
Abstract
Flavins are a unique class of compounds that combine the features of singlet oxygen generators and redox-dependent fluorophores. From a broad family of flavin derivatives, deazaalloxazines are significantly underdeveloped from the point of view of photophysical properties. Herein, we report photophysics of 5-deazaalloxazine (1a) in water, acetonitrile, and some other solvents. In particular, triplet excited states of 1a in water and in acetonitrile were investigated using ultraviolet–visible (UV–Vis) transient absorption spectroscopy. The measured triplet lifetimes for 1a were all on the microsecond time scale (≈ 60 μs) in deoxygenated solutions. The quantum yield of S1 → T1 intersystem crossing for 1a in water was 0.43 based on T1 energy transfer from 1a to indicaxanthin (5) acting as acceptor and on comparative actinometric measurements using benzophenone (6). 1a was an efficient photosensitizer for singlet oxygen in aerated solutions, with quantum yields of singlet oxygen in methanol of about 0.76, compared to acetonitrile ~ 0.74, dichloromethane ~ 0.64 and 1,2-dichloroethane ~ 0.54. Significantly lower singlet oxygen quantum yields were obtained in water and deuterated water (ФΔ ~ 0.42 and 0.44, respectively). Human red blood cells (RBC) were used as a cell model to study the antioxidant capacity in vitro and cytotoxic activity of 1a. Fluorescence-lifetime imaging microscopy (FLIM) data were analyzed by fluorescence lifetime parameters and distribution for different parts of the emission spectrum. Comparison of multidimensional fluorescent properties of RBC under physiological-like and oxidative-stress conditions in the presence and absence of 1a suggests its dual activity as probe and singlet-oxygen generator and opens up a pathway for using FLIM to analyze complex intracellular behavior of flavin-like compounds. These new data on structure–property relationship contribute to the body of information required for a rational design of flavin-based tools for future biological and biochemical applications.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
From a broad family of flavin derivatives, flavins (isoalloxazines), 5-deazaflavins (deazaisoalloxazines) and structurally related alloxazines have become attractive candidates for photochemical and biological studies. Flavins combine the features of photooxygenation via type I and/or type II mechanism being also a pH and redox-dependent fluorophores [1,2,3,4] and an increasing body of structure–property relationship information, which makes flavins well poised for fine tuning of their properties at will. We are undertaking this study to understand the relationship between the structure and singlet-oxygen generation and fluorescence properties to build up a necessary background information for rational design of tools based on flavins for biological and biochemical applications in the future. On the other hand, despite the fact that 5-deazaalloxazines have already been prepared and studied, their photophysical properties are still significantly underdeveloped.
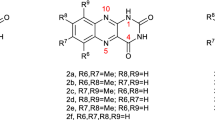
Isoalloxazines and alloxazines and their deaza derivatives differ by only the methine group versus nitrogen in position 5 (see Fig. 1, for the structural difference between 5-deazaalloxazine 1a and alloxazine 2a). Note that this structural feature brings many positive properties in isoalloxazine series and applications in photoredox catalysis. For example, deazaisoalloxazines have higher triplet excited state energies compared to isoalloxazines, which is their advantage in reactions occurring via energy transfer [5]. Deazaisoalloxazines have also more negative reduction potential in comparison to isoalloxazines, and therefore they are more suitable for reductive photocatalysis [6]. Moreover, deazaisoalloxazines have higher photostability. One can expect a similar trend comparing alloxazines to deazaalloxazines.

Structures of 5-deazaalloxazine (1a), alloxazine (2a), lumichrome (2b), 10-ethyl-5-deazaisoalloxazine (3a), 5-deazariboflavin (3b), lumiflavin (4a), riboflavin (4b), indicaxanthin (5), benzophenone (6), and perinaphthenone (7)
Along with characterizing the singlet excited states, a description of triplet excited states is also important for understanding the molecular photophysical properties. Generally, triplet excited states are involved in the generation of reactive oxygen species, including singlet oxygen, and therefore in photooxidation reactions [7,8,9,10,11,12,13,14]. Transient UV–Vis absorption spectra have been recorded upon pulsed laser excitation of various alloxazine derivatives in organic and aqueous solutions [15,16,17,18,19,20]. Triplet excited states have their lifetimes in the microsecond time range, as the T1—S0 transition is spin-prohibited. Theoretical calculations have predicted that the lowest-energy triplet → triplet (T-T) electronic transitions to be of π, π* character in these compounds [9, 21, 22].
The triplet excited states of various alloxazine derivatives, including those of lumichrome (2b) and its derivatives, have been explored in aqueous solutions [20, 23]. In particular, two different species have been detected for lumichrome and 1-methyllumichrome in deoxygenated aqueous solutions at pH ≈ 6 [20, 24]. One of these had a relatively broad absorption band at about 550 nm and 20 µs decay time constant, while the other had a weaker absorption band at about 450 nm and 400 µs decay time. The short-lived species has been identified as the triplet state of the neutral form of lumichrome and 1-methyllumichrome, whereas the long-lived species was the radical anion form of the parent molecule [25, 26]. Note also that the triplet excited state of the lumichrome in oxygen-free solutions was quenched by its ground-state molecules in the concentration range from 1 × 10–5 M to 6 × 10–5 M and higher. The intersystem crossing yields in lumichrome and 1-methyllumichrome were quite high, 0.69 and 0.50, respectively [20]. The presence of the methyl group substituent had little effect on the triplet lifetime 17 µs for lumichrome vs. 18 µs for 1-methyllumichrome in deoxygenated aqueous solutions at pH ≈ 6 [20].
Some flavin derivatives, such as iso- and alloxazines, function as efficient photosensitizers of singlet oxygen [8, 27, 28]. In general, alloxazines were better photosensitizers of singlet oxygen than isoalloxazines. For example, lumichrome (2b, an alloxazine) produced singlet oxygen with the quantum yield ФΔ = 0.85, while lumiflavin (4a, an isoalloxazine) had ФΔ = 0.48 (both values reported in methanol, with similar trends obtained in other solvents) [27]. Note that 5-deazariboflavin (3b) was also an efficient photosensitizer of singlet oxygen, although its ФΔ was lower than that of riboflavin (4b) under the same conditions. Namely, ФΔ values for 3b and 4b were 0.33 and 0.51, respectively, in methanol [9, 16]. However, there is still a lack of information on the singlet oxygen production by deazaalloxazines like 1a and its derivatives.
In the present work, the properties of 1a in its triplet excited state were explored. Transient UV–Vis absorption spectra were recorded in water (pH ~ 6) and acetonitrile. The quantum yield of intersystem crossing (ФISC) was also determined for 1a in aqueous solutions. Finally, singlet oxygen production sensitized by 1a was explored in organic solvents, water, and deuterated water.
In order to demonstrate potential applications of 1a in biological systems, its cytotoxicity (biocompatibility) was evaluated in vitro using human red blood cells (RBC). Hemolysis is the most commonly employed toxicity assessment method [29] and human RBC are used as model cells in biocompatibility studies of chemical compounds with potential biomedical applications [30]. In vivo, RBC are continuously exposed to reactive oxygen species (ROS) in the circulatory system, due to their role as oxygen transporters and to their frequent exposure to different xenobiotics [31]. Therefore, RBC are also an excellent model for the evaluation of the effects of ROS generated from tert-butyl hydroperoxide (TB) in the presence of compounds with potential antioxidant capacity.
2 Experimental
2.1 Materials
1a was synthesized in our group; see ref. [32] for the details of its synthesis and characteristics. Indicaxanthin (5) was used to determine the quantum yield of intersystem crossing for 1a in aqueous solutions; the photophysics of 5 has been recently reported [33]. Benzophenone (6), perinaphthenone (7) and spectroscopic or HPLC-grade solvents (acetonitrile, methanol, dichloromethane, 1,2-dichloroethane and deuterated water) were all purchased from Sigma Aldrich.
The compounds studied were checked for purity and analyzed using TLC, 1H NMR, 13C NMR, HRMS, HPLC, spectral emission excitation matrix, absorption and fluorescence excitation spectra and elemental analysis.
5-Deazaalloxazine, pyrimido[4,5-b]quinoline-2,4(1H,3H)-dione, (1a) (m.p. > 350 °C with decomp, lit. m.p. > 320 °C [34]); 1H NMR (DMSO-d6): δ ppm: 7.55 (m, 1H, H-7); 7.85 (m, 2H, H-8 and H-9); 8.16 (m, 1H, H-6); 9.03 (s, 1H, H-C5); 11.62 (bs, 2H, N(1)-H and N(3)-H). (Atom numbering is shown in Fig. 1). 1H NMR (TFA-d): δ ppm: 8.24 (t, 1H, H-7); 8.22 (d, 1H, H-9); 8.39 (m, 2H, H-8 and H-6); 9.78 (s, 1H, H-C5); 13C NMR (DMSO-d6): 111.6, 125.2, 125.7, 127.3, 130.4, 133.7, 139.6, 150.0, 150.7, 151.2, 162.8; Anal. Calculated for C11H7N3O2:C, 61.97; H, 3.31; N, 19.72. Found: C, 61.68; H, 3.20; N, 19.69; HRMS (ESI +) calculated for C11H7N3O2Na ([M + Na]+): 236.04305, found 236.04310; calculated for C11H8N3O2 ([M + H]+): 214.06110, found 214.06104.
1H NMR spectrum for 1a was recorded in DMSO-d6 and in TFA-d. The 1H NMR spectra were recorded on a Varian Gemini 300 (300 MHz) Spectrometer. 13C NMR spectra were recorded on a JNM-ECZL400S spectrometer (JEOL Ltd., 100.58 MHz for 13C). High-resolution mass spectra were obtained on LTQ Orbitrap Velos (Thermo Scientific), equipped with a linear ion trap and orbitrap analyzer and on Agilent 7200B GC Q-TOF (Agilent Technologies) equipped with a quadrupole and time-of-flight (TOF) analyzer. The internal standard was TMS. 1H NMR spectra of compounds studied were analyzed by comparison to those of similar compounds [34, 35] and by comparison to spectra calculated using the ACD/HNMR predictor.
Purity of the 1a was further confirmed using HPLC analysis (see Fig. 12S) and by emission excitation matrix collected with the A-TEEMs™ (absorbance-transmission and fluorescence excitation-emission matrix), excitation spectra, and synchronous spectra. The EEM data (Fig. 2) indicate that the samples are pure at the spectroscopic and/or fluorescence grade level, according to the detection level of the system used. The purity of the system was also confirmed by the comparison of absorption and fluorescence excitation spectra (Fig. 1SB). The purity was further confirmed by the synchronous scan spectrum, which showed only one maximum overlapping with the edge of the emission spectrum (Fig. 1SB). The data in Fig. 8S also supports the purity, as it shows no differences in the lifetimes recorded at different wavelengths of emission.

Fully corrected Emission Excitation Matrix received with the A-TEEMs™ (absorbance-transmission and fluorescence excitation-emission matrix) method for 1a in acetonitrile (left panel) and in water (right panel). The panels on top and on the right side of each EEM represent emission and fluorescence excitation spectra for selected wavelength of excitation and emission, respectively
2.2 Absorption and emission measurements
UV–Vis absorption spectra were recorded on a Shimadzu UV-2550 spectrophotometer. Emission Excitation Matrix was collected with the A-TEEMs™ (absorbance-transmission and fluorescence excitation-emission matrix) method using a Horiba Scientific Aqualog spectrophotometer. Aqualog integrates its data acquisition functions directly with the Origin software. All EEMs data were corrected for the influence of inner filter effects (IFE) and Rayleigh masking. The measurements in water were performed under unbuffered conditions. However, detailed studies on 1a as a function of pH have been presented in a separate paper, see [36]. The biological part has been conducted using PBS buffer. For general concern we also measured emission spectrum of 1a in water with PBS, using for excitation 405 laser, the same as for FLIM experiments. The results, in particular fluorescence spectrum, were consistent with the experiments in unbuffered water.
2.3 Laser flash photolysis
Transient absorption spectra of 1a in water (pH ~ 6) and acetonitrile were recorded using a nanosecond transient absorption spectrometer, described previously in detail [37], with 0.5 mJ excitation pulses (λexc = 355 nm) to the sample. Transient decays were recorded at individual wavelengths in 10 nm steps from 300 to 700 nm. The 1a solutions in water and acetonitrile were deoxygenated by bubbling argon for 15 min prior to measurements. The experiments were carried out in square (1 cm × 1 cm) fused-silica cells. The same transient absorption spectrometer and procedures were used to record transient absorption spectra of 1a and 5 mixtures, for the intersystem crossing yield measurements.
2.4 Singlet oxygen generation measurements
Characteristic steady-state emission spectra of singlet oxygen were recorded on a Jobin Yvon-Spex Fluorolog 3–221 spectrofluorometer with an H10330B-75 NIR-PMT module, sensitive in the 950–1700 nm NIR range. A xenon lamp with a monochromator was used as an excitation source for the steady-state emission spectra. Phosphorescence decays of 1O2 decays were with a pulsed SpectraLED diode (λexc = 371 nm) at λem = 1270 nm on the same spectrofluorometer. The control experiments, showed that deoxygenation by argon bubbling produced no observable spectra or kinetics.
2.5 Determination of quantum yields of singlet oxygen production (ФΔ)
Quantum yields of singlet oxygen (ФΔ) generated in the presence of 1a in organic solvents (acetonitrile, dichloromethane, and methanol), water and deuterated water were calculated using the results of steady-state measurements. Perinaphthenone (7) in the same solvents was used as a reference, as it has ФΔ = ΦST = 0.95 ± 0.05, independent of the solvent used [38].
Initially, two sets of solutions were prepared as a series of dilutions with optical density maxima in the 0.02 to 0.10 range. These solutions were excited at their respective absorption maxima, containing (i) 1a and (ii) 7 in the same solvent. Then, the characteristic emission spectra of singlet oxygen were recorded for 1a and 7, with the total area under the emission spectrum calculated separately for each of the substances. Finally, ФΔ was calculated according to Eq. (1):
Here, Ф1a is the quantum yield of singlet oxygen production by 1a; Ф7 is the same for the reference sample (7); Grad1a and Grad7 are the slopes of the plots of integrated singlet oxygen intensity versus optical density of 1a and 7, respectively.
3 Hemocompatibility studies of 1a
3.1 Reagents
All reagents were purchased from Avantor Performance Materials Poland SA (Gliwice, Poland). Standard oxidant tert-butyl hydroperoxide (TB) was purchased from Sigma-Aldrich Chemie.
3.2 Human red blood cells
Fresh human red blood cells (RBC) suspensions as concentrates (hematocrit = 65%) were purchased from the blood bank in Poznań, according to the bilateral agreement no. ZP/907/1002/18 between the Blood Bank and Adam Mickiewicz University in Poznań. The RBC suspension was washed three times (3000 rpm, 10 min, + 4 °C) in 7.4 pH buffered phosphate saline (PBS – 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.76 mM KH2PO4) supplemented with 10 mM glucose. After washing, RBC were suspended in PBS buffer supplemented with 10 mM glucose at 1.65 × 109 cells/mL (hematocrit = 15%), stored at + 4 °C and used within 5 h.
3.3 In vitro hemolytic activity assay
The hemolytic potency of 1a was determined by a standard hemolytic assay [39]. Briefly, RBC (1.65 × 108 cells/ml, hematocrit = 1.5%) were incubated in PBS buffer (7.4 pH) supplemented with 10 mM glucose and containing 1a at a concentration equal to 1.0 mg/mL for 1 h and for 24 h at 37 °C in a thermoshaker (BioSan Thermo-Shaker TS-100 C, Biosan, Riga, Latvia). For the positive and negative controls, the RBC were incubated in deionized ice-cold water or in PBS buffer, respectively. Each sample was tested in triplicate and the experiments were repeated with RBC obtained from different blood donors. After incubation, the suspensions RBC were washed (3000 rpm, 10 min, 4 °C) (Sigma 3–30 K Sartorious AG, Gottingen, Germany) and the degree of hemolysis was estimated by measuring the absorbance values (Ab) of the sample supernatant at λ = 540 nm in an EPOLL2000 ECO spectrophotometer (PZ EMCO, Warsaw, Poland). The results were expressed as the percentage (%) of hemolysis, which was determined using the following equation:
where the positive control is Ab of supernatant obtained after incubation of RBC in ice-cold H2O.
The results are presented as the mean values (n = 3) ± standard deviation (SD).
3.4 Fluorescence-lifetime imaging microscopy (FLIM) analysis of RBC
The RBC (1.65 × 108 cells/mL, hematocrit = 1.5%) were incubated in PBS buffer (7.4 pH) supplemented with 10 mM glucose and containing 1a at the final concentration of 1.0 mg/mL for 1 h and for 24 h at 37 °C in a thermoshaker (BioSan Thermo-Shaker TS-100 C, Biosan, Riga, Latvia). To generate oxidative stress conditions within the RBC, cells were incubated with 250 µM tert-butyl hydroperoxide (TB), as the standard inducer of free radicals, for 1.5 h at 37 °C with gentle shaking. Each sample tube was tested in triplicate and the experiments were repeated four times with RBC obtained from different blood donors. After incubation, all RBC suspensions were washed (3000 rpm, 10 min, 4 °C), (Sigma 3–30 K Sartorious AG, Gottingen, Germany). The RBC suspensions (30 µL) were placed on microscope slides and covered with cover slips. A large number of intact RBC were studied in several separate experimental samples using a Abberior combined MINFLUX/STED superresolution laser scanning confocal microscopy system (Abberior Instruments, Germany) equipped with pulsed lasers, 6 high-sensitivity avalanche photodiode (APD) detectors in spectral configuration and dual channel time correlated single photon counting (TCSPC) system (PicoQuant, Germany). The microscope chassis was Olympus IX83 (Olympus, Japan). To avoid unnecessary image aberrations from refractive index mismatch water immersion objective Olympus 60 × NA 1.2 UPLSAPO60XW (Olympus, Japan) was used with optimized coverslip thickness correction collar. Two independent spectral detection channels were used to acquire lifetime information simultaneously. The IRF FWHM was about 200 ps (for complete instrument), the modulation frequency was set to 80 MHz. TCSPC parameters were the following: 25 ns detection range, 1000 bins. Spectral settings for channels were selected based on fluorescent emission spectra acquired at the same microscope with all settings identical, except emission channel). FLIM was applied to study of 1a as a possible redox-sensitive fluorescent probe in intact RBC under normal physiological (PBS buffer, 37 °C) and oxidative stress conditions (250 µM TB in PBS, 37 °C).
The following abbreviations for the differently treated samples will be used from now on:
RBC – control RBC (in PBS buffer),
RBC + TB – RBC incubated with 250 μM TB in PBS (1.5 h, 37 °C),
RBC + 1a – RBC incubated with 1.0 mg/mL 1a (24 h, 37 °C) in PBS buffer,
RBC + 1a + TB – RBC preincubated with 1.0 mg/mL 1a (24 h, 37 °C) in PBS buffer and then incubated with 250 µM TB (1.5 h, 37 °C).
The results and fluorescence lifetime distribution histograms were treated in SymPhoTime64 software (PicoQuant, Germany). IRF (instrument response function) was subtracted before the data fitting.
4 Results and discussion
In order to fully appreciate the implications of our data, it is instructive to place the photophysics of 1a into a wider context. Therefore, the spectroscopic and photophysical properties of 1a in various organic solvents and water are included in Table 1, together with the comparison data of alloxazine itself (2a), based on ref. [28], lumichrome (2b), based on ref. [27], and 10-ethyl-5-deazaisoalloxazine (3a), 5-deazariboflavin (3b), lumiflavin (4a), riboflavin (4b), data based, respectively, on ref. [40]. As this comparison illustrates, that while these isoalloxazines, alloxazines, and their deaza analogs share many structural features, they exhibit divergent spectral and photophysical properties. The difference between the alloxazinic and isoalloxazinic types of structures of molecules lies in the position of their central double bonds. Molecules with the alloxazinic structure have s-cis, whereas molecules with isoalloxazinic structure have an s-trans double-bond configuration. The structurally similar deaza derivatives, where the N-5 of the flavin iso- or alloxazine moiety is replaced by a methine group, are another common way of flavin modifications. These structural changes are mostly responsible for spectral and chemical differences. The longer-wavelength absorption bands (λ1) are particularly sensitive to structural variation (see Fig. 1S). For example, isoalloxazines 4a and 4b, the deazaflavins 3a and 3b, and even alloxazine itself (2a) absorb in the range of 372–443 nm, allowing for convenient visible-light sources for excitation, while the unsubstituted deazaalloxazine 1a absorbs only in the UV region. Shorter-wavelength absorption bands (λ2) follow the same trend, although with a smaller range of variation.
Fluorescence parameters also follow some notable trends. The emission maxima (λF) values for the isoalloxazines (4a and 4b) lie at the longest wavelengths (533 and 517 nm, respectively), while the 5-deaza analogues (3a and 3b) exhibit a hypsochromic shift of 50–60 nm in emission. The lowest wavelength fluorescence emission maxima are from alloxazines (2a and 1a), both of which emit below 435 nm. In terms of fluorescence quantum yields and lifetimes, derivatives 3b, 4a, 4b and 1a have a range of quantum yields (λF) from 0.11 to 0.24, with roughly the same fluorescence lifetimes (~ 4 ns), but shorter compared to lumiflavin (4a), whose fluorescence lifetime is almost double that value (7.7 ns). In contrast, the fluorescence quantum yield of unsubstituted alloxazine (2a) is two orders of magnitude lower (0.009), and its fluorescence lifetime is an order of magnitude lower (0.35 ns).
Figure 2 shows emission excitation matrix for 1a in acetonitrile and in water. Measurements were made on an Aqualog from HORIBA with the ability to simultaneously acquire absorbance, transmittance, and fluorescence of a sample using a technique which combines A-TEEM spectroscopy with simultaneous multichannel CCD detection to provide extremely fast results. The EEMs collected by the instrument were corrected for the inner filter effect. The right-hand side of each EEM matrix shows the fluorescence excitation spectrum, and the top shows the corresponding emission spectrum. The excitation spectrum in each case is virtually the same as the corresponding absorption spectrum for 1a in acetonitrile or in water at pH ~ 6.
According to theoretical calculations of singlet → singlet electronic transitions in alloxazine derivatives, each of the two lowest-energy π,π* transitions is accompanied by a closely-located weak n,π* transition, with the π,π* and n,π* transitions being isoenergetic in some cases [21, 41,42,43,44,45]. On the contrary, the corresponding 5-deaza derivatives (of which 1a is the simplest representative) have the lowest-energy transition of π,π* character, and no accompanying n,π* transition, independent of the substitution pattern, see Fig. 11S [46, 47]. In fact, this feature is one of the most fundamental differences in electronic structure between alloxazines and their 5-deaza counterparts.
To explore the solvent effects, we investigated photophysical properties of 1a in five solvents of different polarity, including protic and aprotic types. The results presented in Table 1 and presented earlier in [46] indicate the importance of intermolecular solute–solvent hydrogen bonding interactions in these compounds. As reported in Ref. [47] the Catalán and Kamlet–Taft parameters show a strong influence of solvent acidity (hydrogen-bond donating ability) on the emission properties of 1a. The absorption properties of 1a in water and acetonitrile did not differ significantly; see Fig. 1S. However, the absorption spectrum in acetonitrile had more structure—there was a double maximum located near 300 nm (296 and 305 nm) in the short-wavelength part of the spectrum, and the long-wavelength maximum at 351 nm was flanked by two shoulders at 340 and 365 nm.
The solvent effect on the position of the emission maxima of 1a is stronger than that on the absorption maxima of the respective compounds. The fluorescence spectrum of 1a revealed slight differences in water vs. acetonitrile. The fluorescence maximum was located at 405 nm in acetonitrile, with a shoulder near 390 nm, while in water the fluorescence maximum was shifted towards the red, with a single peak at 423 nm. Thus, the Stokes shift was slightly higher in water (~ 4600 cm−1) compared to acetonitrile (~ 3800 cm−1). The fluorescence maxima in other solvents fell within the range delimited by those obtained in acetonitrile and water (Table 1). The fluorescence quantum yield had the highest values in methanol and water, while the lowest value was observed in 1,2-dichloroethane. The fluorescence lifetimes were all within the nanosecond timescale, from 2.5 ns in 1,2-dichloroethane to 9.2 ns in water. The fluorescence quantum yields indicate that excited state deactivation for 1a proceeds mainly by non-radiative processes, such as internal conversion to the ground state and intersystem crossing to the triplet states, as well as chemical reactions. The increase of fluorescence of the 1a in protic solvents can be explained by stabilization of the S1 excited state by hydrogen bonding. This could simultaneously reduce the S1 to T1 radiationless intersystem crossing (ISC) rates. In water at pH 7.4 some of the contribution from the anion of isoalloxazine type of structure of 1a can also be noticed [36], leading to stronger emission at longer wavelengths and larger Stokes shift in water comparing to 1a in acetonitrile.
Assuming that ФΔ is expected to be smaller than or equal to the quantum yield of intersystem crossing \({\Phi }_{\mathrm{ISC}}\) in the system under study, most of the deactivation is generally expected to go by intersystem conversion to populate the triplet state, as the singlet oxygen quantum yields indicate. In that context, it is interesting to consider ФF + ФΔ, which for 1a in acetonitrile is equal to 0.92 and in methanol is equal to 1, indicating that intersystem crossing to the triplet state and fluorescence are dominant processes in the relaxation of the singlet excited state. Lower values of ФF + ФΔ were obtained in dichloromethane and 1,2-dichloroethane, but this is largely due to the lower values of ФF (0.03 in dichloromethane and 0.01 in 1,2-dichloroethane), as the values of ФΔ remained relatively high (0.64 in dichloromethane and 0.54 in 1,2-dichloroethane). This points to a larger role of non-radiative internal conversion from the singlet excited state in dichloromethane and 1,2-dichloroethane. The magnitude of ФΔ is much lower (0.43) in water, but the ФF value of 0.21 indicates an important role for non-radiative channel S1 → S0.
4.1 Transient absorption spectra of 1a in water (pH ~ 6) and acetonitrile
Compound 1a upon laser excitation at 355 nm produced transient species in water (pH ~ 6) and in acetonitrile, decaying on the microsecond timescale, as shown in Fig. 3. The experimental transient absorption spectra of 1a showed negative absorbance changes appearing around 360 nm as a result of the ground-state depletion, independent of the solvent. At the same time, positive absorbance changes appeared around 320 and 450 nm. According to previous results drawn from the acid–base equilibria of 1a at different pH, the neutral form of 1a was present exclusively at pH ~ 6, without any other ionic or tautomeric forms, both in the ground and in the singlet excited state [36].

Experimental transient absorption spectra of 1a (A) in water (pH ~ 6) and (B) in acetonitrile, λexc = 355 nm, 0.5 mJ /pulse, argon-flushed solutions
Global analysis of the transient absorption data for 1a in the 300—700 nm spectral range showed the presence of characteristic time-components with decay times of 4.2 µs and 68 µs in water at pH ~ 6, and 12 µs and 59 µs in acetonitrile. The decay-associated spectra are shown in Fig. 2S; Fig. 3S additionally shows the transient absorption kinetics of 1a at λ = 460 nm in the same solvent systems. The transient decay was well described by a bi-exponential function in both solvents, with time constants of 4.2 µs and 53.4 µs in water at pH ~ 6, and 12.5 µs and 60.8 µs in acetonitrile. It is proposed that the longer lifetime belongs to the triplet excited 1a in all of the solvents used, whereas the shorter lifetime is attributed to triplet–triplet annihilation. An experiment with different laser energies was used to explore this hypothesis. An increase in the laser pulse energy resulted in the relative amplitude enhancement of the shorter-lived component, with no effect on the longer-lived component. Thus, Fig. 3 shows the results obtained for a lower laser pulse energy of 0.5 mJ, in order to reduce T-T annihilation while still maintaining a sufficiently high signal-to-noise ratio.
Steady-state absorption spectra were recorded for fresh and photolyzed solutions to verify the stability of 1a in water (pH ~ 6) and acetonitrile solutions upon laser excitation (spectra not shown). No changes were noted in the UV–Vis absorption spectra before and after laser flash photolysis experiments, indicating that 1a was quite stable under the experimental conditions, with no novel persistent photoproducts formed.
4.2 Quantum yield of intersystem crossing (\({\Phi }_{\mathrm{ISC}}\)) for 1a in aqueous solutions
The quantum yield of intersystem crossing (\({\Phi }_{\mathrm{ISC}}\)) for 1a in aqueous solutions can be determined in two types of experiments: (1) transfer of the energy of the T1 state to a reference dye acting as acceptor and (2) comparative actinometric measurements [50].
The spectral properties of 1a and indicaxanthin (5) in water, and the excitation wavelength used (355 nm), were perfectly suited to the donor-1a-acceptor-5 paradigm (see Fig. 4S). As a result of irradiation at 355 nm, an exclusive excitation of 1a was achieved. Figure 4 shows transient absorption spectra of a mixture of 1a and 5 in water (pH ~ 6). 5 is known to produce no triplet excited state through ISC (S1 → T1) upon direct laser excitation at 355 nm [33]. However, when energy transfer was possible between 1a and 5, the value of the molar absorption coefficient of 1a in its triplet excited state was determined (\({\varepsilon }_{T,5DAll}\) = 12.900 M−1 cm−1 at λ = 460 nm), using the extinction coefficient of 5 \({\varepsilon }_{T,IND}\) = 28.000 M−1 cm−1 at λ = 530 nm in water as a reference [33]. Note that the \({\varepsilon }_{T}\) value of 1a is quite close that of the alloxazine derivatives [8].

Transient absorption spectra of the 1a and 5 mixture in water (pH ~ 6); λexc = 355 nm, 0.5 mJ / pulse, argon-flushed solution at different time delays indicated in Figure. At initial delay 0.3 μs 1a in the triplet excited state is observed at 450 nm by T1 → Tn absorption band. At final time at 27 μs, a positive band of 5 in the triplet excited state is observed at 530 nm, as well as the 5 ground state bleaching at about 470 nm. This supports the mechanism of energy transfer from 1a to 5
Next, the quantum yield value of the intersystem crossing quantum yield (\({\Phi }_{\mathrm{ISC}}\)) of 1a was determined in water, using a solution of benzophenone (6) in acetonitrile as a reference, based on Eq. (3):
Here, \({\Phi }_{\mathrm{ISC}}\) is the quantum yield of intersystem crossing of 1a in water, \({\Phi }_{\mathrm{ISC}}^{\mathrm{R}}\) is the same for 6 in acetonitrile, used as a reference with \({\Phi }_{\mathrm{ISC}}^{\mathrm{R}}\)= 1, [50] \({\varepsilon }_{T}^{R}\) is the molar absorption coefficient of 6 in acetonitrile (reference; \({\varepsilon }_{T}^{R}\) = 6500 M−1 cm−1 at λ = 520 nm) [50], \({\varepsilon }_{T}\) is the same for 1a in water (\({\varepsilon }_{T}\)= 12.900 M−1 cm−1), ΔA is the absorbance change of 1a at λ = 460 nm (ΔA = 0.0101), ΔAR is the same for the reference 6 in acetonitrile at λ = 520 nm (ΔAR = 0.0120).
According to Eq. (3), the quantum yield of intersystem crossing of 1a in water (pH ~ 6) was obtained: \({\Phi }_{\mathrm{ISC}}\) = 0.43. This value agreed quite well with the quantum yield of singlet oxygen production for 1a in both water and deuterated water (see the discussion below).
4.3 Deactivation of the singlet excited state of 1a in water
Earlier spectral and photophysical properties of 1a and its 1,3-dimethyl derivative have been explored as a function of pH using different steady-state and time-resolved spectroscopic methods [32, 36], having found that 1a existed in its neutral form at pH ~ 6. This neutral form had its fluorescence quantum yield ФF = 0.21 and fluorescence lifetime τF = 9.2 ns in aqueous solutions. Taking into account currently obtained value of ФISC = 0.43 and using the following definitions:
the following values were calculated: radiative rate constant kr = 2.2 × 107 s−1; sum of non-radiative rate constants ∑knr = 8.6 × 107 s−1; internal conversion rate constant kIC = 3.8 × 107 s−1 and intersystem crossing rate constant kISC = 4.6 × 107 s−1.
It was therefore concluded that non-radiative processes were dominant in the deactivation of the singlet excited states for 1a, as typically happens in organic solvents. When comparing non-radiative processes, the contribution of intersystem crossing (S1 → T1) was slightly higher than that of internal conversion (S1 → S0).
4.4 Singlet oxygen measurements
Isoalloxazine, deazaisoalloxazine, and alloxazine derivatives are interesting photosensitizers of singlet oxygen. However, there was a lack of information on singlet oxygen production by 5-deazaalloxazine 1a and its derivatives. Therefore, photosensitization of singlet oxygen by 1a was explored in organic solvents, including acetonitrile, methanol, dichloromethane, as well as in water and deuterated water. Note that due to the low 1O2 intensity of 1270 nm emission, it was decided to conduct measurements in deuterated water instead regular water, due to the much longer lifetime of singlet oxygen in the latter (69 µs in D2O vs 4.6 µs in H2O). In these experiments, singlet oxygen emission was detected at 1270 nm, which is highly specific to the transition from the singlet excited state to the triplet ground state.
Measurements were performed in air-equilibrated solutions of 1a, maintaining a constant oxygen concentration for a given solvent. It is well known that singlet oxygen may be produced by oxygen quenching of photosensitizer singlet or triplet excited states. When comparing the fluorescence lifetimes of 1a in the nanosecond range with its triplet lifetimes in the microsecond range, it was concluded that 1a should only generate singlet oxygen in solutions through its triplet state, as the probability of quenching of singlet state of 1a should be very low due to its short fluorescence lifetime τF. Table 1 lists the values of the quantum yields of photosensitized production of singlet oxygen ФΔ by 1a in selected organic solvents, as well as in water and deuterated water, with perinaphthenone (7) used as a reference. The values show that 1a is a relatively efficient photosensitizer of singlet oxygen. Comparing data obtained in organic solvents, a higher value of ФΔ was obtained for 1a in methanol (ФΔ ~ 0.76) compared to 1a in acetonitrile (ФΔ ~ 0.74) and in dichloromethane (ФΔ ~ 0.64). A significantly lower value of ФΔ was observed in water and deuterated water (ФΔ ~ 0.42 and 0.44 respectively). Interestingly, these latter values were very close to ФISC = 0.43 of 1a in water. Therefore, it is concluded that the proportion of triplet states quenched by oxygen was 0.96 in water. As a result, the quantum yield of the singlet oxygen for 1a depended on the quantum yield of the formation of T1. The reason for such a lower value of ФΔ (and ISC) in a case of 1a is not clear. Some possibilities to explain the effect of water include proton transfer in the excited singlet state [47], or the energetic order of the 1(n,π*) and 1(π,π*) states [41], or modification of the ISC mechanism based on conical intersection [45].
Note that the concentration of oxygen might be the limiting factor in water, where it equals 0.26 × 10–3 M, while in acetonitrile and methanol it is approximately ten times higher [48]. Thus, apparently all excited triplets were transformed into singlet oxygen; see Table 1. The mechanism of energy transfer from the 1a triplet state to oxygen was also confirmed, as the value of \({k}_{T}^{[{O}_{2}]}\)/kdyf quite close to one-ninth [51], where, \({k}_{T}^{[{O}_{2}]}\) rate constant of quenching of triplet state by oxygen, kdyf rate constants for reaction limited by diffusion.
For comparison, the value ФΔ = 0.36 for 2a in acetonitrile [8] was lower than that for 1a (ФΔ ~ 0.70, this work). Interestingly, lower ФΔ values were also observed in deuterated water compared to organic solvents for monomethyl alloxazine derivatives. Indeed, the ФΔ values for 9-methylalloxazine were 0.74 in acetonitrile, 0.88 in 1,2-dichloroethane, and 0.39 in deuterated water [22].
Table 1 lists also the lifetimes of singlet oxygen generated by 1a in selected organic solvents, as well as in water and deuterated water. The decays recorded with λexc = 371 nm and λem = 1270 nm were all described by a mono-exponential function. The measured lifetimes were typical for singlet oxygen in the solvents used (4.6 µs in H2O, 69 µs in D2O, 11 µs in methanol, and 88 µs in dichloromethane) and are in agreement with the literature reports [51].
Additional measurements were taken to verify the stability of 1a during the time-resolved measurements. Figures 5S and 6S show spectra and singlet oxygen decays generated by 1a and 7 in acetonitrile, with λexc = 371 nm and λem = 1270 nm. Note that the absorbance values at the excitation wavelength remained unchanged during the experimental series for the 1a and 7 solutions. Singlet oxygen emission decays were recorded during 300 s, with the singlet oxygen lifetimes remaining independent of the pre-irradiation time and always equal to 81 µs. Furthermore, singlet oxygen decays were measured at different absorbance values at λexc = 371 nm, ranging from 0.1 to 1.2, with six different samples of 1a in each solvent. Once again, the measured singlet oxygen lifetimes were independent of the 1a concentration, with standard characteristic decay times obtained in each of the solvents used. It was concluded that the SpectraLED diode did not produce any noticeable photodegradation, and that no novel photoproducts were generated. It was also concluded that 1a was a good photosensitizer of singlet oxygen in a broad range of concentrations, independently of the solvent used, and singlet oxygen was never quenched by ground-state 1a. Therefore, 1a would be a convenient 1O2 photosensitizer for studies that require singlet oxygen.
4.5 Hemolytic activity of 1a
In vitro evaluation of hemolytic activity is required before a compound may be deemed biocompatible [29]. The hemolytic activity of 1a was analyzed at a concentration equal to 1.0 mg/mL in standard short-term incubation (1 h at 37 °C) and long-term incubation (24 h at 37 °C). After short-term (1 h) incubation, the degree of hemolysis of control RBC (incubated in PBS buffer) was equal to 2.5 ± 1.0%, vs. 2.8 ± 1.3% obtained for RBC incubated with 1a (1.0 mg/mL). After long-term incubation (24 h), the degree of hemolysis was equal to 3.8 ± 1.6% for control RBC and 4.1 ± 1.7% for RBC incubated with 1a (1.0 mg/mL), respectively. It should be noted that hemolysis values below 5% are assumed to indicate no hemolytic activity (no cell membrane-perturbing activity) of compound studied. Therefore, it can be concluded that 1a at the concentration used (1.0 mg/mL) had no detrimental effects on the molecular structure of the lipid bilayer of the RBC membrane and is a suitable candidate for further evaluation in the cell system.
4.6 Fluorescence lifetime distribution in RBC under physiological and oxidative stress conditions
In search of potential biological applications, 1a was studied in human RBC under physiological-like and oxidative stress conditions. Specifically, FLIM (Fluorescence Lifetime Imaging Microscopy) experiments were performed on RBC after excitation at 405 nm and emission was recorded in two channels (channel 1: 420—550 nm and channel 2: 550—780 nm), enabling finer monitoring of changes in fluorescent properties in cells by separating the emissions from potentially different species. As shown in Fig. 5, RBC treated with 1a at physiological-like conditions (5B – RBC + 1a) are different from untreated cells (5A—RBC). Exposing RBC to oxidative stress by incubation with tert-butyl hydroperoxide further changes the status of RBC, as shown in FLIM images (5C – RBC + 1a + TB and 5D – RBC + TB).

Images of red blood cells (RBC) as obtained by FLIM (λexc = 405 nm emission recorded at two channels; channel 1: 420—550 nm and channel 2: 550—780 nm). RBC incubation condition: A control, in PBS buffer (24 h, 37 °C), B 1.0 mg/mL 1a in PBS (24 h, 37° C), C 1.0 mg/mL 1a (24 h, 37 °C) followed by incubation with 250 μM TB (1.5 h, at 37 °C), D in PBS buffer (24 h, 37 °C) followed by incubation with 250 μM TB (1.5 h, at 37 °C). Representative results are presented. Scale bar 10 μm
It should be noted that the differences are more pronounced in channel 1, which can theoretically record the emission of both endogenous species (such as nicotinamide and flavin derivatives) and 1a itself. The wider emission band in channel 1 of the spectrum collected in cells treated with 1a (RBC + 1a) vs untreated RBC further suggests a potential contribution of 1a to the fluorescence in channel 1 (Fig. 6 red vs green). Channel 2, in turn, covers principally autofluorescence of the cells. These same data also indicate that oxidatively stressed cells, exposed to tert-butyl hydroperoxide (TB), exhibit the same emission spectrum independently of whether they were treated with 1a or not, suggesting that in the case of TB samples, 1a does not substantially contribute to fluorescence emission (Fig. 6 blue and violet).

Fluorescent emission spectra in cells. Excitation was at 405 nm, emission spectra were acquired without gating in the range from 420 to 800 nm with step of 20 nm on the same instrument with the same settings as FLIM acquisitions, except emission windows. RBC incubation condition (as in Fig. 5): green—control, in PBS buffer (24 h, 37 °C), red—1.0 mg/mL 1a in PBS (24 h, 37° C), violet—1.0 mg/mL 1a (24 h, 37 °C) followed by incubation with 250 μM TB (1.5 h, at 37 °C), blue—in PBS buffer (24 h, 37 °C) followed by incubation with 250 μM TB (1.5 h, at 37 °C). Representative results are presented
The differences discussed above were further confirmed by a more detailed analysis of the deconvoluted lifetime distribution (Fig. 7S) and the average values of the fluorescence lifetime parameters (Table 2), all calculated using original FLIM experiments (three images per condition composed of 100 consecutively recorded frames, each image containing more than 100 single cells) by a double-exponential fit after subtraction of the instrument response function.
Lower amplitudes (Table 2) for RBC + 1a vs untreated RBC at physiological-like conditions point at decreasing fluorescence emission intensity in the presence of 1a. It is also accompanied by a lengthening and increase in the amplitude of the longer lifetime component τ2 in channel 1 (rows 1–4 in Table 2: τ2 RBC+1a = 5.72 ns (12%) vs RBC τ2 RBC = 2.64 ns (1.5%)) with a shorter component remaining similar (approx. 0.49 ns). Interestingly, oxidative stress (pre-incubation of cells with TB) leads to a similar value of longer lifetime component τ2 for both RBC + TB and RBC + 1a + TB, placing it in between RBC + 1a and RBC, in a decreasing order: τ2 RBC+1a = 5.72 ns > τ2 RBC+1a+TB = 4.18 ns > τ2 RBC+TB = 4.10 ns > τ2 RBC = 2.64 ns. On the other hand, there was only a small difference in the value of the short components between the RBC treated with or without 1a. In channel 2 (rows 5–8 in Table 2) the longer lifetime components were similarly generally shorter than the analogous parameters in channel 1 but the order was similar and followed the trend τ2 RBC+1a = 3.98 ns > τ2 RBC+1a+TB = 3.16 ns > τ2 RBC+TB = 3.03 ns > τ2 RBC = 1.84 ns. Interestingly, for channel 2, a shorter component seemed to be independent on the oxidative state of the cells, following the order τ1 RBC+1a = 0.43 ns = τ1 RBC+1a+TB = 0.43 ns < τ1 RBC+TB = 0.53 ns = τ1 RBC = 0.54 ns.
Importantly, treatment of cells with TB clearly leads to an increase in the fluorescence intensity, with a larger effect being observed for the cells incubated with 1a (amplitudes in Table 2). In addition, by comparing the FLIM data of the first and last 10 frames of 100 frames series collected for each field of view, it was shown, that the intensity of the fluorescence increases in time of the exposure of the cells to the excitation laser, but this increase is substantially more pronounced for the cells treated with 1a (Table 3). This observation suggests a light-dependent influence of the 1a on the oxidative environment of the cells, in line with its capacity of singlet-oxygen generation. At the same time, fluorescent lifetimes calculated separately for first vs last 10 frames show only modest change proving a better robustness of this parameter in monitoring oxidative state of the cells.
5 Conclusions
On the basis of obtained results, we can state that photolysis of 1a leads to generation of the triplet excited states in high yields both in organic solvents and in water. The 1a triplet lifetime, independently of the solvent, was always on the tens of microsecond time range. Thus, the photophysical behavior of this compound was quite similar to that of 2a and its derivatives.
The characteristic singlet oxygen emission generated by 1a in selected organic solvents and in deuterated water in the NIR spectral range (λem ~ 1270 nm) was recorded using steady-state and time-resolved techniques, showing 1a to be an efficient singlet oxygen photosensitizer. Due to the short fluorescence lifetimes of 1a, its singlet excited state practically did not contribute to singlet oxygen generation.
The ФΔ values in deuterated water were lower than those in organic solvents. The intersystem crossing quantum yield ФISC = 0.43 was obtained for 1a in water, based on experiments involving triplet energy transfer and comparative actinometric measurements. The proposed method of determining ФISC based on the usage of indicaxanthin (5) appeared to be quite useful and could be used to extract ФISC values for a range of alloxazine derivatives in aqueous solutions. Very similar values of singlet oxygen yield generated by 1a (ФΔ = 0.42 and 0.44) were obtained in water and deuterated water, respectively. As far as oxygen quenching of triplet states is concerned, the rate constants for the process are on the order of magnitude 1.2 × 109 M−1 s−1 or higher, therefore, for the triplet state lifetimes exceeding 10 µs most of the triplet states are quenched by oxygen in air saturated solutions, with the respective fraction exceeding 95%.
The detection of characteristic spectra with a maximum at 1270 nm and their emission kinetics directly confirm the existence of singlet oxygen in the case of excitation of 1a in water. The formed triplet was almost entirely quenched, \({P}_{\mathrm{T}}^{\Delta }\) = 0.96, by the molecular oxygen dissolved in water at normal pressure. The intersystem crossing quantum yield of 1a in water was measured as \({\Phi }_{\mathrm{ISC}}\) = 0.43, and the quantum yield of the photosensitized production of singlet oxygen was ФΔ = 0.44, which values are nearly identical. Additionally, in laser flash photolysis experiment, no transient signals from radicals could be detected, while the fluorescence quantum yield was ФF = 0.21.
Regarding the potential biological applications of 1a and similar compounds, the FLIM data were interpreted using the lifetime distribution analysis and comparing the properties of human RBC under oxidative stress conditions in the presence and absence of 1a. Based on the preliminary study using RBC, it was concluded that 1a is a biocompatible, non-hemolytic compound at concentration equal to 1.0 mg/mL in both short-term (1 h) and long-term (24 h) incubation. Differences in fluorescence lifetime parameters obtained from FLIM experiments for pre-incubated RBC with 1a can be used for monitoring the oxidative state of the cells. In addition, analysis of FLIM data from different channels (different emission wavelengths) can provide complementary information to shed light on deciphering the relationship between the cell-derived and probe-driven impact on the cellular environment.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Yeow, J., Kaur, A., Anscomb, M. D., & New, E. J. (2014). A novel flavin derivative reveals the impact of glucose on oxidative stress in adipocytes. Chemical Communications (Cambridge, England), 50, 8181–8184. https://doi.org/10.1039/c4cc03464c
Kaur, A., Haghighatbin, M. A., Hogan, C. F., & New, E. J. (2015). A FRET-based ratiometric redox probe for detecting oxidative stress by confocal microscopy, FLIM and flow cytometry. Chemical Communications (Cambridge, England), 51, 10510–10513. https://doi.org/10.1039/c5cc03394b
Kolanowski, J. L., Kaur, A., & New, E. J. (2016). Selective and reversible approaches toward imaging redox signaling using small-molecule probes. Antioxidants and Redox Signaling, 24, 713–730. https://doi.org/10.1089/ars.2015.6588
Hong, K. I., Lee, S. M., & Jang, W. D. (2018). Flavin-based light-driven fluorescent probe for the detection of antioxidant amino acids. ChemistryOpen, 7, 57–60. https://doi.org/10.1002/open.201700144
Mojr, V., Pitrova, G., Strakova, K., Prukała, D., Brazevic, S., Svobodova, E., Hoskovcova, I., Burdziński, G., Slanina, T., Sikorski, M., & Cibulka, R. (2018). Flavin photocatalysts for visible-light 2+2 cycloadditions: Structure, reactivity and reaction mechanism. ChemCatChem, 10, 849–858. https://doi.org/10.1002/cctc.201701490
Graml, A., Nevesely, T., Jan Kutta, R., Cibulka, R., & König, B. (2020). Deazaflavin reductive photocatalysis involves excited semiquinone radicals. Nature Communications, 11, 3174. https://doi.org/10.1038/s41467-020-16909-y
Cardoso, D. R., Libardi, S. H., & Skibsted, L. H. (2012). Riboflavin as a photosensitizer. Effects on human health and food quality. Food & Function, 3, 487–502. https://doi.org/10.1039/c2fo10246c
Sikorska, E., Sikorski, M., Steer, R. P., Wilkinson, F., & Worrall, D. R. (1998). Efficiency of singlet oxygen generation by alloxazines and isoalloxazines. Journal of the Chemical Society, Faraday Transactions, 94, 2347–2353. https://doi.org/10.1039/a802340i
Sikorska, E., Khmelinskii, I., Komasa, A., Koput, J., Ferreira, L. F. V., Herance, J. R., Bourdelande, J. L., Williams, S. L., Worrall, D. R., Insińska-Rak, M., & Sikorski, M. (2005). Spectroscopy and photophysics of flavin related compounds: Riboflavin and iso-(6,7)-riboflavin. Chemical Physics, 314, 239–247. https://doi.org/10.1016/j.chemphys.2005.03.005
Baier, J., Maisch, T., Maier, M., Engel, E., Landthaler, M., & Baumler, W. (2006). Singlet oxygen generation by UVA light exposure of endogenous photosensitizers. Biophysical Journal, 91, 1452–1459. https://doi.org/10.1529/biophysj.106.082388
Engl, R., Kilger, R., Maier, M., Scherer, K., Abels, C., & Baumler, W. (2002). Singlet oxygen generation by 8-methoxypsoralen in deuterium oxide: Relaxation rate constants and dependence of the generation efficacy on the oxygen partial pressure. The Journal of Physical Chemistry B, 106, 5776–5781. https://doi.org/10.1021/jp013727y
Vakrat-Haglili, Y., Weiner, L., Brumfeld, V., Brandis, A., Salomon, Y., McIlroy, B., Wilson, B. C., Pawlak, A., Rozanowska, M., Sarna, T., & Scherz, A. (2005). The microenvironment effect on the generation of reactive oxygen species by Pd-bacteriopheophorbide. Journal of the American Chemical Society, 127, 6487–6497. https://doi.org/10.1021/ja046210j
Tanielian, C., Schweitzer, C., Mechin, R., & Wolff, C. (2001). Quantum yield of singlet oxygen production by monomeric and aggregated forms of hematoporphyrin derivative. Free Radical Biology & Medicine, 30, 208–212. https://doi.org/10.1016/s0891-5849(00)00460-3
Hanson, K. M., & Simon, J. D. (1998). Epidermal trans-urocanic acid and the UV-A-induced photoaging of the skin. Proceedings of the National academy of Sciences of the United States of America, 95, 10576–10578. https://doi.org/10.1073/pnas.95.18.10576
Lu, C. Y., Han, Z. H., Liu, G. S., Cai, X. C., Chen, Y. L., & Yao, S. (2001). Photophysical and photochemical processes of riboflavin (vitamin B-2) by means of the transient absorption spectra in aqueous solution. Sci. China, B Chem., 44, 39–48. https://doi.org/10.1007/bf02879734
Insińska-Rak, M., Sikorska, E., Bourdelande, J. L., Khmelinskii, I. V., Prukała, W., Dobek, K., Karolczak, J., Machado, I. F., Ferreira, L. F. V., Komasa, A., Worrall, D. R., & Sikorski, M. (2006). Spectroscopy and photophysics of flavin-related compounds: 5-deaza-riboflavin. Journal of Molecular Structure, 783, 184–190. https://doi.org/10.1016/j.molstruc.2005.09.005
Heelis, P. F. (1982). The photophysical and photochemical properties of flavins (isoalloxazines). Chemical Society Reviews, 11, 15–39. https://doi.org/10.1039/cs9821100015
Salzmann, S., Martinez-Junza, V., Zorn, B., Braslavsky, S. E., Mansurova, M., Marian, C. M., & Gärtner, W. (2009). Photophysical properties of structurally and electronically modified flavin derivatives determined by spectroscopy and theoretical calculations. Journal of Physical Chemistry A, 113, 9365–9375. https://doi.org/10.1021/jp905724b
Neiss, C., Saalfrank, P., Parac, M., & Grimme, S. (2003). Quantum chemical calculation of excited states of flavin-related molecules. Journal of Physical Chemistry A, 107, 140–147. https://doi.org/10.1021/jp021671h
Sikorski, M., Sikorska, E., Koziołowa, A., Moreno, R. G., Bourdelande, J. L., Steer, R. P., & Wilkinson, F. (2001). Photophysical properties of lumichromes in water. Journal of Photochemistry and Photobiology A: Biology, 60, 114–119. https://doi.org/10.1016/S1011-1344(01)00134-8
Sikorska, E., Khmelinskii, I. V., Prukała, W., Williams, S. L., Worrall, D. R., Bourdelande, J. L., Bednarek, A., Koput, J., & Sikorski, M. (2004). Spectroscopy and photophysics of 9-methylalloxazine. Experimental and theoretical study. Journal of Molecular Structure, 689, 121–126. https://doi.org/10.1016/j.molstruc.2003.10.28
Sikorska, E., Khmelinskii, I. V., Bourdelande, J. L., Bednarek, A., Williams, S. L., Patel, M., Worrall, D. R., Koput, J., & Sikorski, M. (2004). Spectroscopy and photophysics of mono methyl-substituted alloxazines. Chemical Physics, 301, 95–103. https://doi.org/10.1016/j.chemphys.2004.03.005
Grodowski, M. S., Veyret, B., & Weiss, K. (1977). Photochemistry of flavins.2. Photophysical properties of alloxazines and isoalloxaznies. Photochemistry and Photobiology, 26, 341–352. https://doi.org/10.1111/j.1751-1097.1977.tb07495.x
Encinas, M. V., Bertolotti, S. G., & Previtali, C. M. (2002). The interaction of ground and excited states of lumichrome with aliphatic and aromatic amines in methanol. Helvetica Chimica Acta, 85, 1427–1438. https://doi.org/10.1002/1522-2675(200205)85:5%3c1427::AID-HLCA1427%3e3.0.CO;2-A
Li, H., Jiang, Z. Q., Pan, Y., & Yu, S. Q. (2006). Study on electron transfer of fluorescent probe lumichrome and nucleic acid by laser flash photolysis. Research on Chemical Intermediates, 32, 695–708
Davis, C. A., Erickson, P. R., McNeill, K., & Janssen, E. M. L. (2017). Environmental photochemistry of fenamate NSAIDs and their radical intermediates. Environmental Science. Processes & Impacts, 19, 656–665. https://doi.org/10.1039/c7em00079k
Sikorska, E., Khmelinskii, I. V., Prukała, W., Williams, S. L., Patel, M., Worrall, D. R., Bourdelande, J. L., Koput, J., & Sikorski, M. (2004). Spectroscopy and photophysics of lumiflavins and lumichromes. Journal of Physical Chemistry A, 108, 1501–1508. https://doi.org/10.1021/jp037048u
Sikorski, M., Prukała, D., Insińska-Rak, M., Khmelinskii, I., Worrall, D. R., Williams, S. L., Hernando, J., Bourdelande, J. L., Koput, J., & Sikorska, E. (2008). Spectroscopy and photophysics of dimethyl-substituted alloxazines. Journal of Photochemistry and Photobiology A: Chemistry, 200, 148–160. https://doi.org/10.1016/j.jphotochem.2008.07.006
Greco, I., Molchanova, N., Holmedal, E., Jenssen, H., Hummel, B. D., Watts, J. L., Håkansson, J., Hansen, P. R., & Svenson, J. (2020). Correlation between hemolytic activity, cytotoxicity and systemic in vivo toxicity of synthetic antimicrobial peptides. Sci. Rep. https://doi.org/10.1038/s41598-020-69995-9
Farag, M. R., & Alagawany, M. (2018). Erythrocytes as a biological model for screening of xenobiotics toxicity. Chemico-Biological Interactions, 279, 73–83. https://doi.org/10.1016/j.cbi.2017.11.007
Mohanty, J. G., Nagababu, E., & Rifkind, J. M. (2014). Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Frontiers in Physiology, 5, 84. https://doi.org/10.3389/fphys.2014.00084
Prukała, D., Gierszewski, M., Pędziński, T., & Sikorski, M. (2014). Influence of pH on spectral and photophysical properties of 9-methyl-5-deazaalloxazine and 10-ethyl-5-deaza-isoalloxazine. Journal of Photochemistry and Photobiology A: Chemistry, 275, 12–20. https://doi.org/10.1016/j.jphotochem.2013.10.011
Wendel, M., Szot, D., Starzak, K., Tuwalska, D., Prukała, D., Pędziński, T., Sikorski, M., Wybraniec, S., & Burdziński, G. (2015). Photophysical properties of indicaxanthin in aqueous and alcoholic solutions. Dyes and Pigments, 113, 634–639. https://doi.org/10.1016/j.dyepig.2014.09.036
Taylor, E. C., Jr., & Kalenda, N. W. (1956). The synthesis of pyrimido[4,5-b]quinolines. Journal of the American Chemical Society, 78, 5108–5115. https://doi.org/10.1021/ja01600a079
Kokel, B. (1994). The reaction of N, N-dimethyldichloromethyleniminium chloride (phosgeniminium chloride) with 6-N-arylaminouracils. A new and convenient “one pot” synthesis of l,3-dimethyl-5- dimethylaminopyrimido[4,5-b]quinoline-(1H,3H)-2,4-diones, 1,3-dimethyl-5-chloropyrimido[4,5-b]quinoline-(1H,3H)-2,4-diones and 3-methyl-10-alkyl-5-chloropyrimido[4,5-b]quinoline-(3H,10H)-2,4-diones (3-methyl-10-alkyl-5-chloro-5-deazaflavins). Journal of Heterocyclic Chemistry, 31, 845–855. https://doi.org/10.1002/jhet.5570310427
Prukała, D., Gierszewski, M., Karolczak, J., & Sikorski, M. (2015). Study of photophysical properties of 5-deazaalloxazine and 1,3-dimethyl-5-deazaalloxazine in dependence of pH using different spectral techniques. Physical Chemistry Chemical Physics: PCCP, 17, 18729–18741. https://doi.org/10.1039/c5cp01566a
Burdziński, G., Maciejewski, A., Buntinx, G., Poizat, O., & Lefumeux, C. (2004). Ultrafast quenching of the excited S2 state of benzopyranthione by acetonitrile. Chemical Physics Letters, 384, 332–338. https://doi.org/10.1016/j.cplett.2003.12.029
Schmidt, R., Tanielian, C., Dunsbach, R., & Wolff, C. (1994). Phenalenone, a universal reference compound for the determination of quantum yields of singlet oxygen O2(1Δg) sensitization. Journal of Photochemistry and Photobiology A: Chemistry, 79, 11–17. https://doi.org/10.1016/1010-6030(93)03746-4
Mrówczyńska, L., & Hägerstrand, H. (2009). Platelet-activating factor interaction with the human erythrocyte membrane. Journal of Biochemical Toxicology, 23, 345–348. https://doi.org/10.1002/jbt.20297
Sikorski, M., Khmelinskii, I., & Sikorska, E. (2021). Spectral Properties of Flavins. In R. Cibulka & M. Fraaije (Eds.), Flavin-Based Catalysis (pp. 67–96). Weinheim, Germany: Wiley-VCH.
Salzmann, S., & Marian, C. M. (2009). The photophysics of alloxazine: A quantum chemical investigation in vacuum and solution. Photochemical & Photobiological Sciences, 8, 1655–1666. https://doi.org/10.1039/b9pp00022d
Moyon, N. S., & Mitra, S. (2011). Fluorescence solvatochromism in lumichrome and excited-state tautomerization: a combined experimental and DFT study. Journal of Physical Chemistry A, 115, 2456–2464. https://doi.org/10.1021/jp1102687
Sikorska, E., Szymusiak, H., Khmelinskii, I. V., Koziołowa, A., Spanget-Larsen, J., & Sikorski, M. (2003). Spectroscopy and photophysics of alloxazines studied in their ground and first excited singlet states. Journal of Photochemistry and Photobiology A: Chemistry, 158, 45–53. https://doi.org/10.1016/s1010-6030(03)00104-7
Sikorska, E., Khmelinskii, I. V., Nowacka, G., Koput, J., & Sikorski, M. (2005). Spectroscopy and photophysics of cyanoalloxazines. Theoretical study. Journal of Molecular Structure: THEOCHEM, 722, 51–56. https://doi.org/10.1016/j.theochem.2004.11.058
Chang, X. P., Xie, X. Y., Lin, S. Y., & Cui, G. L. (2016). QM/MM Study on mechanistic photophysics of alloxazine chromophore in aqueous solution. Journal of Physical Chemistry A, 120, 6129–6136. https://doi.org/10.1021/acs.jpca.6b02669
Prukała, D., Taczkowska, M., Gierszewski, M., Pędziński, T., & Sikorski, M. (2014). Spectroscopy and photophysics of monomethyl-substituted derivatives of 5-deazaalloxazine and 10-ethyl-5-deaza-isoalloxazine. Journal of Fluorescence, 24, 505–521. https://doi.org/10.1007/s10895-013-1320-9
Prukała, D., Khmelinskii, I., Koput, J., Gierszewski, M., Pędziński, T., & Sikorski, M. (2014). Photophysics, excited-state double-proton transfer and hydrogen-bonding properties of 5-deazaalloxazines. Photochemistry and Photobiology, 90, 972–988. https://doi.org/10.1111/php.12289
Quaranta, M., Murkovic, M., & Klimant, I. (2013). A new method to measure oxygen solubility in organic solvents through optical oxygen sensing. The Analyst, 138, 6243–6245. https://doi.org/10.1039/c3an36782g
Sato, T., Hamada, Y., Sumikawa, M., Araki, S., & Yamamoto, H. (2014). Solubility of oxygen in organic solvents and calculation of the Hansen solubility parameters of oxygen. Industrial and Engineering Chemistry Research, 53, 19331–19337. https://doi.org/10.1021/ie502386t
Carmichael, I., & Hug, G. L. (1986). Triplet-triplet absorption-spectra of organic-molecules in condensed phases. Journal of Physical and Chemical Reference Data, 15, 1–250. https://doi.org/10.1063/1.555770
Wilkinson, F., Helman, W. P., & Ross, A. B. (1993). Quantum yields for the photosensitized formation of the lowest electronically excited singlet-state of molecular-oxygen in solution. Journal of Physical and Chemical Reference Data, 22, 113–262. https://doi.org/10.1063/1.555934
Acknowledgements
This paper is dedicated to the memory of David R. Worrall. This study was supported by the research grants 2017/27/B/ST4/02494 (OPUS) and NCN CEUS-UNISONO 2020/02/Y/ST4/00042, both from The National Science Centre of Poland (NCN) and by grant 21-14200K from the Czech Science Foundation. MG would like to express his thanks to The National Science Centre of Poland (NCN) for financial support from grant 2013/08/T/ST4/00640 (Etiuda). JLK would like to acknowledge the financial support of the grant no POIR.04.04.00-00-441F/17-00 within HOMING programme of the Foundation for Polish Science co-financed by the European Union under the European Regional Development Fund and financial support from the grant 2018/29/B/ST4/01498 (Opus) for the National Science Centre of Poland (NCN). JLK, DK and VCh would like to acknowledge the financial support of the POIR.04.02.00-00-C004/19-00 project funded under the Smart Growth Operational Programme 2014-2020, co-financed by the European Union through the European Regional Development Fund and financial support from the Polish Ministry of Education and Science (previously MNiSW, decision no DIR/WK/2018/06) for involvement in the joint international project entitled European Infrastructure of Open Screening Platforms of Chemical Biology European Research Infrastructure Consortium (EU-OPENSCREEN ERIC). We are grateful Dr. S. Wybraniec for providing purified indicaxanthin used in actinometric measurements.
Author information
Authors and Affiliations
Contributions
The following co-authors contributed in particular with; MI-R and AG: steady state and time resolved experiments with 1a in different solvents. AG: singlet oxygen measurements, JLK, VCh, DK- FLIM experiments, its analysis and figures related to FLIM, MG steady state and time resolved experiments with 1a GB: laser flash photolysis experiments and its analysis, interpretation and respective figures preparation. ZA: singlet oxygen measurements. ES: conceptualization, methodology, formal analysis, A-TEEMs™ experiments. IK: conceptualization. RC, JLM, LM and MS: supervision, funding acquisition, conceptualization, methodology, formal analysis, investigation, writing original draft. Four lead scientists RC, JLK, LM, and MS supervised respective areas of research: RC – organic chemistry, JLK – FLIM, LM – biochemistry, MS – photochemistry and total coverage of the project. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Insińska-Rak, M., Golczak, A., Gierszewski, M. et al. 5-Deazaalloxazine as photosensitizer of singlet oxygen and potential redox-sensitive agent. Photochem Photobiol Sci 22, 1655–1671 (2023). https://doi.org/10.1007/s43630-023-00401-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43630-023-00401-9