
Abstract
The photon upconversion based on triplet–triplet annihilation (TTA) is a mechanism that converts the absorbed low-energy electromagnetic radiation into higher energy photons also at extremely low excitation intensities, but its use in actual technologies is still hindered by the limited availability of efficient annihilator moieties. We present here the results obtained by the synthesis and application of two new fluorinated chromophores based on phenazine and acridine structures, respectively. Both compounds show upconverted emission demonstrating their ability as TTA annihilator. More interesting, the acridine-based chromophore shows an excellent TTA yield that overcomes the one of some of best model systems. By correlating the experimental data and the quantum mechanical modeling of the investigated compound, we propose an alternative efficient pathway for the generation of the upconverted emissive states involving the peculiar high-energy triplet levels of the dye, thus suggesting a new development strategy for TTA annihilators based on the fine tuning of their high-energy excited states properties.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The photon upconversion (UC) is a mechanism that converts the absorbed low-energy electromagnetic radiation into higher energy photons. A broad and deep research on UC has been motivated for its application in several fields such as photocatalysis, photodynamic therapy, optogenetics, bioimaging and anti-counterfeiting [1,2,3,4,5]. Moreover, UC has been foreseen as potential method to recover the low-energy tail of the solar emission to enhance the light-harvesting ability of solar devices. Classical mechanisms as multistep excitation of lanthanides ions [6], two-photon absorption [7], or second harmonic generation [8], require, however, a too high excitation intensity to work efficiently with respect to the solar irradiance, or even the use of coherent radiation to be efficient. The sensitized photon UC based on triplet–triplet annihilation (sTTA-UC) has recently attracted attention due to its unique efficiency of non-coherent light UC at low powers [9, 10]. The sTTA-UC process typically involves two components: an energy harvester/triplet sensitizer and a triplet annihilator/emitter (Fig. 1a) [11]. The sensitizer absorbs low-energy photons and transfers the harvested energy to the triplet’s annihilator through non-radiative energy transfer (ET). Subsequently, if two emitters in their triplet state encounter, they can annihilate generating a high-energy emissive excited singlet state, with an energy larger than the one of the absorbed photons. For optimized sensitizer/annihilator pairs, UC output quantum yield QYuc, larger than 20% can be obtained at low excitation intensities similar to the solar irradiance [12]. This excellent yield has been obtained thanks to the efforts spent in the past 15 years to develop efficient sensitizers [13,14,15]. However, to further improve the overall efficiency of sTTA-UC is still a challenge because of the shortage of highly efficient annihilators [12].

a Outline of the photophysical processes involved in sTTA-UC. Dashed lines represent radiation less transitions. b Molecular structures of fluorinated conjugated chromophores testes as annihilators/emitters in this study
A part for limited examples where perylene bisimide systems are used as acceptor, there are no reports where other electron poor nitrogen-containing heteroaromatic systems are used in this context. Large part of the reported examples deals with their use as triplet sensitizer systems [16]. One of the main reasons for this limited use is related to the fact that suitable systems for this application are not commercially available and specific synthetic strategies need to be developed. Recently, we have developed the synthesis of acridine [17] and phenazine [18] based systems, which show interesting optical and electronic properties that seem to match the prerequisites for being suitable acceptor for upconversion processes. The developed strategies allow also the synthesis of these with a different level of fluorination along the aromatic skeleton. Usually, the substitution of hydrogen with fluorine has a remarkable impact on the properties of the heterocycle, such as the enhancement of the thermal and photochemical stability. In addition, the fluorine atom bound to these systems undergoes easy aromatic nucleophilic substitution opening to a further functionalization of these systems to obtain a fine tuning of the optical and electronic properties. As a consequence of these peculiar properties, the investigation of fluorinated aromatic heterocycles as acceptor in TTA processes appears particularly promising. In this work, we show the results obtained on the synthesis and application of two new fluorinated chromophores, namely the 1,2,3,4,tetrafluoro-11H-indeno[1,2-b]phenazine (1) and the 1,2,3,4-tetrafluoro-13-(pentafluorophenyl)-11H-indeno[2,1-b]acridine (2), as annihilator/emitters for the TTA-UC processes (Fig. 1b). Both moieties work as annihilators/emitters for sTTA-UC and, importantly, the obtained results suggest that fluorinated acridine-based chromophore show excellent properties as annihilators because of its peculiar electronic properties that positively affect the efficiency of the singlet generation by the TTA mechanism.
2 Materials and methods
2.1 Synthesis
The synthesis of compounds 1 (Scheme 1) was achieved starting from decafluoroazobenzene (3), which was obtained by the mechanochemical oxidation of pentafluoroaniline with phenyl iodide(III)diacetate [19]. Afterwards, 3 was subjected to a two-step sequential procedure to construct the phenazine system to give the desired compounds 1 [18]. The first step concerns the regioselective ortho nucleophilic aromatic substitution (SNAr) of fluorine by 2-aminofluorene performed in decalin at 170 °C for 36 h. The absence of H-bonding sites in decalin allows direct interaction between the lone pair of the nitrogen of the azo group and the N–H hydrogens of the aromatic amines, thus directing the SNAr preferentially to the fluorine in ortho respect to the azo function. After removal of all volatiles and filtration through a short pad of silica gel, the crude obtained from the previous step was reacted with trifluoroacetic acid (TFA) at room temperature overnight to perform the acid catalyzed electrocyclization-aromatization. In the case of 1, the electrocyclization can occur in two different positions of the fluorene moiety, thus producing a mixture of desired isomer 1 and undesired isomer 4 in a 2:1 ratio. The two isomers were easily separated by chromatography on silica gel. By following the same chemical approach, we were able to synthesize also a dimeric system based on spirobifluorene-core using the 9,9′-spirobi[fluorene]-2,2′-diamine [20] as reactant. Due to the steric hindrance induced by the presence of the spiro-core, we did not observe the formation of regioisomers during the electrocyclization step to give the dimeric compound (Supporting Scheme 1). See Supporting Information (SI) for further details.

Synthesis of 1 and 2. Reagents and conditions: [a] PhI(OAc)2 (2 equiv), SiO2, ball milling (15 Hz), 1 h; [b] 2-aminofluorene (0.9 equiv), decahydronaphthalene, 170 °C, 36 h, N2; [c] TFA, r.t., 12 h, N2; [d] 2-aminofluorene (0.9 equiv), decahydronaphthalene, 170 °C, 36 h, N2
The synthesis of acridine 2 (Scheme 1) is similar to that used for compounds 1 [17]. Sequential regioselective ortho SNAr in decalin at 170 °C on decafluorobenzophenone 5 by 2-aminofluorene and TFA promoted electrocyclization-aromatization. As for the synthesis of phenazine 1, two regioisomers were produced, i.e., target acridine 3 and acridine 6 in a 1.5:1 ratio and sufficient yields. The two isomers were isolated in a pure form by chromatography on silica gel. Detailed synthetic procedures and characterization of synthesized compounds are reported in the Supporting Information.
2.2 Photophysical studies
Steady-state PL spectra were acquired with a nitrogen cooled charge coupled device (Spex ≈ 2000) coupled to a polychromator Triax 190 from J-Horiba. A Xe lamp coupled to a Gemini doubled monochromator was employed to have monochromatic excitation light at 370 nm. Upconversion spectra were recorded using a 532 nm solid-state laser diode from Roithner Lasertechnik as excitation source. The excitation light was removed using line filters, while for detection the laser stray light was attenuated with a notch filter. The 1/e2 excitation beam diameter was measured by the knife-edge method. The laser intensity was varied using reflective power density neutral filters and measured with an optical power meter (Thorlabs PM100USB, power sensor S120VC). All spectra were corrected for the instrumental optical response. The fluorescence and upconversion photoluminescence quantum yields were measured by relative methods using 9,10-diphenylanthracene (DPA) and the upconverting solution of PtOEP/DPA (10−4 M/10−2 M) in THF as standard [21]. For time-resolved photoluminescence measurements, the samples were excited at 340 nm with an EPL solid-state laser Edinburgh (pulse width 150 ps). For time-resolved upconversion measurements, the samples were excited at 532 nm by modulating the laser intensity with a TTi TG5011 wavefunction generator. The spectra were recorded by nitrogen cooled photomultiplier (Hamamatsu R5509-73) coupled with a high-speed amplifier (Hamamatsu C5594), a 74 100 Cornerstone 2601/4 (ORIEL) monochromator, and a PCI plug-in multichannel scaler ORTEC 9353 100 ps time digitizer/MCS in a photon counting acquisition mode. Fluorescence and upconversion quantum yield have been measured by relative methods as described in the Supporting Information file.
2.3 Quantum chemical modeling
In this work, the energy of the electronic states and the photophysical properties of the investigated systems 1 and 2 were computed employing Density Functional Theory (DFT) as well as full time-dependent DFT methods (TDDFT), to deeply explore the occurrence of intra-TTA processes. Geometry optimization of ground and excited states were calculated with Gaussian16 [22], meta-hybrid M06-2X functional [23] and 6-31+G* basis set [24]. The default ultra-fine grid for all numerical integrations and convergence criterion of 10–8 Hartree were applied. Solvent effects (dichloromethane, ε = 9.1) were included employing the Conductor-like polarized continuum model (CPCM) [25, 26] as implemented in Gaussian16. Starting from the minimum energy geometry calculated for the ground state S0 at the level of theory mentioned above, the TDDFT vertical excitations energies (10 for singlets and 10 for triplets) were computed and the geometries of the excited states S1, S2, T1, T2, T3 and T4 were optimized at full TDDFT level. The vibrational frequencies were calculated on the minimum geometries of the excited states, to verify the nature of the stationary points obtained. The S–S, S–T and T–T energy gap were calculated including the zero-point energy correction (ZPE). Spin–orbit coupling constants (SOCC) between singlets and triplets excited states were calculated for 2 at TDDFT level using ADF2020.10 [27] with the ZORA [28, 29] (zero-order regular approximated Hamiltonian) formalism, where scalar relativistic effects are included. The ZORA Hamiltonian can be written as the sum of a scalar relativistic (SR) part and a SOC part:
With this partitioning, SOC can be included self-consistently during the SCF and TDDFT calculations (SOC-TDDFT) [30]. Alternatively, SOC can be included as a perturbation based on the scalar relativistic orbitals (pSOC-TDDFT) [30]. As with other perturbative approaches, pSOC-TDDFT is best applied to systems where the perturbation (SOC) is small. The latter is the approach employed in this work. The M06-2X functional was used with all electron Slater-type orbital basis sets: triple-zeta + polarization (TZP) for all atoms. Starting from the previously optimized excited state geometries, we computed 10 singlet and 10 triplet excitations, which are used as the basis for the perturbative expansion in the pSOC-TDDFT. Solvation effects for dichloromethane were included employing the Conductor-like Screening Model (COSMO) as implemented in ADF2020.10. The calculated SOCC values are reported in Fig. 4b.
3 Results and discussion
3.1 Photophysical properties of annihilators
The chromophores electronic properties have been investigated by means of optical absorption and photoluminescence experiments. It is worth to note that the photophysical properties of the dimeric spiro-based phenazine system were very similar to the phenazine parent system 1, so we will focus the following discussion only on compounds 1 and 2. Figure 2a shows the normalized absorption and photoluminescence spectra of compounds 1 and 2 in diluted solution (10–5 M) of tetrahydrofuran (THF). Both dyes show an absorption peaked in the near UV-blue spectral region at ca. 420 nm that marks the S0–S1 transition. Under 370 nm excitation, the dyes show a blue fluorescence peaked at 480, 450 nm for compounds 1 and 2, respectively, with similar emission profiles. The observed absorption maxima energies are in good agreement with the predicted ground state electronic energies, reported in Table 1. The quantum mechanical modeling enables also to estimate the energies of the optically dark T1 states, which are found around 1.6 eV. This means that both the proposed dyes can work as annihilators/emitters for TTA, having satisfied the minimum necessary energetic constraint that ∆ETTA = (2T1 − S1) > 0 (Table 1). Conversely, the fluorescence quantum yield (\({\phi }_{\mathrm{fl}}\)), defined as the ratio between the number emitted photons on the number of the absorbed ones is quite different. Compounds 1 shows a low \({\phi }_{\mathrm{fl}}\) ̴ 6%, while the compound 2 yield grows as high as 34%. The significantly improved emission efficiency of 2 is reflected also by the observed fluorescence characteristic lifetime, measured under pulsed laser excitation, that is reported in Fig. 2c. The fluorescence recombination kinetics becomes much slower from 1 to 2 (2.9 ns). Specifically, the five times increment observed for the fluorescence lifetime τfl from 0.6 to 2.9 ns is in excellent agreement with the one observed for \({\phi }_{\mathrm{fl}}\) (Table 1), thus suggesting the effective reduction of competitive non-radiative intramolecular recombination channels upon the introduction of the lateral phenyl ring with steric hindrance that rigidify the 2 system with respect to 1.

a Absorption and photoluminescence (PL) spectra in THF-diluted solution of compouds 1 (top) and 2 (bottom). PL spectra are recorded under 355 nm excitation. b Time-resolved PL spectrum recorded at the emission maximum under pulsed excitation at 370 nm. Solid lines are the fit of the data with a single exponential decay function with characteristic decay time τfl
3.2 Upconversion properties in solution with triplet sensitizers
The property of 1 and 2 as TTA-UC annihilators are tested in THF solution (3.5 × 10–5 M) with platinum (II) octaethyl porphyrin (PtOEP, 10–4 M) as triplet sensitizer. Figure 3a reports the photoluminescence spectrum of the bicomponent solution under cw excitation at 532 nm. The laser stray light has been partially removed using a notch filter. A low-energy emission at 645 is detectable in both samples, due to incomplete energy transfer (ET) from excited sensitizers to annihilators triplets at the employed concentrations. These latter are kept low to avoid possible intermolecular interactions between annihilators that can affect the UC performance, but large enough to have a measurable ET yield \({\phi }_{\mathrm{ET}}\), which is assessed at 27% and 25% for compound 1 and 2, respectively (Figure S3). Both samples also present a blue upconverted photoluminescence that matches their fluorescence spectrum. The occurrence of sTTA-UC mechanism is demonstrated by time-resolved experiments performed under modulated excitation at 532 nm (Fig. 3b). The upconverted emission intensity shows a single exponential decay kinetic in the microsecond time range, thus order of magnitudes slower than the prompt fluorescence, because of the involvement of the long living annihilating triplets that generate the emissive state by TTA [12]. These data allow also to estimate the triplet state lifetime τT, which can be calculated as twice the observed UC emission lifetime τuc. We therefore obtain a τT of 4 µs for compound 1 and 8 µs for compound 2. These findings highlight again the positive effect of the introduction of the lateral phenyl ring in the molecular structure on the excited states properties.

a Photoluminescence (PL) spectrum of upconverting solution of chromophores 1 and 2 in THF (3.5 × 10−5 M) with the sensitizer PtOEP (10–4 M). PL spectra are recorded under 532 nm excitation. For clarity, the laser stray light is partially removed using a 532 nm notch filter. b Time-resolved PL spectrum of UC solutions recorded at 460 nm under modulated excitation at 532 nm. Solid lines are the fit of the data with a single exponential decay function. c UC quantum efficiency \({\mathrm{QY}}_{\mathrm{uc}}\) as a function of the excitation intensity at 532 nm
The upconversion performance of the solution is tested as a function of the excitation intensity to evaluate the maximum upconversion yield (\({\mathrm{QY}}_{\mathrm{uc}}\)) achievable. We report here the \({\mathrm{QY}}_{\mathrm{uc}}\) defined as the number of emitted UC photons with respect to the number of absorbed ones thus with a theoretical maximum value of 50% [31]. Figure 3c shows the obtained results. In both cases, we observe an upconverted emission, but using 1 as annihilator the conversion efficiency remains lower than 0.01 even at high excitation intensities. This is in agreement with its very low fluorescence efficiency, given that the system maximum yield cannot be higher than the theoretical maximum value of \({\mathrm{QY}}_{\mathrm{uc}}^{1}\)[max] = 0.5 \({\phi }_{\mathrm{ET}}^{1}{\phi }_{\mathrm{fl}}^{1}\) = 0.008. On the other side, the compound 2 shows a significantly better performance. As a function of the excitation intensity, the \({\mathrm{QY}}_{\mathrm{uc}}\) value increases until a saturation plateau at 4% is reached. The excitation intensity threshold, defined at the intensity at which half of the maximum yield is reached, is a assessed at 1017 ph cm−2 s−1, a value larger than the typical observed in best liquid upconverters because of the short triplet lifetime that increases the excitons density required to maximize the TTA yield \({\phi }_{\mathrm{TTA}}\) [32]. At excitation powers above 2 × 1018 ph s−1 cm−2 (ca. 750 mW cm−2) the upconversion efficiency decreases because of the appearance homo-molecular annihilation between PtOEP triplets enabled by the incomplete transfer [33]. On the other hand, the process yield is constant in the region between 7 × 1017 ph s−1 cm−2 and 2 × 1018 ph s−1 cm−2. This suggest that the \({\phi }_{\mathrm{TTA}}\) is unity, and therefore, we can analyze more in detail the sTTA-UC mechanism. Specifically, in this regime, we can directly estimate from experimental data the statistical parameter \(f\) that marks the probability of generation a singlet upon the TTA step, as
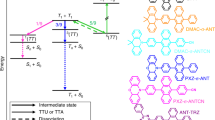
Using parameters measured independently, by means of Eq. 2, we estimate a f as large as 0.94 ± 0.08 for compound 2. This means that all the annihilating triplet pairs generate a singlet state, a peculiar condition that has been observed only in very few cases [12, 34]. The quantum mechanical modeling allows us to speculate on the origin of this high efficiency in the triplet-to-singlet conversion. Figure 4 depicts the simplified Jablonski diagram for the compound 2 showing the series of calculated singlet and triplet energies in optimized geometry and the calculated spin–orbit coupling constants (SOCC) values between S1 and the triplets state T1, T2 and T3. The T3 level of the annihilator is not easily accessible at room temperature by TTA since (T3 − 2T1) > 25 meV. Moreover, its coupling with the S1 state is the weakest (Fig. 4b), thus in first approximation we suppose that it does not participate in the formation of emitting singlets upon TTA. Conversely, the state T2 is fully accessible by TTA, possessing an energy 0.35 eV lower than twice the T1. This means that, from pure statistical arguments, the expected f factor should be 0.4, according to the partial recovering of the electrons in the T2 state that repopulate T1 by ultrafast non-radiative internal conversion that creates additional states available for TTA [34]. Nevertheless, it should be noted that in compound 2 the T2 is in very good resonance with the S1 state, almost isoenergetic, and that the two states show a not negligible SOCC, comparable to that one between the S1 → T1 transition that limit the dye fluorescence efficiency (Figure S4). Their high resonance and their intrinsic coupling suggests that a fast efficient delocalization of the molecular exciton between the two states is possible, thus enabling an efficient interplay between S1 and T2, as recently discussed for other systems [35, 36]. Moreover, the calculations suggest that at the thermodynamic equilibrium the average population of the S1 and T2 should be unbalanced toward the singlet manifold, considering the exothermic transition T2 → S1. This picture allows us to propose a global model for the fate of the molecular exciton to support the experimental evidences. As depicted in Fig. 4c, the exciton created on T2 upon TTA rapidly delocalizes on the high vibrational states of the S1 level before dropping to T1. Subsequently, it quickly thermalizes to the lowest vibrational level, from which the endothermic backward transition to T2 is improbable and, moreover, it has to compete with the fast recombination to the ground state. Therefore, upconverted fluorescence can be generated from the additional S1 states populated by recovering the energy stored on the T2 level.

a Calculated Jablonski diagram of compound 2. Excited states were optimized in dichloromethane (CPCM model) at M06-2X/6-31+G* level of theory in Gaussian16. Adiabatic energies are reported in eV including Zero Point Energy (ZPE) correction. b Electronic transitions, spin–orbit coupling constants (SOCC, cm−1) computed employing ADF2020.10 with M06-2X meta-hybrid functional and TZP basis set and S–T energy gap (ΔE, eV) calculated as the difference between the adiabatic energies of singlet and triplet excited states including zero-point energy correction (ZPE). Solvation effects for dichloromethane were included. SOCC values were calculated starting from singlet and triplet optimized geometries, respectively. c Sketch of the proposed energy pathway that allow to recover the energy stored on the T2 level thanks to the resonance with the S1 level
The proposed energetic pathway can fully justify the observed optimal efficiency in the generation of upconverted emitting states in compound 2 in strict correlation with his electronic structure. If further experimental studies will confirm it, for example by monitoring the kinetics of the formation of singlet states with transient experiment in the ultrafast time scale, the proposed mechanism can be, therefore, considered as new general guideline for the design and synthesis of conjugates systems possessing strictly resonant S1 and Tn levels optimized for TTA. This can open a completely new strategy for the development of sTTA-UC annihilators, in opposition to the more straightforward approach to search for annihilators with inaccessible high-energy triplets that has been successful in very few cases.
4 Conclusion
To summarize, we demonstrated how newly synthesized non-symmetric fluorinated dyes can work as annihilator/emitters in bicomponent systems for sTTA-UC. The employed molecular design strategy improves the photoluminescence properties of the proposed acridine-based dye by suppressing the intramolecular quenching mechanism. Moreover, the system shows an efficiency in the generation of upconverted singlets by TTA that matches the one of the best model annihilators. The fluorinated dye shows indeed a triplet to singlet conversion yield close to unity, which is a very rare condition. The results of the quantum mechanical analysis of the molecular electronic properties strongly suggest that this occurs thanks to its peculiar electronic structure, which shows a low-energy T2 state accessible by TTA and strongly resonant with the emissive S1 energy. This energetic scenario could enable a fast delocalization of the molecular exciton in the triplet state on the singlet orbital, where it relaxes to the lower vibronic level and successively recombines by producing upconverted excited singlets with an efficiency that surpasses the limit imposed by classical spin-statistics in the best polyacene-based annihilators. This model, which involves the interplay of excited singlet and triplet states, suggests, therefore, a new design strategy to make efficient TTA annihilators involving high-energy triplets by exploiting the attitude of aromatic fluorinated compounds to further functionalization and fine tuning of electronic and emission properties.
References
Kim, H.-I., Kwon, O. S., Kim, S., Choi, W., & Kim, J.-H. (2016). Harnessing low energy photons (635 nm) for the production of H2O2 using upconversion nanohybrid photocatalysts. Energy & Environmental Science, 9, 1063–1073.
Kim, J.-H., & Kim, J.-H. (2012). Encapsulated triplet–triplet annihilation-based upconversion in the aqueous phase for sub-band-gap semiconductor photocatalysis. Journal of the American Chemical Society, 134, 17478–17481.
Liu, Q., Xu, M., Yang, T., Tian, B., Zhang, X., & Li, F. (2018). Highly photostable near-IR-excitation upconversion nanocapsules based on triplet–triplet annihilation for in vivo bioimaging application. ACS Applied Materials & Interfaces, 10, 9883–9888.
Schulze, T., & Schmidt, T. (2015). Photochemical upconversion: Present status and prospects for its application to solar energy conversion. Energy & Environmental Science, 8, 103–125.
Chen, S., Weitemier, A. Z., Zeng, X., He, L., Wang, X., Tao, Y., Huang, A. J., Hashimotodani, Y., Kano, M., & Iwasaki, H. (2018). Near-infrared deep brain stimulation via upconversion nanoparticle-mediated optogenetics. Science, 359, 679–684.
Zhou, J., Liu, Z., & Li, F. (2012). Upconversion nanophosphors for small-animal imaging. Chemical Society Reviews, 41, 1323–1349.
Pawlicki, M., Collins, H. A., Denning, R. G., & Anderson, H. L. (2009). Two-photon absorption and the design of two-photon dyes. Angewandte Chemie International Edition, 48, 3244–3266.
Mao, C., Min, K., Bae, K., Cho, S., Xu, T., Jeon, H., & Park, W. (2019). Enhanced upconversion luminescence by two-dimensional photonic crystal structure. ACS Photonics, 6, 1882–1888.
Baluschev, S., Miteva, T., Yakutkin, V., Nelles, G., Yasuda, A., & Wegner, G. (2006). Up-conversion fluorescence: Noncoherent excitation by sunlight. Physical Review Letters, 97, 143903.
Oldenburg, M., Turshatov, A., Busko, D., Wollgarten, S., Adams, M., Baroni, N., Welle, A., Redel, E., Wöll, C., & Richards, B. S. (2016). Photon upconversion at crystalline organic–organic heterojunctions. Advanced Materials, 28, 8477–8482.
Keivanidis, P. E., Baluschev, S., Miteva, T., Nelles, G., Scherf, U., Yasuda, A., & Wegner, G. (2003). Up-conversion photoluminescence in polyfluorene doped with metal (II)–octaethyl porphyrins. Advanced Materials, 15, 2095–2098.
Sun, W., Ronchi, A., Zhao, T., Han, J., Monguzzi, A., & Duan, P. (2021). Highly efficient photon upconversion based on triplet–triplet annihilation from bichromophoric annihilators. Journal of Materials Chemistry C, 9, 14201–14208.
Xia, P., Raulerson, E. K., Coleman, D., Gerke, C. S., Mangolini, L., Tang, M. L., & Roberts, S. T. (2020). Achieving spin-triplet exciton transfer between silicon and molecular acceptors for photon upconversion. Nature Chemistry, 12, 137–144.
Ronchi, A., Capitani, C., Pinchetti, V., Gariano, G., Zaffalon, M. L., Meinardi, F., Brovelli, S., & Monguzzi, A. (2020). High photon upconversion efficiency with hybrid triplet sensitizers by ultrafast hole-routing in electronic-doped nanocrystals. Advanced Materials, 32, 2002953.
Wu, W., Wu, X., Zhao, J., & Wu, M. (2015). Synergetic effect of C* N^ N/C^ N^ N coordination and the arylacetylide ligands on the photophysical properties of cyclometalated platinum complexes. Journal of Materials Chemistry C, 3, 2291–2301.
Manna, M. K., Shokri, S., Wiederrecht, G. P., Gosztola, D. J., & Ayitou, A.J.-L. (2018). New perspectives for triplet–triplet annihilation based photon upconversion using all-organic energy donor & acceptor chromophores. Chemical Communications, 54, 5809–5818.
Del Buttero, P., Gironda, R., Moret, M., Papagni, A., Parravicini, M., Rizzato, S., & Miozzo, L. (2011). Orthogonal synthesis of fluorinated acridones and acridines from perfluorobenzophenone. European Journal of Organic Chemistry, 2011, 2265–2271.
Vaghi, L., Coletta, M., Coghi, P., Andreosso, I., Beverina, L., Ruffo, R., & Papagni, A. (2019). Fluorine substituted non-symmetric phenazines: A new synthetic protocol from polyfluorinated azobenzenes. ARKIVOC, 2019, 340–351.
Andreosso, I., Papagni, A., & Vaghi, L. (2018). Mechanochemical oxidation of fluorinated anilines to symmetric azobenzenes. Journal of Fluorine Chemistry, 216, 124–127.
Matsumoto, T., Murakami, T., Schlüter, F., Murata, H., Vohra, V., & Rizzo, F. (2022). Water‐soluble organic dyes as efficient anode interlayer materials for PEDOT:PSS‐free inverted bulk heterojunction solar cells, Solar RRL, 6, 2100661.
Saenz, F., Ronchi, A., Mauri, M., Kiebala, D., Monguzzi, A., & Weder, C. (2021). Block copolymer stabilized liquid nanodroplets facilitate efficient triplet fusion-based photon upconversion in solid polymer matrices. ACS Applied Materials & Interfaces, 13, 43314–43322.
Frisch, M., Trucks, G., Schlegel, H., Scuseria, G., Robb, M., Cheeseman, J., Scalmani, G., Barone, V., Petersson, G., & Nakatsuji, H. (2016). Gaussian 16, in, Gaussian, Inc. Wallingford, CT
Zhao, Y., & Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts, 120, 215–241.
Mitin, A. V., Baker, J., & Pulay, P. (2003). An improved 6-31G* basis set for first-row transition metals. The Journal of Chemical Physics, 118, 7775–7782.
Barone, V., & Cossi, M. (1998). Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. Journal of Physical Chemistry A, 102, 1995–2001.
Cossi, M., Rega, N., Scalmani, G., & Barone, V. (2003). Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. Journal of Computational Chemistry, 24, 669–681.
te Velde, G., Bickelhaupt, F. M., Baerends, E. J., Fonseca-Guerra, C., van Gisbergen, S. J. A., Snijders, J. G., & Ziegler, T. (2001). Chemistry with ADF. Journal of Computers and Chemistry, 22, 931–967.
van Lenthe, E., Baerends, E. J., & Snijders, J. G. (1993). Relativistic regular two-component Hamiltonians. The Journal of Chemical Physics, 99, 4597–4610.
van Lenthe, E., Baerends, E. J., & Snijders, J. G. (1994). Relativistic total energy using regular approximations. The Journal of Chemical Physics, 101, 9783–9792.
Wang, F., Ziegler, T., van Lenthe, E., van Gisbergen, S., & Baerends, E. J. (2005). The calculation of excitation energies based on the relativistic two-component zeroth-order regular approximation and time-dependent density-functional with full use of symmetry. The Journal of Chemical Physics, 122, 204103.
Zhou, Y., Castellano, F. N., Schmidt, T. W., & Hanson, K. (2020). On the quantum yield of photon upconversion via triplet–triplet annihilation. ACS Energy Letters, 5, 2322–2326.
Meinardi, F., Ballabio, M., Yanai, N., Kimizuka, N., Bianchi, A., Mauri, M., Simonutti, R., Ronchi, A., Campione, M., & Monguzzi, A. (2019). Quasi-thresholdless photon upconversion in metal–organic framework nanocrystals. Nano Letters, 19, 2169–2177.
Monguzzi, A., Tubino, R., Hoseinkhani, S., Campione, M., & Meinardi, F. (2012). Low power, non-coherent sensitized photon up-conversion: Modelling and perspectives. Physical Chemistry Chemical Physics, 14, 4322–4332.
Hoseinkhani, S., Tubino, R., Meinardi, F., & Monguzzi, A. (2015). Achieving the photon up-conversion thermodynamic yield upper limit by sensitized triplet–triplet annihilation. Physical Chemistry Chemical Physics, 17, 4020–4024.
Meroni, D., Monguzzi, A., & Meinardi, F. (2020). Photon upconversion in multicomponent systems: Role of back energy transfer. The Journal of Chemical Physics, 153, 114302.
Bossanyi, D. G., Sasaki, Y., Wang, S., Chekulaev, D., Kimizuka, N., Yanai, N., & Clark, J. (2021). Spin statistics for triplet–triplet annihilation upconversion: Exchange coupling, intermolecular orientation, and reverse intersystem crossing, JACS Au
Acknowledgements
This work was financially supported by the Italian Ministry of University and Research (MIUR) through grant Dipartimenti di Eccellenza—2017 “Materials for Energy” and by “Fondo di Ateneo (FA)”. We acknowledge CINECA for the availability of high-performance computing resources as part of the agreement with the University of Milano-Bicocca. We would like to thank Giorgio Patriarca for the NMR analyses and Ivan Andreosso for laboratory support.
Funding
Open access funding provided by Università degli Studi di Milano - Bicocca within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Contributions
LV, FR and AP designed and synthesized the conjugated systems employed in this study as sTTA-UC annihilators/emitters. AM, UC and CG performed the quantum mechanical modeling. JP, FM and AM designed and performed the photoluminescence spectroscopy experiments.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
This article is dedicated to Professor Angelo Albini in the occasion of his 75th birthday.
The original online version of this article was revised: The funding note was missing.
Supplementary Information
Below is the link to the electronic supplementary material.
43630_2022_225_MOESM1_ESM.docx
Supplementary Information are available online and include details and additional data on the synthesis and characterization of materials and photophysical studies. Data request should be addressed to corresponding authors (DOCX 4780 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vaghi, L., Rizzo, F., Pedrini, J. et al. Bypassing the statistical limit of singlet generation in sensitized upconversion using fluorinated conjugated systems. Photochem Photobiol Sci 21, 913–921 (2022). https://doi.org/10.1007/s43630-022-00225-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43630-022-00225-z