °C (Table 2) probably played an important role in determining the performance of CS materials by reducing the content of LSF-SrLaFeO4 phases (Tables 2,3) and blocking the access of methane to active sites associated with O-vacancies [1]. Therefore, inhibition of catalyst activity (methane conversion) is reversible, as reported in [34].
Besides CS-3 and CS-6 prepared with the same complexant and different Fe2O3 content, two other core–shell catalysts, CS-4 and CS-5, were prepared with different complexants and the same Fe2O3 as CS-3. Their performance in the reduction step is shown in Fig. 5. The methane conversion (Fig. 5a) is very similar for all three catalysts. The differences in the CO, CO2 and H2 selectivity as well as coke selectivity are minor compared with the different performance of LSF, illustrated in Fig. 2. In all the CS catalysts, despite the different level of core coverage,thickness and structure of the shell, the shell is porous, as reported in [23]. It enables access of methane to iron oxide and formation of combustion products. As reflected in the similar phase composition listed in Table 3, a different amount of combustion products has an almost identical effect on the performance. Therefore, the configuration has a significant effect on the performance.

Performance of CS-3, CS-4 and CS-5 in the reduction step measured at 900 °C and WHSV = 3 h−1. a methane conversion; b H2 selectivity and H2/CO molar ratio; c CO and CO2 selectivity; d coke selectivity
The performance of the two components of core–shell (CS-6), LSF and Fe2O3, was further tested in two other configurations with the same composition as CS-6 (45 wt% LSF and 55 wt% Fe2O3). One configuration consisted of packing a uniform mixture (UM) of LFS and Fe2O3 and another of packing Fe2O3 in front of LSF (PIS). In the latter, WHSV related to Fe2O3 was 2.75 h−1, meaning that the gas composition fed to LSF changed with time, as depicted in Fig. 4. XRD data of LSF measured after the reduction and oxidation steps in the PIS configuration are listed in Table 4. It indicates that LSF changed significantly after the oxidation step, in contrast to CS-6 (Table 2). Furthermore, the phase composition after the reduction step did not change significantly after oxidation. XRD data of LSF measured after the reduction and oxidation steps in the UM configuration are also listed in Table 4. It indicates that the phase composition after reduction step did not change significantly after oxidation.
In PIS, the reductive decomposition of LSF phase was completely different from that in pure LSF. It was completely converted to other phases, mainly to an amorphous phase (La-Sr-Fe–O) (46%wt). Other phases were Fe2O3 (17%wt) and SrLaFeO4 (8%wt). Furthermore, phase La2O2CO3 and mainly the carbonate enriched phase La2C2O7, that was not identified in the CS catalysts, were formed because of the presence of CO2 on the surface.
The reduction step, depicted in Fig. 6a, started and ended at about 15% and 25% methane conversion, respectively, for the three configurations. The methane conversion between 5 and 18 min, was different for the three configurations. UM configuration produced significantly higher values of CO2 (1.54 mmol) than CS-6 (0.98 mmol) and PIS (0.19 mmol). The total CO2 formed in UM and in CS-6 during the reduction step was much higher than that expected to be formed from combustion of methane on Fe2O3 (0.22 mmol). It means that LSF in these configurations produced more CO2, in comparison with PIS that produced a similar amount of CO2 as expected from the performance of Fe2O3 packed in the reactor (Fig. 4). PIS displayed much higher values of CO selectivity than CS-6 (Fig. 6c), starting at 70%, reaching 95% and decreasing to 70% at the end of the reduction step. Similarly, the hydrogen selectivity (Fig. 6b) of PIS started at 70%, increasing rapidly to 100%. Thus, the H2/CO molar ratio started at 2 and increased to about 3.5. Accordingly, the coke selectivity (Fig. 5d) of PIS was low at about 10% for 11 min then increased to 30%. UM started at lower CO selectivity than CS-6 but increased rapidly to 70% after 8 min, remaining constant. Different than the other two configurations, both H2/CO ratio (Fig. 5b) and coke selectivity (Fig. 5d) were constant over the entire reduction time, about 2.3 and 20%, respectively.

Performance of CS-6, UM and PIS in the reduction step measured at 900 °C and WHSV = 3 h−1. a methane conversion; b H2 selectivity and H2/CO molar ratio; c CO and CO2 selectivity; d coke selectivity
The oxidation step further underlined the distinctive behavior of the UM configuration. While the oxidation of CS-6 and PIS configurations yielded very similar behavior (Fig. 7a), the oxidation of UM occurred at significantly lower rate. Furthermore, coke combustion on CS-6 yielded mostly CO while PIS produced 65% CO2. Surprisingly, in contrast with all other experiments conducted with the various catalysts in different configuration where coke was fully oxidized, only about 30% of the coke was combusted in UM. Therefore, 70% of the coke in each cycle was accumulated on the surface.

Performance of CS-6, UM and PIS in the oxidation step measured at 900 °C and WHSV = 3 h−1. a oxygen conversion; b coke conversion
The improved performance of PIS configuration was further studied in two other cases, packing lower and higher amounts of Fe2O3, changing WHSVFe2O3 to 30 h−1 and 1.25 h−1, respectively, denoted as PISL and PISH. The results of the reduction step are shown in Fig. 8. Although the amount of CO2 and steam calculated from Fig. 4 increases from 0.033 and 0.066 mmol, respectively, to 0.23 and 0.46 mmol, as WHSV decreases from 30 to 1.25 h−1, the performance of PISL, PIS and PISH was relatively similar but very different from the performance of LSF, CS-6 and UM. Furthermore, it appears that injecting to LSF an almost pulse of steam and CO2 changes the performance of LSF much more significantly than injecting them uniformly along the bed, adjacent to LSF (core–shell and UM), as illustrated in Fig. 6, and reflected by different phase composition after reduction step (Tables 2 and 4).

Performance of PIS, PISL and PISH in the reduction step measured at 900 °C and WHSV = 3 h−1. a methane conversion; b H2 selectivity and H2/CO molar ratio; c CO and CO2 selectivity; d coke selectivity
Combining Fe2O3 with LSF and applying a specific configuration of the two components are the two dominant factors affecting the LSF performance. Fe2O3 combusts methane to produce CO2 and steam. Those two compounds are expected to have a significant effect on the LSF. Furthermore, interaction of CO2 and steam with LSF depends on whether they are introduced in the gas fed to the LSF packing or produced in-situ either by the core inside the shell or in a uniform mixture of Fe2O3 and LSF. It is imperative to determine the significance of this effect by feeding CO2 and steam, each one of them separately and the two combined, mixed with methane and nitrogen.
3.4 Effect of CO2 and H2O on performance of LSF
Packing Fe2O3 (converting methane to carbon dioxide and water) in front of LSF rendered significant phase changes in LSF measured by XRD (Table 4) after the oxidation step, in contrast to LSF or the core–shell materials exposed only to methane in the feed (Table 2). The effect of CO2 and steam fed with methane and nitrogen on LSF performance was tested in two types of experiments operated at standard conditions. Experiment CO2F was conducted with gas containing 0.4 mol% CO2 mixed with 10 mol% methane and nitrogen. Experiment CO2SF was performed with a gas feed containing 1 mol% steam and 0.4 mol% CO2 mixed with 10 mol% methane and nitrogen. It is specifically significant that the XRD phase composition (Table 5) after the reduction step in experiment CO2SF is very similar to the phase composition after the oxidation step, which has not been observed in other experiments or configurations.
The results of the two experiments are compared with those of the reference run (10 mol% methane in nitrogen) called RR experiment and the PIS experiment in Figs. 9 and 10 for the reduction and oxidation steps, respectively. The methane conversion in the reduction step (Fig. 9) started at a high level (55%) in experiment CO2F, decreasing to 30%, like in RR. Adding steam to the feed decreased the methane conversion. No CO2 was detected in the product of CO2F run. It implies that CO2 reacts with LSF, partially reoxidizing it, to maintain a certain level of active oxygen to be available for reaction with methane in contrast with the gradually diminishing oxygen in LSF that ultimately was depleted. Overoxidation by CO2 and steam in the reduction step may block oxygen vacancies. This explains the inhibition at the initial stage of reduction step measured with CS configurations compared with LSF (Fig. 2). The CO selectivity in the PIS, CO2F and CO2SF experiments is relatively similar, higher than that in RR. Very little or no CO2 was measured in the experiments.

Performance of LSF in the reduction step measured at 900 °C and WHSV = 3 h−1 in the three experiments. a methane conversion; b H2 selectivity and H2/CO molar ratio; c CO and CO2 selectivity; d coke selectivity

Performance of LSF in the oxidation step measured at 900 °C and WHSV = 3 h−1. a oxygen conversion; b coke conversion
There is a very significant difference in the oxygen consumption during the oxidation step, related mainly to the amount of oxygen from LSF consumed during the reduction step. The oxygen transferred to LSF during the oxidation step in RR experiment is 14.7 mmol compared to 7.0 mmol and 6.0 mmol in CO2F and CO2SF experiments, respectively. This may be a result of significant partial catalyst reoxidation during reduction step combined with the different phase composition of LSF after oxidation step in RR and PIS experiments. CO2 and H2O in the feed inhibit combustion of methane (Fig. 9c). It retains a high amount of Fe2O3 phase in reduced-oxidized catalyst (Table 5), consistent with low oxygen consumption in oxidation step (Fig. 10).
4 Concluding remarks
Partial oxidation of methane to syngas was conducted in the chemical looping mode employing LSF and combinations of Fe2O3 and LSF in three configurations: core–shell (CS), packed in series (PIS) and uniformly mixed (UM). LSF display a significant change in the methane conversion, CO selectivity and coke formation with time in the reduction step, starting at high methane conversion and CO selectivity then decreasing after about 5 min forming mainly coke. On the opposite, the core–shell catalysts started at low methane conversion and CO selectivity, increasing with time and forming significantly less coke. The different behavior is related to the active oxygens that were gradually depleted in LSF while core–shell catalysts generate CO2 and H2O by combustion of methane on Fe2O3 and formed a different phases composition. Relatively little difference in performance was found among the three core–shell catalysts. A major difference was recorded between the three configurations tested at the same operating conditions. Specifically, PIS displayed the best performance among the three because of the CO2 and H2O formed by combustion of methane on Fe2O3, over 1–2 min at the start of the reduction step, that were fed to LSF. It changed the phases composition and enhanced the content of active oxygens. Feeding continuously CO2 with the methane and nitrogen mixture yielded an improved and stable performance, probably due to formation of active oxygens on LSF. In contrast to all other phases compositions found in the other experiments, in this case the solid was about 50% amorphous and the crystalline phases did not change between the reduction and oxygenation step.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Elbadawi AH, Ge L, Li Z, Liu S, Wang S, Zhu Z. Catalytic partial oxidation of methane to syngas: review of perovskite catalysts and membrane reactors. Catal Rev Sci Eng. 2021;63:1–67. https://doi.org/10.1080/01614940.2020.1743420.
York APE, Xiao T, Green MLH. Brief overview of the partial oxidation of methane to synthesis gas. Top Catal. 2003;22:345–58. https://doi.org/10.1023/A:1023552709642.
Fan LS. Chemical looping partial oxidation: gasification, reforming, and chemical syntheses. Cambridge series in chemical engineering. Cambridge: Cambridge University Press; 2017.
Zhu X, Imtiaz Q, Donat F, Müller CR, Li F. Chemical looping beyond combustion-a perspective. Energy Environ Sci. 2020;13:772–804. https://doi.org/10.1039/c9ee03793d.
Li D, Xu R, Gu Z, Zhu X, Qing S, Li K. Chemical-looping conversion of methane: A Review. Energy Technol. 2020;8:1–28. https://doi.org/10.1002/ente.201900925.
Mishra A, Galinsky N, He F, Santiso EE, Li F. Perovskite-structured AMn: XB1- xO3 (A = Ca or Ba; B = Fe or Ni) redox catalysts for partial oxidation of methane. Catal Sci Technol. 2016;6:4535–44. https://doi.org/10.1039/c5cy02186c.
Chen S, Zeng L, Tian H, Li X, Gong J. Enhanced lattice oxygen reactivity over Ni-modified WO3-based redox catalysts for chemical looping partial oxidation of methane. ACS Catal. 2017;7:3548–59. https://doi.org/10.1021/acscatal.7b00436.
Kang Y, Han Y, Tian M, Huang C, Wang C, Lin J, Hou B, Su Y, Li L, Wang J, Wang X. Promoted methane conversion to syngas over Fe-based garnets via chemical looping. Appl Catal B Environ. 2020;278: 119305. https://doi.org/10.1016/j.apcatb.2020.119305.
Yan Y, Xu L, Wang L, Fu K, Tang M, Fan M, Ma X. Syngas production from chemical-looping reforming of methane using iron-doped cerium oxides. Energy Technol. 2018;6:1610–7. https://doi.org/10.1002/ente.201700884.
Zhang L, Zhang L, Hu Y, Xu W, Huang C, Su Y, Tian M, Zhu Y, Gong H, Wang X. Anti-coke BaFe1- xSnxO3-δ oxygen carriers for enhanced syngas production via chemical looping partial oxidation of methane. Energy Fuels. 2020;34:6991–8. https://doi.org/10.1021/acs.energyfuels.0c00951.
Zagaynov IV. active components of catalysts of methane conversion to synthesis gas: brief perspectives. Energy Fuels. 2021;35:9124–36. https://doi.org/10.1021/acs.energyfuels.1c00219.
Donat F, Müller CR. CO2-free conversion of CH4 to syngas using chemical looping. Appl Catal B Environ. 2020;278:119328. https://doi.org/10.1016/j.apcatb.2020.119328.
Iftikhar S, Jiang Q, Gao Y, Liu J, Gu H, Neal L, Li F. LaNixFe1-xO3−δ as a robust redox catalyst for CO2 splitting and methane partial oxidation. Energy Fuels. 2021;35:13921–9. https://doi.org/10.1021/acs.energyfuels.1c02258.
Yang J, Bjørgum E, Chang H, Zhu KK, Sui ZJ, Zhou XG, Holmen A, Zhu YA, Chen D. On the ensemble requirement of fully selective chemical looping methane partial oxidation over La-Fe-based perovskites. Appl Catal B Environ. 2022;301:120788. https://doi.org/10.1016/j.apcatb.2021.120788.
Shafiefarhood A, Hamill JC, Neal LM, Li F. Methane partial oxidation using FeOx@La0.8Sr0.2FeO3-δ core-shell catalyst-transient pulse studies. Phys Chem Chem Phys. 2015;17:31297–307. https://doi.org/10.1039/c5cp05583k.
Shafiefarhood A, Zhang J, Neal LM, Li F. Rh-promoted mixed oxides for “low-temperature” methane partial oxidation in the absence of gaseous oxidants. J Mater Chem A. 2017;5:11930–9. https://doi.org/10.1039/c7ta01398a.
Jiang B, Li L, Zhang Q, Ma J, Zhang H, Yu K, Bian Z, Zhang X, Ma X, Tang D. Iron-oxygen covalency in perovskites to dominate syngas yield in chemical looping partial oxidation. J Mater Chem A. 2021;9:13008–18. https://doi.org/10.1039/d1ta02103f.
Neal LM, Shafiefarhood A, Li F. Dynamic methane partial oxidation using a Fe2O3@La0.8Sr0.2FeO3-δ core-shell redox catalyst in the absence of gaseous oxygen. ACS Catal. 2014;4:3560–9. https://doi.org/10.1021/cs5008415.
Shafiefarhood A, Galinsky N, Huang Y, Chen Y, Li F. Fe2O3@LaxSr1-xFeO3 core-shell redox catalyst for methane partial oxidation. ChemCatChem. 2014;6:790–9. https://doi.org/10.1002/cctc.201301104.
Neal L, Shafiefarhood A, Li F. Effect of core and shell compositions on MeOx@LaySr1-yFeO3 core-shell redox catalysts for chemical looping reforming of methane. Appl Energy. 2015;157:391–8. https://doi.org/10.1016/j.apenergy.2015.06.028.
Donat F, Xu Y, Müller CR. Combined partial oxidation of methane to synthesis gas and production of hydrogen or carbon monoxide in a fluidized bed using lattice oxygen. Energy Technol. 2020;8:1–10. https://doi.org/10.1002/ente.201900655.
Zhong Z, Chen K, Ji Y, Yan Q. Methane combustion over B-site partially substituted perovskite-type LaFeO3 prepared by sol-gel method. Appl Catal A Gen. 1997;156:29–41. https://doi.org/10.1016/S0926-860X(97)00003-3.
Dahan HO, Landau MV, Nehemya RV, Edri E, Herskowitz M, Ruan C, Li F. Core-shell Fe2O3@La1−xSrxFeO3−δ material for catalytic oxidations: coverage of iron oxide core, oxygen storage capacity and reactivity of surface oxygens. Materials. 2021;14:7355. https://doi.org/10.3390/ma14237355.
Chen D, Yang G, Ciucci F, Tadé MO, Shao Z. 3D core-shell architecture from infiltration and beneficial reactive sintering as highly efficient and thermally stable oxygen reduction electrode. J Mater Chem A. 2014;2:1284–93. https://doi.org/10.1039/c3ta13253f.
Wang W, Qu J, Zhao B, Yang G, Shao Z. Core-shell structured Li0.33La0.56TiO3 perovskite as a highly efficient and sulfur-tolerant anode for solid-oxide fuel cells. J Mater Chem. 2015. https://doi.org/10.1039/c5ta01213a.
Berry FJ, Marco JF, Ren X. Berry FJ; Marco JF; Ren X. Reduction properties of phases in the system La0.5Sr0.5MO3 (M=Fe, Co). J Solid State Chem. 2005;178:961–9. https://doi.org/10.1016/j.jssc.2004.10.003.
Albrecht M, Rodemerck U, Schneider M, Bröring M, Baabe D. Kondratenko EV Unexpectedly efficient CO2 hydrogenation to higher hydrocarbons over non-doped Fe2O3. Appl Catal B Environ. 2017;204:119–26. https://doi.org/10.1016/j.apcatb.2016.11.017.
Feng Y, Wang N, Guo X, Zhang S. Characteristics of dopant distribution and surface oxygen vacancy formation for modified Fe2O3 in chemical looping combustion. Fuel. 2020;276:117942. https://doi.org/10.1016/j.fuel.2020.117942.
Jozwiak WK, Kaczmarek E, Maniecki TP, Ignaczak W, Maniukiewicz W. Reduction behavior of iron oxides in hydrogen and carbon monoxide atmospheres. Appl Catal A Gen. 2007;326:17–27. https://doi.org/10.1016/j.apcata.2007.03.021.
Bakiz B, Guinneton F, Arab M, Benlhachemi A, Villain S, Satre P. Gavarri JR Carbonatation and decarbonatation kinetics in the La2O3-La2O2CO3 system under CO2 gas flows. Adv Mater Sci Eng. 2010. https://doi.org/10.1155/2010/360597.
Svoboda K, Leitner J, Havlica J, Hartman M, Pohořelý M, Brynda J, Šyc M, Chyou YP, Chen PC. Thermodynamic aspects of gasification derived syngas desulfurization, removal of hydrogen halides and regeneration of spent sorbents based on La2O3/La2O2CO3 and cerium oxides. Fuel. 2017;197:277–89. https://doi.org/10.1016/j.fuel.2016.12.035.
Zhao H, Li H, Gu Y, Zheng T, Zhao D, Xia W, Zhao Y, Chen H. La2O2CO3-Induced phase composition oscillation in La-Cu mixed oxides during repeated catalytic soot combustion. Catal Sci Technol. 2019;9:5100–10. https://doi.org/10.1039/c9cy01061k.
Burch R, Urbano FJ, Loader PK. Methane combustion over palladium catalysts: The effect of carbon dioxide and water on activity. Appl Catal A Gen. 1995;123:173–84. https://doi.org/10.1016/0926-860X(94)00251-7.
Wang CH, Chen CL, Weng HS. Surface properties and catalytic performance of La1-xSrxFeO3 perovskite-type oxides for methane combustion. Chemosphere. 2004;57:1131–8. https://doi.org/10.1016/j.chemosphere.2004.08.031.
Acknowledgements
This work was supported by US-Israel Binational Science Foundation, grant No. 2019613. The authors are grateful to Dr. A. Erenburg, Dr. N. Froumin and Dr.V.Ezersky for XRD, XPS and HRTEM characterization.
Funding
This work was supported by US-Israel Binational Science Foundation, grant No. 2019613.
Author information
Authors and Affiliations
Contributions
HOD, MVL and MH have developed the original ideas, designed the experimental program and have written the manuscript. HOD and GS have conducted the experimental program and analyzed the results. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Additional file 1: Figure. S1
HRTEM - EELS image of CS-6
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ohayon Dahan, H., Sror, G., Landau, M.V. et al. Chemical looping reaction of methane with oxygen from La0.8Sr0.2FeO3-δ and La0.8Sr0.2FeO3-δ -Fe2O3 systems to syngas. Discov Chem Eng 2, 3 (2022). https://doi.org/10.1007/s43938-022-00010-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43938-022-00010-5