
Abstract
In situ IR detection of photoreactions induced by the light of LEDs at appropriate wavelengths provides a simple, cost-effective, and versatile method to get insight into mechanistic details. In particular, conversions of functional groups can be selectively followed. Overlapping UV–Vis bands or fluorescence from the reactants and products and the incident light do not obstruct IR detection. Compared with in situ photo-NMR, our setup does not require tedious sample preparation (optical fibers) and offers a selective detection of reactions, even at positions where 1H-NMR lines overlap or 1H resonances are not clear-cut. We illustrate the applicability of our setup following the photo-Brook rearrangement of (adamant-1-yl-carbonyl)-tris(trimethylsilyl)silane, address photo-induced α-bond cleavage (1-hydroxycyclohexyl phenyl ketone), study photoreduction using tris(bipyridine)ruthenium(II), investigate photo-oxygenation of double bonds with molecular oxygen and the fluorescent 2,4,6-triphenylpyrylium photocatalyst, and address photo-polymerization. With the LED/FT-IR combination, reactions can be qualitatively followed in fluid solution, (highly) viscous environments, and in the solid state. Viscosity changes during the reaction (e.g., during a polymerization) do not obstruct the method.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Over the past decade, the interest in organic photochemistry experienced a renaissance, and more and more scientists have opted to develop a large amount of novel photo-induced reactions [1,2,3,4,5,6,7,8,9]. This leads to an increasing need for tools and methods to monitor the molecular nature and kinetics of intrinsically complex photochemical transformations.
A large arsenal of advanced methods [10, 11] has become available to achieve a mechanistic understanding of photochemistry such as time-resolved X‑ray methods [12], ultrafast electronic spectroscopy [13,14,15,16], and (multi-dimensional) vibrational spectroscopy even at very fast time scales [17,18,19,20,21]. In terms of an easy and convenient reaction monitoring, nuclear magnetic resonance (NMR) is often the method of choice, but although several methodologies have been developed to combine sample irradiation inside the NMR magnet [22,23,24], they can hardly be run on a routine basis. IR spectroscopy, on the other hand, provides an extensive amount of information on molecular structure particularly to identify certain functional groups [25,26,27]; furthermore, standard Fourier transform infrared (FT-IR) spectrometers are not exceedingly costly and are simple to operate. The benefits of IR spectroscopy for studying the dynamics of photochemical systems are evident from the development of time-resolved infrared spectroscopy (TR-IR) [28,29,30,31].The TR-IR systems offer a temporal window ranging from fs to ms. To perform the TR-IR measurements, depending on the desired timescale, pump-probe [32,33,34,35,36], stop-scan [37, 38], or rapid-scan [39,40,41,42,43] setups are employed. The progress and the selectivity of photo-induced reactions have been also promoted by the development of LEDs. They provide a narrow emission spectrum (line width of 10–30 nm) and cover a range between ca. 350 and 750 nm. The light intensity can be easily controlled and the output stability is much higher than that of other light sources, e.g., high-pressure lamps. Additional features such as fast turn-on–off times, long lifetime, low prices, and wide availability have made LEDs extensively utilized in the field of photo-catalysis [44,45,46].
Here, we report in situ monitoring of diverse photo reactions. By implementing LED irradiation into a readily available FT-IR spectrometer, we are able to analyze the migration of a TMS (trimethylsilyl) group from a Si atom to adjacent carbonyl oxygen in a Brook rearrangement. Moreover, we detected follow-up reactions with atmospheric oxygen and hydrolysis. IR detection allows an essentially background-free analysis of the reaction sequence at a sub-minute resolution. The experimental effort is low. We have added examples on the α-bond cleavage in the radical photoinitiator 1-hydroxycyclohexyl phenyl ketone, photo-catalysis utilizing 2,4,6-triphenylpyrylium tetrafluoroborate, photoreduction with tris(bipyridine)ruthenium(II) chloride, and photo-polymerization of styrene using phenyl-bis(2,4,6-trimethylbenzoyl)phosphine oxide photoinitiator in a viscous reaction mixture. The common feature of the above reactions is that they can hardly be followed by standard optical or NMR detection owing to overlapping bands and signals, fluorescence, and high viscosity.
2 Experimental
2.1 Design of the IR cell with LED irradiation
The 3D model of the IR cell was designed with the AutoCAD 2022 software, the file is sliced using Chitubox64 software, and the designed cell is 3D printed using black Elegoo water washable photopolymer resin on an Anycubic Photon DLP printer equipped with 405 nm high power LED as the irradiation source (files available upon request). Two IR cells are designed, one for the irradiation of solid samples prepared as a thin film and the second for the irradiation of liquid samples consisting of a commercially available Specac® Omni IR cell with a 3D printed LED adapter (Fig. S1).
2.2 FT-IR spectroscopy with in situ LED irradiation
The FT-IR spectra were recorded on a Bruker Alpha spectrometer running OPUS 7.5 software in transmission mode. The custom-made IR cell was placed inside the spectrometer and the IR spectra were averaged over 22 scans (25 s each) in the range from 500 to 4000 cm−1 at a resolution of 4 cm−1. If necessary, the Savitzky Golay smoothing (filtering) and baseline correction were applied to the spectra. The deconvolution of IR spectra was performed using OriginPro 2021, 9.8.0.200 (Academic) OriginLab Corporation software. Tool: peak and baseline → multiple peak fit (nonlinear curve fit) using Levenberg Marquardt iteration algorithm with Lorentz model, adj. R2 > 0.98. The optical path thickness (b) for the liquid samples was calculated to be (0.02 cm, Fig. S2) based on the following equation: \(b= \frac{m}{2}\times \frac{1}{{\nu }_{1}- {\nu }_{2}},\) where m is the number of interference maximums between wavenumbers \({\nu }_{1}\) and \({\nu }_{2}\). The low-power LEDs (> 50 mW) were purchased from Roithner Laser Tehnik Gmbh, and the emission spectra of the used LEDs are presented in Fig. S3. The LED is driven in a constant current mode using Keithley 224 programmable current source, allowing for precise control and high stability of the light output.
Background scans with an empty IR cell equipped with desired LED eliminated distortions based on light scattering (Fig. S4). The low reflectivity for wavelengths < 800 nm of the IR system caused no measurable influence of the LED irradiation on the operation of the FT-IR spectrometer or the resulting IR spectrum.
2.3 Advantages and limitations
2.3.1 Advantages
Owning to the specific vibrational bands of functional groups, it is possible to extract qualitative information about their reactions. Basically, any types of sample (gasses, liquids, and solids) are attainable. Quantitative analysis of several components in the mixture is possible when there is at least one isolated and well-defined band found in the IR spectra for each of the components being analyzed [47, 48].
2.4 Limitations
When performing quantitative analysis using IR spectroscopy, it is important to have an appropriate standard to perform calibration. Often the band shape in the IR spectra changes with concentration, thus simple peak height vs concentration plot is not always a straight line. For that reason, an integrated peak area should be used while performing calibration for quantitative analysis. Even then, overlap between the bands might permit accurate analysis. Deviations from Lambert–Beer law are occasionally encountered as a result of intermolecular interactions, of the sample molecules, affecting the absorption coefficient of the band [47].
Quantum yields of photochemical reactions are a fundamental quantity describing the photochemical process and they are mostly determined by actinometry [49]. It is crucial that the molar photon flux through the sample and the actinometer is identical. For that reason, a monochromatic light source should be collimated and the beam should enter the wall of the cuvette at 90° (thus minimizing reflections caused by the air wall and wall solution interfaces) [50]. In our system, the light source (LED) is not collimated (the light path is diverging from the point source) and the LED is positioned at ≈ 45° vs. the window of the cell, causing reflections and decreasing the light intensity hitting the sample. The refractive properties of the solutions (actinometer and the sample) additionally lead to scattering. This leads to a large difference in the molar photon flux between a sample and the actinometer (Figure S5). Accordingly, photochemical quantum yields are not accessible by our method.
2.5 Chemicals
(Adamant-1-yl-carbonyl)-tris(trimethylsilyl)silane was prepared according to [51]. All other chemicals were commercially available and were used as received unless otherwise noted. Detailed experimental details are given in the Supporting Information.
3 Results and discussion
3.1 Photo-Brook rearrangement in solution and in the solid state
In 1982, Brook had shown a photo-induced shift of the trimethylsilyl (TMS) group from silicon to oxygen in a tris(trimethylsilyl)acylsilane. Such a photo-rearrangement also holds for (adamant-1-yl-carbonyl)-tris(trimethylsilyl)silane (1), which was reported to rearrange to 1a, the first isolable (solid) and room temperature stable, silaethene (Fig. 1) [51]. The widespread interest in photo-Brook rearrangement is evident from its recent application in photo-affinity labeling as a method to investigate noncovalent protein-small molecule interactions [52].

Experimental IR spectra for the photo-Brook rearrangement of 1, a in CCl4 solution, b calculated concentration (left side) and conversion (right side) of 1 vs time, c rearrangement performed in a solid state (a thin film of 1), d changes in the absorbances at 1710 cm−1, 1692 cm−1, and 1620 cm−1
Herein, we show that we can follow the Brook photo-rearrangement of 1 in solution and in the solid state by IR. To that end, the solution of 1 in CCl4 was irradiated at 360 nm while simultaneously recording IR spectra (low-power LED, λmax = 360 nm, for UV–vis spectra see Fig. S6). The tris(trimethyl silan)-carbonyl group band at 1618 cm−1 of 1 decreases during the photolysis (Fig. 1a). The exponential decrease in the concentration of 1 and its conversion during the irradiation was modeled (Fig. 1b) with the help of the calibration of the C=O stretching band of 1 (at 1618 cm–1, Fig. S7). The emerging band at 1051 cm−1 is attributed to the Si=C stretching in 1a (Fig. S8) indicating the 1 → 1a photo-rearrangement.
The 1 → 1a photo-rearrangement was monitored up to ≈ 50% conversion (to avoid by-products) in dry and degassed CCl4, in a well-sealed IR cell, thus the decomposition of highly reactive 1b is not expected. Nevertheless, a band at 1810 cm−1 appears indicating the formation of a decomposition product pointing to the formation of a new carbonyl group (possibly 1-adamantanecarbonyl chloride presumably by the reaction with Cl· radical from CCl4).
For the reaction in the solid phase, we prepared a film of 1 by dissolving it in a small amount of CCl4, placing a drop on the KBr window, and evaporating the solvent under an argon atmosphere. The KBr window was inserted into the IR-photolysis cell under argon. Analogous to the experiment in solution, the C=O band (at 1620 cm−1 in a solid state, Fig. 1c) decreases and the band attributed to Si=C stretching vibration is observed (1041 cm−1 in a solid state, Fig. S9). The small amount of ambient oxygen, which gradually diffuses into the IR cell, converts the highly reactive 1a into trimethylsilyl ester of adamantane-1-carboxylic acid (1b) indicated by the band at 1710 cm−1. Traces of water, rapidly convert 1b further to adamantane carboxylic acid 1c, as indicated by the formation of the C=O stretching band at 1692 cm−1 (Fig. 1c). A closer look at the kinetic curves (Fig. 1d) supports the 1b to 1c conversion (1710 and 1692 cm−1 curves), but it also indicates that there is a process, from 1 to the products, that cannot be observed. This can be seen as the advantage of the method as it simplifies the data processing and analysis. Owing to the unknown concentrations of H2O and O2 diffusing into the sample a kinetic analysis is not straight forward.
3.2 Photochemical α-cleavage of 1-hydroxycyclohexyl phenyl ketone
1-Hydroxycyclohexyl phenyl ketone (2, Irgacure 184®) is a widely used non-yellowing UV curing type I photoinitiator for photo-polymerization of acrylates. It is used in the production of coatings, adhesives, and electronic products. The photo-polymerization process is initiated by α-carbonyl cleavage of 1 upon excitation producing primary benzoyl and cyclohexanoyl radicals [53]. In the absence of monomers, photolysis of 2 produces benzaldehyde (3) and cyclohexanone (4). The UV–vis absorption spectrum of 2 is characterized by a strong π \(\to\) π* absorption at 250 nm and weak n \(\to\) π*absorption at 290 nm with a shoulder at 330 nm extending to ca. 380 nm (Fig. S10). Irradiation of 1 at 360 nm in dry and degassed CCl4 yields the IR spectra shown in Fig. 2. The band at 1668 cm−1 corresponding to the α-hydroxy carbonyl group in parent 2 decreased with the appearance of two new vibrations at 1717 cm−1 and 1776 cm−1 attributable to 3 and 4, respectively (for deconvolution of the C=O and changes in the OH stretching vibration bands, see Fig. S11). The kinetic curves (Fig. 2 inset) of 3 and 4 display similar slopes confirming that their origin is 2 and no side reactions occur (Fig. S12).

Experimental IR spectra during the photolysis of 2. Changes in the absorbances at 1668 cm−1, 1717 cm−1, and 1776 cm−1 corresponding to 2, 3, and 4 C=O stretching bands are presented as an insert
3.3 E-Stilbene photo-oxygenation using molecular oxygen and 2,4,6-triphenylpyrylium (TPP) as the photocatalyst
TPP has been extensively used as an electron transfer photosensitizer [54, 55]. The first excited singlet and triplet states of TPP are strong oxidants [56]. TPP absorbs in the visible range (λmax = 417 nm in dichloromethane), exhibits high molar absorptivity (εM = 29 500 M−1 cm−1 @ 417 nm in dichloromethane), a fluorescence quantum yield of 0.52, emission at 465 nm) [57, 58]. Herein, we demonstrate that IR monitoring is not obstructed by the high absorptivity (intense color) and fluorescence of TPP [59].
Photooxidation of E-stilbene (5) with TPP (as BF4− salt) in dry DCM under oxygen using a low-power LED emitting at 420 nm (for UV–vis spectrum of 5, and TPP see Fig. S13) leads to the IR spectra shown in Fig. 3a. The C=C stretching band of the central ethene double bond at 1599 cm−1 decreases during the irradiation coinciding with the appearance of a new band at 1700 cm−1 assigned to benzaldehyde carbonyl stretching vibration reaching a steady state after ca. 120 s at identical rates (Fig. 3b, for the deconvolution of bands see Fig. S14). This is in line with the experimental setup: at the beginning of the reaction, the solution was saturated once with oxygen just before irradiation without further O2 being supplied. The use of an appropriately constructed flow system would be beneficial in this example as it would provide a continuous oxygen supply to the reaction mixture. This oxygen-saturated reaction mixture can then be circulated through the IR cell equipped with LED providing a setup for monitoring photo-induced gas–liquid reactions (Fig. S15).

Photo-oxygenation of 5 using molecular oxygen and TPP photocatalyst a experimental IR spectra, b changes in the absorbances at 1700 cm−1, 1622 cm−1, and 1599 cm−1, c proposed general mechanism for photo-oxidations using TPP as a photocatalyst, producing donor (D) radical cation D·+ and TP· radical, later of which reacts further with ground-state molecular oxygen forming TPOO· radical [54]
Another noteworthy band is at 1622 cm−1. It is assigned to the pyrylium-ring C=C stretching vibration [60]. Its decrease suggests that the TPP pyrylium ring is decomposed. It is established that the first excited state of TPP does not produce singlet oxygen or the superoxide radical anion, thus both are excluded to cause the destruction of the pyrylium ring. However, it was demonstrated that the 2,4,6-triphenylpyranyl radical (TP·) reacts with ground-state oxygen on the time scale of seconds producing 2,4,6-triphenylpyranyl-peroxyl radical (TPOO·) [59, 61] annihilating the aromatic character of the pyrylium ring (Fig. 3c). This side reaction explains why oxygen consumption has a higher rate than the formation of benzaldehyde (Fig. 3b).
3.4 Photoreduction of benzil to benzoin with [Ru(bpy)3]2
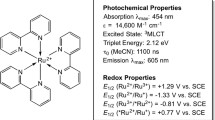
Visible-light-induced transition-metal photo-catalysis has been frequently used over the past decade [4, 8]. Amongst the wide variety of available transition-metal photocatalysts, tris(bipyridine)ruthenium(II) has been one of the most extensively investigated and widely applied [62,63,64]. Excitation of [Ru(bpy)3]2+ leads to the first excited triplet metal to ligand charge transfer state (3MLCT) characterized by \({\lambda }_{\mathrm{max}}\) 450 nm in acetonitrile (Fig. S16). The excited 3MLCT displays phosphorescence (\({\lambda }_{\mathrm{max}}\) = 620 nm, \({\tau }_{\mathrm{P}}\) = 0.75 µs in ethanol at room temperature, \({\Phi }_{\mathrm{P}}\) = 0.045) with slight solvent and temperature dependence [65]. The excited 3MLCT state is a good energy donor, electron donor, and electron acceptor and is the reason for the versatile use of [Ru(bpy)3]2+in photo-catalysis.
Photoreduction of benzil (6) with triethylamine (TEA) as an electron donor and methanol (CH3OD) as a proton source was performed in the presence of [Ru(bpy)3]2+in acetonitrile (ACN) [66]. Upon irradiation of this multi-component reaction mixture with a 450 nm LED we have recorded the IR spectra displayed in Fig. 4 (together with reference spectra of 6 and the product benzoin). The C=O stretching bands of 6 are at 1683 cm−1, and 1673 cm−1 whereas the product, benzoin (7), has a C=O stretching band at 1686 cm−1. Despite the substantial overlap between the C=O bands, their individual time profiles can be extracted owing to the different relative intensities and widths of the respective bands. The decrease of the 1673 cm−1 band has a larger slope compared to the 1683 cm−1 since the latter is the sum of two C=O bands, the decreasing C=O in 6 and the increasing C=O band in 7. Here, deconvolution of the C=O region (Fig. S17), reveals two overlapping bands at 1673 and 1683 cm−1. A plot of the area of these deconvoluted bands vs time (Fig. 4d) shows that the band of starting compound 6 (1673 cm−1) decreases. On the other hand, the band at 1683 cm−1 increases revealing the formation of 7. Further insight is provided by two-dimensional correlation spectroscopy [67]. The observed correlation patterns in synchronous and asynchronous spectra show that the decrease in the overall band has two components of which one of the components causes the band width decrease coupled with overall intensity decrease, whereas the other component increases in intensity [68] (Fig. S18).

Reduction of 6 to 7 employing Ru[bpy]3 photocatalyst, a experimental IR spectra during the irradiation (with deconvolution of C=O band for experimental spectra at 500 s) and the reference IR spectra of 6 (0.136 mol L−1) and 7 (0.136 mol L−1) in the solvent mixture (CH3OD and TEA in ACN), b evolution of the OH stretching band during irradiation, c changes of 3538 cm−1, 1673 cm−1, and 1683 cm−1 bands during the irradiation and d plotted area of peak at 1673 cm−1 (C=O band of 5) and 1683 cm−1 (C=O band of 6, and 7) vs time (error bars represented by the shaded gray and blue area)
The use of CH3OD should in principle yield the 7 with two deuteriums, which would be possible to conveniently monitor using IR spectroscopy. Unfortunately, the benzoin C–D vibration falls within the range of solvent (ACN) C–H stretching vibrations, and as such is difficult to observe in this example. The CH3OD and water from the ACN and photocatalyst (Ru[bpy]3Cl2 × 6 H2O) undergo virtually instantaneous proton–deuterium exchange equilibrium CH3OD + H2O \(\leftrightarrow\) CH3OH + DOH, with all of the present OH stretching vibrations from the initial equilibrium observable as a broad and low-intensity band in the 3300–3600 cm−1 region in reference IR spectra of 6 (the OH(D) groups present in the solvent are forming C=O–H(D)O bonds, from which only H-bonds are observable in a depicted region of IR spectra). Upon the photoreduction, all four of the equilibrium components are used as the proton source producing 7 with a statistically controlled distribution of proton and deuterium in both CH(D) and OH(D) positions. This manifests as an increase of the 3532 cm−1 band present in 7 (OH stretching, Fig. 4b) during the irradiation and the decrease of the OD stretching vibration at 2617 cm−1 (Fig. S19).
3.5 Radical photopolymerizations of styrene using bis(mesitoyl)phenylphosphine oxide as the photoinitiator
Radical photo-polymerization is a well-established but still growing technique enabling the fast and simple production of polymeric materials, e.g., for 3D printing. BAPO (Bis(Acyl)Phosphine Oxide, phenyl-bis-(2,4,6-trimethylbenzoyl)phosphine oxide) is widely investigated and employed photoinitiator [69, 70]. We have used BAPO to photo-polymerize styrene (8) while monitoring the reaction by IR. To that end, a mixture of BAPO and 8 (bulk polymerization conditions) was placed in the IR cell and irradiated at 405 nm (Fig. 5a, for UV–vis spectra of BAPO, see Fig. S20). The first step is the α-cleavage of the phosphorus-carbon bond resulting in the phosphanoyl (9a) and mesitoyl radicals (9b) [71, 72]. Both radicals add to 8, resulting in products of type 9d (or the analogous product carrying two 9b end groups). Besides the mesitoyl and phosphinoyl end groups, it carries a polystyrene chain of variable length [73]. The IR spectra identify a C=C stretching band (vinyl C=C) of 8 at 1629 cm−1, which decreases during the irradiation, the observation is in line with consumption of 8 during the irradiation (for the kinetics curve see Fig. 5b). The appearance of two new bands in the carbonyl region at 1729 cm−1 and 1700 cm−1 is observed. The first band is assigned to the C=O stretching of the 9a radical fragment whereas the second one is attributed to the C=O stretching present in the 9b radical fragment of 9d. The kinetic curve of 9a radical fragment C=O band (1729 cm−1) is observed to increase in intensity (concentration) until the maximum value followed by a slight decrease. This is in contrast to the 9b radical fragment C=O band (1700 cm−1) which continues to increase with irradiation time (Fig. 5b). The cleavage of the 9a fragment from 9d during the irradiation providing another 9b radical for further polymerization explains both C=O stretching bands. Performing deconvolution of the bands in the 1550–1785 cm−1 region helps to better visualize the dynamics of decreasing vinyl C=C band (1629 cm−1) and the formation of new bands at 1700 and 1729 cm−1 (C=O bands in 9d and follow-up products) overlapping with phenyl ring C=C vibration bands (Fig. S21). A characteristic band for the (Ph)C–C(O) stretching was found at 1167 cm−1 and the (Ph)C–C(O)–C deformation in mixed aryl–alkyl ketone was observed at 1260 cm−1. The kinetic curves of these bands overall coincide well with the formation of C=O stretching bands (Fig. 5b/c). In addition, we have detected changes in the P=O stretching bands upon polymerization. The P=O band (often split in two overlapping bands) [74, 75] of parent BAPO at 1200 cm−1 decreases in intensity whereas the 1218 cm−1 band decreases as well as shifts to higher wavenumbers (up to 1223 cm−1) during the irradiation. The observation is in line with the formation of 9d (carrying only one 9b fragment, Fig. 5a/c). The P=O band kinetic curve shows steady decrease of the band (formation of 9b) until the ca. 160 s, after which the slope increases, indicating the 9d consumption in the follow-up reactions (Fig. 5c, even though P = O bond stays intact, cleavage of another acyl group causes the further shift of the band to the higher wavenumbers manifested as the decrease in intensity at 1200 cm−1). The deconvolution of bands in the 1120–1350 cm−1 region helps to better visualize the formation of (Ph)C–C(O) stretching band at (1167 cm−1) and (Ph)C–C(O)–C deformation band (1260 cm−1) overlapped with the complex dynamics of P=O band (Fig. S22). The proposed mechanism for forming product 9b and follow-up products is supported by comparing the kinetic curves of C=O and P=O bands (absorbance vs time and area of deconvoluted peak vs time, Fig. S23).

Photopolymerization of 9 using BAPO as a photoinitiator a experimental IR spectra during irradiation using LED emitting at 405 nm. Reference IR spectra of pure BAPO and 8 are offset for clarity, b changes in the absorbances at 1729 cm−1, 1700 cm−1, and 1629 cm−1, c changes in the absorbances at 1260 cm−1, 1200 cm−1, and 1167 cm−1
4 Conclusion
Our results illustrate that we can follow the photo-Brook rearrangement and additional photo-induced reactions at high detail at a molecular level using a simple LED/FT-IR setup. A variety of reaction types and even multi-component mixtures and nonuniform products are distinguishable by this approach. The method works for reactions in fluid solutions, viscous environments, and the solid state. Moreover, optical absorption phenomena like overlapping bands of the reactants or highly dominating absorbances of photocatalysts do not obstruct the clear-cut information of the IR spectra. The conversion efficiencies and observable time scales can be shifted by adjusting LED intensities. This allows observing the reactions at time scales suitable for the detection of the spectrometer system. Moreover, following the kinetic curves may disclose yet underestimated side reactions (e.g., conversions of the pyrylium cation in Sect. 3.3). Methods for deconvolution, 2D plots, and machine learning procedures should enhance the analysis of even more complex IR spectra extending the attainable information to the fingerprint range [76,77,78,79,80,81,82,83]. In summary, this simple and cost-effective setup has the potential to be applied in a wide variety of photo-induced reactions providing evidence at the molecular scale.
Data availability
The data associated with this manuscript is available from the corresponding author upon reasonable request.
References
Zhang, W., & Cao, R. (2022). Water oxidation with polymeric photocatalysts. Chemical Reviews, 122(6), 5408–5410. https://doi.org/10.1021/acs.chemrev.2c00175
Juliá, F., Constantin, T., & Leonori, D. (2022). Applications of halogen-atom transfer (XAT) for the generation of carbon radicals in synthetic photochemistry and photocatalysis. Chemical Reviews, 122(2), 2292–2352. https://doi.org/10.1021/acs.chemrev.1c00558
Capaldo, L., Ravelli, D., & Fagnoni, M. (2022). Direct photocatalyzed hydrogen atom transfer (HAT) for aliphatic C-H bonds elaboration. Chemical Reviews, 122(2), 1875–1924. https://doi.org/10.1021/acs.chemrev.1c00263
Pitre, S. P., & Overman, L. E. (2022). Strategic use of visible-light photoredox catalysis in natural product synthesis. Chemical Reviews, 122(2), 1717–1751. https://doi.org/10.1021/acs.chemrev.1c00247
Chang, L., An, Q., Duan, L., Feng, K., & Zuo, Z. (2022). Alkoxy radicals see the light: new paradigms of photochemical synthesis. Chemical Reviews, 122(2), 2429–2486. https://doi.org/10.1021/acs.chemrev.1c00256
Großkopf, J., Kratz, T., Rigotti, T., & Bach, T. (2022). Enantioselective photochemical reactions enabled by triplet energy transfer. Chemical Reviews. https://doi.org/10.1021/acs.chemrev.1c00272
Genzink, M. J., Kidd, J. B., Swords, W. B., & Yoon, T. P. (2022). Chiral photocatalyst structures in asymmetric photochemical synthesis. Chemical Reviews, 122(2), 1654–1716. https://doi.org/10.1021/acs.chemrev.1c00467
Cheung, K. P. S., Sarkar, S., & Gevorgyan, V. (2022). Visible light-induced transition metal catalysis. Chemical Reviews, 122(2), 1543–1625. https://doi.org/10.1021/acs.chemrev.1c00403
Wu, C., Corrigan, N., Lim, C. H., Liu, W., Miyake, G., & Boyer, C. (2022). Rational design of photocatalysts for controlled polymerization: effect of structures on photocatalytic activities. Chemical Reviews, 122(6), 5476–5518. https://doi.org/10.1021/acs.chemrev.1c00409
Gao, Y., Nie, W., Wang, X., Fan, F., & Li, C. (2020). Advanced space—And time-resolved techniques for photocatalyst studies. Chemical Communications, 56(7), 1007–1021. https://doi.org/10.1039/c9cc07128h
Johnston, L. J., & Scaiano, J. C. (1989). Time-resolved studies of biradical reactions in solution. Chemical Reviews, 89(3), 521–547. https://doi.org/10.1021/cr00093a004
Chergui, M., & Collet, E. (2017). Photoinduced structural dynamics of molecular systems mapped by time-resolved X-ray methods. Chemical Reviews, 117(16), 11025–11065. https://doi.org/10.1021/acs.chemrev.6b00831
Tamai, N., & Miyasaka, H. (2000). Ultrafast dynamics of photochromic systems. Chemical Reviews, 100(5), 1875–1890. https://doi.org/10.1021/cr9800816
Biswas, S., Kim, J. W., Zhang, X., & Scholes, G. D. (2022). Coherent two-dimensional and broadband electronic spectroscopies. Chemical Reviews, 122(3), 4257–4321. https://doi.org/10.1021/acs.chemrev.1c00623
Soorkia, S., Jouvet, C., & Grégoire, G. (2020). UV photoinduced dynamics of conformer-resolved aromatic peptides. Chemical Reviews, 120(7), 3296–3327. https://doi.org/10.1021/acs.chemrev.9b00316
Fujii, K., Nakano, H., Sato, H., & Kimura, Y. (2021). Experimental observation of the unique solvation process along multiple solvation coordinates of photodissociated products. Physical Chemistry Chemical Physics, 23(8), 4569–4579. https://doi.org/10.1039/d0cp06588a
Wang, Y., & Wöll, C. (2017). IR spectroscopic investigations of chemical and photochemical reactions on metal oxides: Bridging the materials gap. Chemical Society Reviews, 46(7), 1875–1932. https://doi.org/10.1039/c6cs00914j
Kraack, J. P., & Hamm, P. (2017). Surface-sensitive and surface-specific ultrafast two-dimensional vibrational spectroscopy. Chemical Reviews, 117(16), 10623–10664. https://doi.org/10.1021/acs.chemrev.6b00437
Schoonover, J. R., & Strouse, G. F. (1998). Time-resolved vibrational spectroscopy of electronically excited inorganic complexes in solution. Chemical Reviews, 98(4), 1335–1355. https://doi.org/10.1021/cr950273q
Kuramochi, H., & Tahara, T. (2021). Tracking ultrafast structural dynamics by time-domain raman spectroscopy. Journal of the American Chemical Society, 143(26), 9699–9717. https://doi.org/10.1021/jacs.1c02545
Buhrke, D., & Hildebrandt, P. (2020). Probing structure and reaction dynamics of proteins using time-resolved resonance raman spectroscopy. Chemical Reviews, 120(7), 3577–3630. https://doi.org/10.1021/acs.chemrev.9b00429
Nitschke, P., Lokesh, N., & Gschwind, R. M. (2019). Combination of illumination and high resolution NMR spectroscopy: Key features and practical aspects, photochemical applications, and new concepts. Progress in Nuclear Magnetic Resonance Spectroscopy, 114–115, 86–134. https://doi.org/10.1016/j.pnmrs.2019.06.001
Paululat, T., Rabe, M., & Berdnikova, D. V. (2021). Modification of an NMR probe for monitoring of photoreactions. Journal of Magnetic Resonance, 327, 106990. https://doi.org/10.1016/j.jmr.2021.106990
Ji, Y., DiRocco, D. A., Hong, C. M., Wismer, M. K., & Reibarkh, M. (2018). Facile quantum yield determination via NMR actinometry. Organic Letters, 20(8), 2156–2159. https://doi.org/10.1021/acs.orglett.8b00391
Perakis, F., Marco, L. D., Shalit, A., Tang, F., Kann, Z. R., Kühne, T. D., Torre, R., Bonn, M., & Nagata, Y. (2016). Vibrational spectroscopy and dynamics of water. Chemical Reviews, 116(13), 7590–7607. https://doi.org/10.1021/acs.chemrev.5b00640
Nihonyanagi, S., Yamaguchi, S., & Tahara, T. (2017). Ultrafast dynamics at water interfaces studied by vibrational sum frequency generation spectroscopy. Chemical Reviews, 117(16), 10665–10693. https://doi.org/10.1021/acs.chemrev.6b00728
Nibbering, E. T. J., & Elsaesser, T. (2004). Ultrafast vibrational dynamics of hydrogen bonds in the condensed phase. Chemical Reviews, 104(4), 1887–1914. https://doi.org/10.1021/cr020694p
Vlček, A., Farrell, I. R., Liard, D. J., Matousek, P., Towrie, M., Parker, A. W., Grills, D. C., & George, M. W. (2002). Early photochemical dynamics of organometallic compounds studied by ultrafast time-resolved spectroscopic techniques. Dalton Transactions, 5, 701–712. https://doi.org/10.1039/b106249m
Pattengale, B., Ostresh, S., Schmuttenmaer, C. A., & Neu, J. (2022). Interrogating light-initiated dynamics in metal-organic frameworks with time-resolved spectroscopy. Chemical Reviews, 122(1), 132–166. https://doi.org/10.1021/acs.chemrev.1c00528
Mezzetti, A., Schnee, J., Lapini, A., & Di Donato, M. (2022). Time-resolved infrared absorption spectroscopy applied to photoinduced reactions: How and why. Photochemical and Photobiological Sciences, 21(4), 557–584. https://doi.org/10.1007/s43630-022-00180-9
Nibbering, E. T. J., Fidder, H., & Pines, E. (2005). Ultrafast chemistry: Using time-resolved vibrational spectroscopy for interrogation of structural dynamics. Annual Review of Physical Chemistry, 56(1), 337–367. https://doi.org/10.1146/annurev.physchem.56.092503.141314
Phelps, R., & Orr-Ewing, A. J. (2022). Direct observation of the dynamics of ylide solvation by hydrogen-bond donors using time-resolved infrared spectroscopy. Journal of the American Chemical Society, 144(21), 9330–9343. https://doi.org/10.1021/jacs.2c01208
Zhang, J., Grills, D. C., Huang, K. W., Fujita, E., & Bullock, R. M. (2005). Carbon-to-metal hydrogen atom transfer: Direct observation using time-resolved infrared spectroscopy. Journal of the American Chemical Society, 127(45), 15684–15685. https://doi.org/10.1021/ja0555724
Ritter, E., Puskar, L., Bartl, F. J., Aziz, E. F., Hegemann, P., & Schade, U. (2015). Time-resolved infrared spectroscopic techniques as applied to channelrhodopsin. Frontiers in Molecular Biosciences, 2(JUL), 1–7. https://doi.org/10.3389/fmolb.2015.00038
Yoneda, Y., Arsenault, E. A., Yang, S. J., Orcutt, K., Iwai, M., & Fleming, G. R. (2022). The initial charge separation step in oxygenic photosynthesis. Nature Communications, 13(1), 1–9. https://doi.org/10.1038/s41467-022-29983-1
Lewis-Borrell, L., Sneha, M., Clark, I. P., Fasano, V., Noble, A., Aggarwal, V. K., & Orr-Ewing, A. J. (2021). Direct observation of reactive intermediates by time-resolved spectroscopy unravels the mechanism of a radical-induced 1,2-metalate rearrangement. Journal of the American Chemical Society, 143(41), 17191–17199. https://doi.org/10.1021/jacs.1c07964
Uhmann, W., Becker, A., Taran, C., & Siebert, F. (1991). Time-resolved FT-IR absorption spectroscopy using a step-scan interferometer. Applied Spectroscopy, 45(3), 390–397. https://doi.org/10.1366/0003702914337128
Mezzetti, A., & Spezia, R. (2008). Time-resolved step scan FTIR spectroscopy and DFT investigation on triplet formation in peridinin-chlorophyll-a-protein from Amphidinium carterae at low temperature. Spectroscopy, 22(4), 235–250. https://doi.org/10.3233/SPE-2008-0355
Griffiths, P. R., Hirsche, B. L., & Manning, C. J. (1999). Ultra-rapid-scanning Fourier transform infrared spectrometry. Vibrational Spectroscopy, 19(1), 165–176. https://doi.org/10.1016/S0924-2031(99)00006-5
Barth, A., Corrie, J. E. T., Gradwell, M. J., Maeda, Y., Mäntele, W., Meier, T., & Trentham, D. R. (1997). Time-resolved infrared spectroscopy of intermediates and products from photolysis of 1-(2-nitrophenyl)ethyl phosphates: Reaction of the 2- nitrosoacetophenone byproduct with thiols. Journal of the American Chemical Society, 119(18), 4149–4159. https://doi.org/10.1021/ja964430u
Süss, B., Ringleb, F., & Heberle, J. (2016). New ultrarapid-scanning interferometer for FT-IR spectroscopy with microsecond time-resolution. Review of Scientific Instruments. https://doi.org/10.1063/1.4953658
Kazlauskas, R. J., & Wrighton, M. S. (1982). Application of rapid-scan Fourier transform infrared spectroscopy to characterize the monodentate intermediate in the photochemical formation of tetracarbonyl(4,4′-dialkyl-2,2′-bipyridine)metal from hexacarbonylmetal. Journal of the American Chemical Society, 104(21), 5784–5786. https://doi.org/10.1021/ja00385a039
Mezzetti, A., Alexandre, M., Thurotte, A., Wilson, A., Gwizdala, M., & Kirilovsky, D. (2019). Two-step structural changes in orange carotenoid protein photoactivation revealed by time-resolved fourier transform infrared spectroscopy. Journal of Physical Chemistry B, 123(15), 3259–3266. https://doi.org/10.1021/acs.jpcb.9b01242
Ward, C. P., Bowen, J. C., Freeman, D. H., & Sharpless, C. M. (2021). Rapid and reproducible characterization of the wavelength dependence of aquatic photochemical reactions using light-emitting diodes. Environmental Science and Technology Letters, 8(5), 437–442. https://doi.org/10.1021/acs.estlett.1c00172
Marzo, L., Pagire, S. K., Reiser, O., & König, B. (2018). Visible-light photocatalysis: does it make a difference in organic synthesis? Angewandte Chemie-International Edition, 57(32), 10034–10072. https://doi.org/10.1002/anie.201709766
Chen, J. R., Hu, X. Q., Lu, L. Q., & Xiao, W. J. (2016). Exploration of visible-light photocatalysis in heterocycle synthesis and functionalization: reaction design and beyond. Accounts of Chemical Research, 49(9), 1911–1923. https://doi.org/10.1021/acs.accounts.6b00254
Larkin, P. J. (2011). IR and Raman spectroscopy, principles and spectral interpretation (1st ed). Elsevier. https://doi.org/10.3390/rel9100297
Schrader, B. (1995). Infrared and Raman spectroscopy. Springer. https://doi.org/10.1016/0924-2031(00)00065-5
Klán, P., & Wirz, J. (2009). Photochemistry of organic compounds, from concepts to practice. Wiley-Blackwell. https://doi.org/10.1021/cr60290a001
Kuhn, H. J., Braslavsky, S. E., & Schmidt, R. (2004). Chemical actinometry (IUPAC technical report). Pure and Applied Chemistry, 76(12), 2105–2146. https://doi.org/10.1351/pac200476122105
Brook, A. G., Nyburg, S. C., Abdesaken, F., Gutekunst, B., Gutekunst, G., Kallury, R. K. M. R., Poon, Y. C., Chang, Y., & Wong-ng, W. (1982). Stable solid silaethylenes. Journal of the American Chemical Society, 104(21), 5667–5672.
Page, A. C. S., Scholz, S. O., Keenan, K. N., Spradlin, J. N., Belcher, B. P., Brittain, S. M., Tallarico, J. A., McKenna, J. M., Schirle, M., Nomura, D. K., & Toste, F. D. (2022). Photo-Brook rearrangement of acyl silanes as a strategy for photoaffinity probe design. Chemical Science, 13(13), 3851–3856. https://doi.org/10.1039/d2sc00426g
Eibel, A., Fast, D. E., & Gescheidt, G. (2018). Choosing the ideal photoinitiator for free radical photopolymerizations: Predictions based on simulations using established data. Polymer Chemistry, 9(41), 5107–5115. https://doi.org/10.1039/c8py01195h
Miranda, M. A., & Garcia, H. (1994). 2,4,6-Triphenylpyrylium tetrafluoroborate as an electron-transfer photosensitizer. Chemical Reviews, 94, 1063–1089.
Akaba, R., Sakuragi, H., & Tokumaru, K. (1991). Triphenylpyrylium-salt-sensitized electron transfer oxygenat ion of adamantylideneadamantane. Product, fluorescence quenching, and laser flash photolysis studies. Journal of Chemical Society Perkin Transactions, 1(2), 291–297.
Romero, N. A., & Nicewicz, D. A. (2016). Organic photoredox catalysis. Chemical Reviews, 116(17), 10075–10166. https://doi.org/10.1021/acs.chemrev.6b00057
Ramamurthy, P., Parret, S., Morlet-Savary, F., & Fouassier, J. P. (1994). Spin-orbit-coupling-induced triplet formation of triphenylpyrylium ion: A flash photolysis study. Journal of Photochemistry and Photobiology, A: Chemistry, 83(3), 205–209. https://doi.org/10.1016/1010-6030(94)03826-0
Wintgens, V., Pouliquen, J., Kossanyi, J., Williams, J. L. R., & Doty, J. C. (1985). Emission of substituted pyrylium and thiapyrylium salts: Phosphorescence and delayed fluorescence emission in polymeric matrices. Polymer Photochemistry, 6(1), 1–20. https://doi.org/10.1016/0144-2880(85)90002-8
Akaba, R., Ohshima, K., Kawai, Y., Obuchi, Y., Negishi, A., Sakuragi, H., & Tokumaru, K. (1991). Triplet-mediated electron transfer oxygenation of stilbene derivatives with 2,4,6-triphenylpyrylium tetrafluoroborate. Tetrahedron Letters, 32(1), 109–112. https://doi.org/10.1016/S0040-4039(00)71231-7
Balaban, A. T., Mateescu, G. D., & Elian, M. (1962). Infra-red absorption spectra of pyrylium salts. Tetrahedron, 18(10), 1083–1094. https://doi.org/10.1016/S0040-4020(01)99274-9
Akaba, R., Aihara, S., Sakuragi, H., & Tokumaru, K. (1987). Triphenylpyrylium salt as sensitizer for electron transfer oxygenation not involving superoxide anion. Journal of the Chemical Society, Chemical Communications, 16, 1262–1263.
Chan, A. Y., Perry, I. B., Bissonnette, N. B., Buksh, B. F., Edwards, G. A., Frye, L. I., Garry, O. L., Lavagnino, M. N., Li, B. X., Liang, Y., Mao, E., Millet, A., Oakley, J. V., Reed, N. L., Sakai, H. A., Seath, C. P., & MacMillan, D. W. C. (2022). Metallaphotoredox: The merger of photoredox and transition metal catalysis. Chemical Reviews, 122(2), 1485–1542. https://doi.org/10.1021/acs.chemrev.1c00383
Narayanam, J. M. R., & Stephenson, C. R. J. (2011). Visible light photoredox catalysis: Applications in organic synthesis. Chemical Society Reviews, 40(1), 102–113. https://doi.org/10.1039/b913880n
Hopkinson, M. N., Tlahuext-Aca, A., & Glorius, F. (2016). Merging visible light photoredox and gold catalysis. Accounts of Chemical Research, 49(10), 2261–2272. https://doi.org/10.1021/acs.accounts.6b00351
Kalyanasundaram, K. (1982). Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coordination Chemistry Reviews, 46(C), 159–244. https://doi.org/10.1016/0010-8545(82)85003-0
Willner, I., Tsfania, T., & Eichen, Y. (1990). Photocatalyzed and electrocatalyzed reduction of vic-dibromides and activated ketones using ruthenium(I) tris(bipyridine) as electron-transfer mediator. Journal of Organic Chemistry, 55(9), 2656–2662. https://doi.org/10.1021/jo00296a023
Noda, I., & Ozaki, Y. (2005). Two-dimensional correlation spectroscopy—Applications in vibrational and optical spectroscopy. Wiley. https://doi.org/10.1002/0470012404
Czarnecki, M. A. (2000). Two-dimensional correlation spectroscopy: Effect of band position, width, and intensity changes on correlation intensities. Applied Spectroscopy, 54(7), 986–993. https://doi.org/10.1366/0003702001950463
Rutsch, W., Dietliker, K., Leppard, D., Köhler, M., Misev, L., Kolczak, U., & Rist, G. (1996). Recent developments in photoinitiators. Progress in Organic Coatings, 27(1–4), 227–239. https://doi.org/10.1016/0300-9440(95)00539-0
Yagci, Y., Jockusch, S., & Turro, N. J. (2010). Photoinitiated polymerization: Advances, challenges, and opportunities. Macromolecules, 43(15), 6245–6260. https://doi.org/10.1021/ma1007545
Jockusch, S., & Turro, N. J. (1998). Phosphinoyl radicals: Structure and reactivity. A laser flash photolysis and time-resolved ESR investigation. Journal of the American Chemical Society, 120(45), 11773–11777. https://doi.org/10.1021/ja982463z
Eibel, A., Schmallegger, M., Zalibera, M., Huber, A., Bürkl, Y., Grützmacher, H., & Gescheidt, G. (2017). Extending the scope of bis(acyl)phosphane oxides: Additional derivatives. European Journal of Inorganic Chemistry, 2017(18), 2469–2478. https://doi.org/10.1002/ejic.201700140
Fast, D. E., Zalibera, M., Lauer, A., Eibel, A., Schweigert, C., Kelterer, A. M., Spichty, M., Neshchadin, D., Voll, D., Ernst, H., Liang, Y., Dietliker, K., Unterreiner, A. N., Barner-Kowollik, C., Grützmacher, H., & Gescheidt, G. (2016). Bis(mesitoyl)phosphinic acid: Photo-triggered release of metaphosphorous acid in solution. Chemical Communications, 52(64), 9917–9920. https://doi.org/10.1039/c6cc05219c
Thomas, L. C., & Chittenden, R. A. (1965). Characteristic infra-red absorption frequencies of organophosphorus compounds-V. Phosphorus-carbon bonds. Spectrochimica Acta, 21(11), 1905–1914. https://doi.org/10.1016/0371-1951(65)80238-7
Thomas, L. C., & Chittenden, R. A. (1964). Characteristic infrared absorption frequencies of organophosphorus compounds-II. PO(X) bonds. Spectrochimica Acta, 20(3), 489–502. https://doi.org/10.1016/0371-1951(64)80044-8
Joung, J. F., Han, M., Hwang, J., Jeong, M., Choi, D. H., & Park, S. (2021). Deep learning optical spectroscopy based on experimental database: potential applications to molecular design. JACS Au, 1(4), 427–438. https://doi.org/10.1021/jacsau.1c00035
Gastegger, M., Behler, J., & Marquetand, P. (2017). Machine learning molecular dynamics for the simulation of infrared spectra. Chemical Science, 8(10), 6924–6935. https://doi.org/10.1039/c7sc02267k
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O., & Walsh, A. (2018). Machine learning for molecular and materials science. Nature, 559(7715), 547–555. https://doi.org/10.1038/s41586-018-0337-2
Han, R., Ketkaew, R., & Luber, S. (2022). A concise review on recent developments of machine learning for the prediction of vibrational spectra. Journal of Physical Chemistry A, 126(6), 801–812. https://doi.org/10.1021/acs.jpca.1c10417
Meuwly, M. (2022). Atomistic simulations for reactions and vibrational spectroscopy in the era of machine learning—Quo Vadis? Journal of Physical Chemistry B, 126(11), 2155–2167. https://doi.org/10.1021/acs.jpcb.2c00212
Jansen, T. L. C. (2021). Computational spectroscopy of complex systems. Journal of Chemical Physics. https://doi.org/10.1063/5.0064092
Henschel, H., Andersson, A. T., Jespers, W., Mehdi Ghahremanpour, M., & Van Der Spoel, D. (2020). Theoretical infrared spectra: Quantitative similarity measures and force fields. Journal of Chemical Theory and Computation, 16(5), 3307–3315. https://doi.org/10.1021/acs.jctc.0c00126
Kananenka, A. A., Yao, K., Corcelli, S. A., & Skinner, J. L. (2019). Machine learning for vibrational spectroscopic maps. Journal of Chemical Theory and Computation, 15(12), 6850–6858. https://doi.org/10.1021/acs.jctc.9b00698
Acknowledgements
The authors would like to gratefully acknowledge NAWI Graz funding. We thank Univ.-Prof. Dr. Paolo Falcaro (TU Graz) for inspiring and fruitful discussions.
Funding
Open access funding provided by Graz University of Technology.
Author information
Authors and Affiliations
Contributions
The manuscript was written through the contributions of all authors and all authors have read and agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Glotz, G., Püschmann, S., Haas, M. et al. Direct detection of photo-induced reactions by IR: from Brook rearrangement to photo-catalysis. Photochem Photobiol Sci 22, 1683–1693 (2023). https://doi.org/10.1007/s43630-023-00406-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43630-023-00406-4