
Abstract
The feasibility of using advanced oxidation processes (AOPs) for abatement of ammonia from livestock buildings was examined in a series of pilot plant experiments. In this study, all the experiments were conducted in a two-step unit containing a dry photolytic reactor (UV185/UV254/O3) and a photochemical scrubber (UV254/H2O2). The unit efficiency was tested for two initial ammonia concentrations (20 and 35 ppmv) and three different air flows (150, 300 and 450 m3·h−1). While the first step removes mainly organic pollutants that are often present together with ammonia in the air and ammonia only partially, the second step removes around 90% of ammonia emissions even at the highest flow rate of 450 m3·h−1. Absorbed ammonia in the aqueous phase can be effectively removed without adjusting the pH (i.e. without the addition of other additives) using UV and ozone. Complete removal of ammonia was achieved after 15 h of irradiation. In order to assess the price efficiency of the suggested technology and to be able to compare it with other methods the figures-of-merit were determined. The price needed for lowering ammonia emission by one order of magnitude is 0.002 € per cubic meter of treated air at the highest flow rate of 450 m3·h−1 and for initial ammonia concentrations of 20 ppmv. These findings demonstrate that AOPs are a promising method for ammonia abatement from livestock buildings which are rarely using any waste air treatment method.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
It is not only greenhouse gas or organic compounds emissions that raise serious concerns. For example, nitrogen containing bases, such as ammonia, have significant impact on the atmosphere. Ammonia emissions are primarily associated with two environmental issues leading to ecosystem disruption: acidification and eutrophication. Gaseous ammonia reacts easily with acid oxides (SO2, NOx) in the atmosphere to form ammonium salts aerosols. These aerosols condensate on surfaces or they are removed from atmosphere by wet deposition to cause acidification of soil and water and eutrophication of aquatic ecosystems—algal and plant growth due to excess nutrients. An imbalance in nutrition cycling leads to a change in ecosystem diversity [1,2,3].
Ammonia exposure is also harmful for human (and animal) health. According to European chemicals agency (ECHA), ammonia is classified in Acute toxicity category 3—H331: Toxic if inhaled and in Skin corrosion category 1B—H314: Causes severe skin burns and eye damage. Long-term Exposure Limit (LTEL) value for ammonia is 14 mg·m−3 a and Short-term Exposure Limit (STEL) value is 36 mg·m−3 [4].
Directive on the reduction of national emissions of certain atmospheric pollutants for EU members came into force in 2016. The Czech Republic is obligated to reduce ammonia annual emissions by 7% from 2020 and 22% from 2030 (compared to the 2005 level) [5]. The levels of ammonia emissions in the Czech Republic were 170, 99 and 85 Gg per annum in 1990, 2005, and 2019, respectively [6]. Values of ammonia emissions in EU decreased throughout all EU countries, except Ireland, Luxembourg, Austria, and Spain between 1990 and 2019 [6]. However, the decrease is still getting slower and therefore it is necessary to find and implement innovative and effective techniques to meet the limits and contribute to Earth´s health. EU member states have to take into account the relevant Ammonia Guidance Document and have to use the best available techniques (BAT) in accordance with Directive 2010/75/EU, which covers also low-emission animal housing systems [5]. The limits for national NH3 emissions are also established and the reporting of annual emission inventories to demonstrate compliance is required. The relevant legislation is very comprehensive and complex. To mention an emission limit example: in intensive rearing of pigs with more than 2 000 places for production pigs (over 30 kg), the NH3 emissions shall not exceed the limit 2.6 kg NH3/animal place/year for fattening pigs [7].
Agriculture was responsible for most (94%) of the ammonia emissions in the EU in 2019 and the manure management represented the main emitter of ammonia in this field [6]. Livestock excreta emit ammonia rapidly to the atmosphere, immediately after deposition and also during decomposition by urease enzyme or microbial activity. Ammonia emissions are often unnaturally increased by overuse of the high-protein feeds—the nitrogen unconsumed in animal gastrointestinal tract is excreted in the urine and the feces [8, 9].
The animal housing outlet air contains a broad mixture of pollutants (ammonia, greenhouse gases, odors, hydrogen sulfide, volatile organic compounds, particulate matter, and bio-aerosols including pathogenic microorganisms) [10]. Polluted outlet air is most often treated by various air filtration systems, especially by air scrubbers, biofilters and dry filters [11, 12]. Chemical air scrubbers usually use the sulfuric acid for showering the waste air and achieve the ammonia removal efficiency of 70–90%; while biological scrubbers (nitrification by ammonia-oxidizing bacteria) remove approximately 70% of ammonia [10, 13, 14]. Biofilters contain a special filling substrate to ensure the environmental and nutritional conditions for microbial growth (wood bark, wood chip, peat, compost, gravel etc.) [10, 15]. Air pollutants are then oxidized by the microbial biofilm growing on the substrate with a very variable degradation efficiency, but often about 50–60% [10, 14, 16]. Dry filters are usually made from polyethylene, polypropylene foil, glass fiber, etc. and their ammonia degradation capacity is negligible, therefore, they are often combined with other techniques [17]. Air filtration systems have a few significant disadvantages, primarily weather-dependent efficiency, high consumption of sulfuric acid in the chemical scrubber or large space demands and clogging of biofilters [15, 18].
Very few articles describing photochemical AOPs for the removal of ammonia have been published. Rockafellow et al., 2012 almost completely reduced ammonia from the gas phase using VUV (vacuum ultraviolet) photolysis (185 nm) in a laboratory scale [19]. Zhu et al. [20] achieved a significant decrease on ammonia content in the aqueous phase using TiO2 photocatalytic oxidation at pH = 10.2 [20]. Deng and Ezyske, 2011 successfully used sulfate radical-AOP for the removal of ammonia in landfill leachate [21].
This work presents the usage of advanced oxidation processes (AOPs) as a possible alternative method for the treatment of air stream containing ammonia to imitate the exhaust of livestock building. AOPs use the production of powerful transitory species—highly reactive radicals (especially hydroxyl radicals), which are able to oxidize wide range of organic and inorganic substances. AOPs are primarily used in waste-air and wastewater treatment, where the hydroxyl radicals can attack harmful pollutant molecules and oxidatively decompose them into simple innocuous compounds (mineralization) [22, 23].
2 Materials and methods
The chemicals used in this study were purchased from commercial companies with these purities: hydrogen peroxide (30%, p.a., Mach chemikálie Ltd.), ammonium hydroxide (25%, p.a., Mach chemikálie Ltd.).
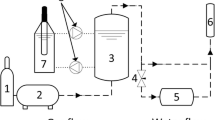
Experiments were carried out in a two-step pilot plant unit. The first step (R1) was a dry photolytic reactor with UV lamps (λmax = 185 and 254 nm) producing ozone. The second step (R2) was a photochemical scrubber with UV lamps (λmax = 254 nm) and solution of hydrogen peroxide. A detailed description of the pilot plant unit is given in the Supplementary Materials.
Experiments were carried out at two different initial ammonia concentration (20 and 35 ppmv) and with three different air flows (150, 300 and 450 m3·h−1). These concentrations were chosen based on the long-term exposure limit, which is 14 mg·m−3 (20 ppmv). The second concentration was chosen between long-term and short-term exposure limit. The determination of the ammonia concentration is described in the Supplementary Materials. Lamps in R1, R2 and the circulation of hydrogen peroxide solution were turned on separately in consecutive steps so the effectiveness of each of technology step could be evaluated.
The degree of conversion was chosen as a determining factor for the degradation of ammonia [Eqs. (1) and (2)].
X stands for the degree of conversion (%), n0 for the initial substance amount of ammonia (mol), n for the substance amount of ammonia (mol), c0 for the initial concentration of ammonia (ppmv) and c for the concentration of ammonia (ppmv).
3 Results and discussion
3.1 Degradation of ammonia in the first step (R1) and in both steps (R1 + R2)
The first step (R1) of the technology consists of dry photolytic reactor where all reactions proceeds only in gas phase. The oxidation of ammonia by hydroxyl radical in gas phase has been investigated in the past [24]. There are several degradations pathways of ammonia in gas phase.
Strong UV irradiation, which is produced by the UV lamps, decomposes oxygen molecules in the air into oxygen atoms [Eq. (3)] [25]. Then the ozone is created from the oxygen atom and the oxygen molecule [Eq. (4)] [26]. Ozone can be photolytically decomposed to form an electronically excited singlet oxygen atom [Eq. (5)] [25]. The ammonia reacts with single oxygen atom O(1D) according to the Eq. (6) [27].
The second degradation pathway of ammonia is the reaction with hydroxyl radicals which can be formed either by reaction of singlet oxygen with water/humidity present in the air resulting in two hydroxyl radicals (Eq. (7)) [28], or by the direct photolysis of water under 185 nm [Eq. (8)] [25]. The reaction of ammonia with HO• provides NH2• and water [Eq. (9)] [28].
The last possible ammonia oxidation pathway is simply by direct photolysis under 185 nm [Eq. (10)] [28,29,30].
Even though, the reaction of ammonia with singlet oxygen [Eq. (6)] is much faster (by 3 orders of magnitude) [27] than the reaction with hydroxyl radicals [Eq. (9)] [28], it most likely does not represent the main oxidation pathway of ammonia in gas phase. There are two reasons for that, first of all formation of singlet oxygen [Eq. (5)] is much slower [25] and second of all there is a competing reaction utilizing singlet oxygen for reaction with water molecule [Eq. (7)] [28]. Therefore, it seems the main oxidation pathway of ammonia proceeds according the Eq. (9). The kinetic constant of this reaction is not the fastest one, but there is no competing reaction that would consume hydroxyl radicals and in addition hydroxyl radicals are formed by several ways [Eqs. (6–8)].
Nonetheless, in spite of everything described above the oxidation of ammonia in gas phase is relatively slow reaction and the conversion of ammonia in the first step of the technology (R1) was around 30% for flow rate of 150 m3·h−1 with initial ammonia concentration of 20 ppmv and even lower for higher flow rates (Fig. 1). Even though, the photolytic step (R1) proved to have low efficiency toward ammonia oxidation its main purpose in this technology is different. The first step (R1) is for pretreatment of the waste air. Waste air contains variety of compounds including organic compounds such as carboxylic acids, saccharides, aldehydes, fats and others. The presence of these compounds depends where does the waste air comes from (biogas stations, composting plants, animal housings). Since the hydroxyl radical is much more likely to attack weaker C–H bond in organic compounds than N–H bond [24] these organic impurities are being oxidized in the first step (R1) and are not consuming hydroxyl radicals in liquid phase in second step (R2). Nonetheless, it is important to mention this study used air without organic pollutants, however, it was proved in our previous studies that the first step (R1) is effective for organic pollutants removal [31,32,33].

Average ammonia conversions after passing first step (R1 ON) and both steps (R1+R2 ON) with initial ammonia concentration a 20 ppmv and b 35 ppmv
The benefit of using AOP technology is that it can lead to complete degradation of ammonia into nitrogen and water which was proofed by the FT-IR, where nitrous oxide and nitrogen dioxide were found only in trace amounts and their sum never exceeded 2.5 ppmv during the experiments. FT-IR library and experimental spectra of nitrous oxide and nitrogen dioxide are included in the Supplementary materials as Fig. S6 and Fig. S7. No traces of nitric oxide were found, probably due to its rapid reaction with many reactive species in the irradiated photolytic reactor [Eqs. (11–15)] [28, 34].
The majority of ammonia is removed from the waste air in the second step (R2) which is connected behind the first step (R1). The conversion of ammonia reaches over 90% for the flow rate of 150 m3·h−1 with initial ammonia concentration of 20 ppmv at the output of the whole technology (Fig. 1). The very high ammonia removal from waste air is mainly caused by the absorption of ammonia to the hydrogen peroxide solution in the second step (R2) which is photochemical wet scrubber. FT-IR library spectra of ammonia as well as comparison of spectra of ammonia at the inlet of the technology and at the outlet of the technology after both steps were turned on are included in Supplementary materials as Fig. S4 and Fig. S5. In order to confirm the ammonia was removed by absorption and not by oxidation a theoretical calculation of absorbed amount of ammonia was conducted and compared with experimental determination of ammonia in hydrogen peroxide solution by photometric measurements (see Supplementary materials). The theoretical and experimental values are in a very good agreement. Therefore, we can assume no ammonia oxidation is happening in the gas phase in the second step (R2). Additionally, it is known ammonia in the gas phase cannot undergo the direct photolysis at 254 nm (wavelength of the lamps in R2), because at this wavelength the absorption coefficient of ammonia is practically nonexistent [35]. Based on the information above, it is clear the ammonia oxidation has to proceed in liquid phase. Since the ammonia is dissolved in the hydrogen peroxide solution in the presence of UV irradiation, there is a surplus of hydroxyl radicals in liquid phase that can oxidize ammonia. However, reaction of ammonia with HO• is pH dependent. The pKa value for ammonia is 9.25 [36], therefore ammonia is occurring in its protonated form (NH4+) at lower pH. Also, the reaction of dissolved ammonia with hydroxyl radicals from the photolysis of hydrogen peroxide is not very fast or even non-measurable [37], in addition this reaction is even diminished by the presence of other dissolved compounds in the solution, especially carbonates which serve as scavengers of HO• [38]. Hydroxyl radicals are strong electrophilic species [39], thus the reaction with neutral form of ammonia (NH3) is preferred and faster [24]. Yang et al. determined the rate constants in order of magnitude 106 dm3·mol−1·s−1 for reaction of HO• with NH4+ and 108 dm3·mol−1·s−1 for reaction of HO• with NH3 [24].
3.2 Reaction kinetics of ammonia in the water phase
Several 24-h experiments were carried out to examine the reaction pathways and kinetics of ammonia removal from the water phase. The airflow rate through the unit was set to 150 m3·h−1 and the initial concentration of ammonia in the water phase was 20 ppm, following the amount of absorbed ammonia after 1 h in previous experiments. The time range of the experiments was 0–24 h and samples were taken at 2-h intervals. Different experiments with different reaction conditions were conducted: with irradiation by the lamps in the second step—UV254nm; with the UV lamps in first step turned on for the production of ozone and circulation of water in the second step turned on—O3; with the circulation of hydrogen peroxide solution turned on and with the irradiation by the lamps in the second step—UV254nm + H2O2; with the UV lamps in first step turned on for the production of ozone and the circulation of water turned on and with irradiation by the lamps in the second step—UV254nm + O3; with the UV lamps in first step turned on for the production of ozone and the circulation of hydrogen peroxide solution turned on and with the irradiation by the lamps in the second step—UV254nm + H2O2 + O3. Initial concentration of hydrogen peroxide for experiments with H2O2 was 0.1 mol·dm−3.
Due to the difficulties to prepare exactly the same initial concentration of ammonia (Fig. 2a) the results were recalculated as c/c0 ratio (Fig. 2b). It is clear the removal of ammonia from liquid phase is highly dependent on the used oxidation agent. Ozone or UV irradiation alone have little effect and even after 24 h there is still 50% of ammonia left. The situation is better if H2O2 is used, no matter what other oxidation agent is used (UV, O3). The concentration of ammonia was almost 0 after 24 h. The best results were obtained when UV and O3 was used. Ammonia was completely removed after 15 h in this case.

Time dependence of a NH4+ concentration and b c/c0 for the kinetic study
The decrease of ammonia concentration followed linear dependence in all cases. The experimental results showed, that ammonia decomposition in the water phase followed zero-order reaction kinetics. Thereby the differential [Eq. (17)] and integral [Eq. (18)] rate law can be expressed as:
where d[NH3/NH4+]/dt is the ammonia removal rate (mol·dm−3·h−1), k is the rate constant (mol·dm−3·h−1), [NH3/NH4+]0 is the initial ammonia concentration (mol·dm−3) and t is the time (h).
The highest rate was obtained by using a combination of UV254nm + O3. The determined rate constants for different conditions were rising as in the following Table 1.
It is necessary to mention: when the UV irradiation was not used (experiment “O3”), the ozone absorption to the water phase in the scrubber was negligible. The decrease of the ammonia concentration was caused almost exclusively by the desorption, similar situation was in experiment with UV254nm. The photolysis of ammonia at 254 nm is also negligible, because of its weak absorption at wavelengths greater than 220 nm [40]. The ammonia removal rate in the experiments with hydrogen peroxide “UV254nm + H2O2” and “UV254nm + H2O2 + O3” was much faster which agrees with results found in corresponding literature [37, 41]. The highest reaction rate was determined for the “UV254nm + O3” reaction. In this case, ozone is photolytically decomposed to O(1D) and O2 [Eq. (4)] where O(1D) reacts with H2O to form another two HO• [Eq. (7)]. Considering the above-mentioned results, the reaction of O(1D) with NH3/NH4+ and H2O2 is also assumed.
It is important to mention, removal of ammonia from liquid phase has been documented very well in the literature [24, 37] and it is well known this reaction proceeds the best at basic pH. All the reactions above were conducted at natural pH of 8.2 (no additives). The idea was to test how different oxidation agents (used in this technology) influence the removal of ammonia from liquid phase without the need of adjusting the pH. In order to determine the difference, removal of ammonia at basic pH of 10.1 was conducted in presence of UV254nm + H2O2 + O3 (Fig. S8). In case of natural pH ammonia was not completely removed even after 24 h, however, in case of basic pH ammonia was completely removed after 4 h as experimentally confirmed.
3.3 Figures-of-merit
The feasibility of various technologies for air treatment is very hard to determine, especially if different principles and different reactor geometry are used. The best way how to compare different technologies are money, specifically how much it cost to treat 1 m3 of polluted air. For that reason, figures-of-merit have been calculated. The information about the price of the technology operational cost can be gained from a parameter EEO (electric demand for degradation of pollutants). EEO is defined as the amount of energy that must be provided to the system to decrease the concentration of the pollutant in one order of magnitude. For low concentrations of pollutants, the equation goes as follows:
where EEO is the electric demand for the degradation of pollutants (kW·h·m−3·order−1), P is the rated power (kW), F is the air flow through the unit (m3·h−1), cin is the inlet concentration of pollutants (ppmv) and cout is the outlet concentration of pollutants (ppmv).
Table 2 shows that the cost of electric energy for 1-h use of the technology would range between 0.6 and 1.2 € under the experimental conditions and with considering the average cost of 1 kWh (0.2 €, 2022, Czech Republic). The technology is more cost-convenient for higher flow rates.
4 Conclusions
This study demonstrates that advanced oxidation processes (AOPs) are an effective technology for the abatement of ammonia from a livestock building. The two-step pilot plant unit containing a dry photolytic reactor (UV185/UV254/O3) and a photochemical scrubber (UV254/H2O2) was tested for two initial ammonia concentrations (20 and 35 ppmv) and for three different air flows (150, 300 and 450 m3·h−1). The presented technology consists of two steps utilizing different advanced oxidation processes. The first step has little to none effect on ammonia removal, however, it is extremely important for the removal of organic pollutants which are present together with ammonia in real waste air from animal housing, and for the production of ozone, which is then used together with UV to oxidize ammonia in the liquid phase in a photochemical scrubber. Even at the highest flow rate of 450 m3·h−1 the ammonia removal from waste air was around 90%. Majority of the ammonia is dissolved in the aqueous solution of H2O2 and further oxidized by combination of oxidizing agents (H2O2, UV, O3). The rate of dissolved ammonia oxidation depends on the present oxidizing agents. The fastest removal was achieved in the presence of UV254nm and ozone (15 h). The ammonia is completely oxidized to nitrogen and hydrogen and only trace amounts of N2O and NO2 in the output air were detected using FT-IR analyzer. In order to compare presented technology with other commonly used technologies, the cost for lowering the ammonia concentration by order of magnitude was calculated. Based on the average price of electricity in Czech Republic the cost for treating one cubic meter of ammonia polluted air is 0.002 € at the highest flow rate of 450 m3·h−1 and for initial ammonia concentrations of 20 ppmv.
Data availability
The data supporting the findings of this study are available upon request at corresponding author (e-mail: tomas.prostejovsky@vsb.cz).
References
Galloway, J. N., et al. (2004). Nitrogen cycles: past, present, and future. Biogeochemistry, 70(2), 153–226. https://doi.org/10.1007/s10533-004-0370-0
Rodhe, H., Dentener, F., & Schulz, M. (2002). The global distribution of acidifying wet deposition. Environmental Science & Technology., 36(20), 4382–4388. https://doi.org/10.1021/es020057g
Dentener, F., et al. (2006). Nitrogen and sulfur deposition on regional and global scales: A multimodel evaluation. Global Biogeochemical Cycles. https://doi.org/10.1029/2005GB002672
Substance information: Ammonia, anhydrous. (2022). https://echa.europa.eu/cs/substance-information/-/substanceinfo/100.028.760. Accessed 5 June 2022
(2016). Directive (EU) 2016/2284 of the European Parliament and of the Council of 14 December 2016 on the reduction of national emissions of certain atmospheric pollutants, amending Directive 2003/35/EC and repealing Directive 2001/81/EC (Text with EEA relevance ), in OJ L 344. p. p. 1–31. http://data.europa.eu/eli/dir/2016/2284/oj
(2021). EEA Report No 5/2021: European Union emission inventory report 1990–2019 under the UNECE Convention on Long-range Transboundary Air Pollution (Air Convention). https://doi.org/10.2800/701303.
(2017). Consolidated text: Commission Implementing Decision (EU) 2017/302 of 15 February 2017 establishing best available techniques (BAT) conclusions, under Directive 2010/75/EU of the European Parliament and of the Council, for the intensive rearing of poultry or pigs (notified under document C(2017) 688) (Text with EEA relevance). http://data.europa.eu/eli/dec_impl/2017/302/2017-02-21.
Waldrip, H. M., Cole, N. A., & Todd, R. W. (2015). Review: nitrogen Sustainability and beef cattle feedyards: II. Ammonia emissions. The Professional Animal Scientist., 31(5), 395–411. https://doi.org/10.15232/pas.2015-01395
Insausti, M., et al. (2020). Advances in sensing ammonia from agricultural sources. Science of The Total Environment., 706, 135124. https://doi.org/10.1016/j.scitotenv.2019.135124
Guo, L., et al. (2022). Mitigation strategies of air pollutants for mechanical ventilated livestock and poultry housing-a review. Atmosphere. https://doi.org/10.3390/atmos13030452
Loyon, L., et al. (2016). Best available technology for European livestock farms: Availability, effectiveness and uptake. Journal of Environmental Management., 166, 1–11. https://doi.org/10.1016/j.jenvman.2015.09.046
Aarnink, J. A., et al. (2011). Scrubber capabilities to remove airborne microorganisms and other aerial pollutants from the exhaust air of animal houses. Transactions of the ASABE, 54(5), 1921–1930. https://doi.org/10.13031/2013.39833
Melse, R. W., Ploegaert, J. P. M., & Ogink, N. W. M. (2012). Biotrickling filter for the treatment of exhaust air from a pig rearing building: Ammonia removal performance and its fluctuations. Biosystems Engineering., 113(3), 242–252. https://doi.org/10.1016/j.biosystemseng.2012.08.010
Maurer, D. L., et al. (2016). Summary of performance data for technologies to control gaseous, odor, and particulate emissions from livestock operations: Air management practices assessment tool (AMPAT). Data in Brief., 7, 1413–1429. https://doi.org/10.1016/j.dib.2016.03.070
Kafle, G. K., et al. (2015). Field evaluation of wood bark-based down-flow biofilters for mitigation of odor, ammonia, and hydrogen sulfide emissions from confined swine nursery barns. Journal of Environmental Management., 147, 164–174. https://doi.org/10.1016/j.jenvman.2014.09.004
Wang, Y.-C., et al. (2021). Emissions, measurement, and control of odor in livestock farms: A review. Science of The Total Environment, 776, 145735. https://doi.org/10.1016/j.scitotenv.2021.145735
Winkel, A., et al. (2015). Evaluation of a dry filter and an electrostatic precipitator for exhaust air cleaning at commercial non-cage laying hen houses. Biosystems Engineering., 129, 212–225. https://doi.org/10.1016/j.biosystemseng.2014.10.006
Ogink, N. W. M., Melse, R. W., & Mosquera, J. (2008). Multi-pollutant and one-stage scrubbers for removal of ammonia, odor, and particulate matter from animal house exhaust air. American Society of Agricultural and Biological Engineers. https://doi.org/10.13031/2013.25508
Rockafellow, E. M., Koziel, J. A., & Jenks, W. S. (2012). Laboratory-scale investigation of UV treatment of ammonia for livestock and poultry barn exhaust applications. Journal of Environmental Quality., 41(1), 281–288. https://doi.org/10.2134/jeq2010.0536
Zhu, X., et al. (2005). Effects of pH and catalyst concentration on photocatalytic oxidation of aqueous ammonia and nitrite in titanium dioxide suspensions. Environmental Science & Technology., 39(10), 3784–3791. https://doi.org/10.1021/es0485715
Deng, Y., & Ezyske, C. M. (2011). Sulfate radical-advanced oxidation process (SR-AOP) for simultaneous removal of refractory organic contaminants and ammonia in landfill leachate. Water Research., 45(18), 6189–6194. https://doi.org/10.1016/j.watres.2011.09.015
Munter, R. (2001). Advanced oxidation processes-current status and prospects. Proceedings of the Estonian Academy of Sciences. Chemistry., 50, 59–80. https://doi.org/10.3176/chem.2001.2.01
Litter, M. I. (2005). Introduction to photochemical advanced oxidation processes for water treatment. In P. Boule, D. W. Bahnemann, & P. K. J. Robertson (Eds.), Environmental photochemistry part II (pp. 325–366). Berlin, Heidelberg: Springer. https://doi.org/10.1007/b138188
Yang, X., Tao, Y., & Murphy, J. (2021). Kinetics of the oxidation of ammonia and amines with hydroxyl radicals in the aqueous phase. Environmental Science: Processes and Impacts. https://doi.org/10.1039/d1em00317h
Burkholder, J. B., et al. (2015). Chemical kinetics and photochemical data for use in atmospheric studies, evaluation number 18. Pasadena, California: Jet Propulsion Laboratory, National Aeronautics and Space Administration. https://doi.org/10.13140/RG.2.1.2504.2806
Atkinson, R., et al. (1997). Evaluated kinetic and photochemical data for atmospheric chemistry: supplement VI. IUPAC subcommittee on gas kinetic data evaluation for atmospheric chemistry. Journal of Physical and Chemical Reference Data., 29, 167–266. https://doi.org/10.1063/1.556010
DeMore, W., et al. (1997). Chemical Kinetics and Photochemical Data for Use in Stratospheric Modeling, Evaluation Number 12 (Vol. 90, p. 23). JPL Publication.
Atkinson, R., et al. (2004). Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I - gas phase reactions of Ox, HOx, NOx and SOx species. Atmospheric Chemistry and Physics., 4(6), 1461–1738. https://doi.org/10.5194/acp-4-1461-2004
Manap, H., et al. (2009) Ammonia Detection in the UV Region Using an Optical Fiber Sensor. pp. 140–145. https://doi.org/10.1109/ICSENS.2009.5398215
McDonald, C. C., Kahn, A., & Gunning, H. E. (1954). The photolysis of ammonia at 1849A in a flow system. The Journal of Chemical Physics., 22(5), 908–916. https://doi.org/10.1063/1.1740214
Prostějovský, T., et al. (2021). Advanced oxidation processes for elimination of xylene from waste gases. Journal of Photochemistry and Photobiology A: Chemistry., 407, 113047. https://doi.org/10.1016/j.jphotochem.2020.113047
Prostějovský, T., et al. (2022). Photochemical treatment (UV/O3+UV/H2O2) of waste gas emissions containing organic pollutants in pilot plant unit. Process Safety and Environmental Protection., 163, 274–282. https://doi.org/10.1016/j.psep.2022.05.032
Kočí, K., et al. (2019). Degradation of Styrene from Waste Gas Stream by Advanced Oxidation Processes. Clean-Soil Air Water. https://doi.org/10.1002/clen.201900126
Tsang, W., & Herron, J. T. (1991). Chemical kinetic data base for propellant combustion I. Reactions involving NO, NO2, HNO, HNO2, HCN and N2O. Journal of Physical and Chemical Reference Data, 20(4), 609–663. https://doi.org/10.1063/1.555890
Keller-Rudek, H., et al. (2013). The MPI-Mainz UV/VIS spectral atlas of gaseous molecules of atmospheric interest. Earth System Science Data., 5(2), 365–373. https://doi.org/10.5194/essd-5-365-2013
Haynes, W. M., Lide, D. R., & Bruno, T. J. (2016). CRC handbook of chemistry and physics (97th ed., p. 2670). CRC Press.
Huang, L., et al. (2008). Removal of ammonia by OH radical in aqueous phase. Environmental Science & Technology., 42(21), 8070–8075. https://doi.org/10.1021/es8008216
Hoigne, J., & Bader, H. (1978). Ozonation of water: Kinetics of oxidation of ammonia by ozone and hydroxyl radicals. Environmental Science & Technology., 12(1), 79–84. https://doi.org/10.1021/es60137a005
Marusawa, H., et al. (2002). Hydroxyl radical as a strong electrophilic species. Bioorganic & Medicinal Chemistry., 10(7), 2283–2290. https://doi.org/10.1016/S0968-0896(02)00048-2
Zhang, X., et al. (2015). UV/chlorine process for ammonia removal and disinfection by-product reduction: Comparison with chlorination. Water Research., 68, 804–811. https://doi.org/10.1016/j.watres.2014.10.044
Wang, J., et al. (2017). Effects of pH and H2O2 on ammonia, nitrite, and nitrate transformations during UV254nm irradiation: Implications to nitrogen removal and analysis. Chemosphere, 184, 1003–1011. https://doi.org/10.1016/j.chemosphere.2017.06.078
Acknowledgements
This work was supported by EU structural funding in Operational Programme Research, Development and Education, project No. CZ.02.1.01/0.0/0.0/17_049/0008419 “COOPERATION” and by using Large Research Infrastructure ENREGAT supported by the Ministry of Education, Youth and Sports of the Czech Republic under project No. LM2023056.
Funding
Open access publishing supported by the National Technical Library in Prague.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prostějovský, T., Kulišťáková, A., Reli, M. et al. Innovative technology for ammonia abatement from livestock buildings using advanced oxidation processes. Photochem Photobiol Sci 22, 1603–1610 (2023). https://doi.org/10.1007/s43630-023-00400-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43630-023-00400-w