
Abstract
We investigated the influence of a range of factors—temperature, redox midpoint potential of an electron carrier, and protein dynamics—on nanosecond electron transfer within a protein. The model reaction was back electron transfer from a bacteriopheophytin anion, HA−, to an oxidized primary electron donor, P+, in a wild type Rhodobacter sphaeroides reaction center (RC) with a permanently reduced secondary electron acceptor (quinone, QA−). Also used were two modified RCs with single amino acid mutations near the monomeric bacteriochlorophyll, BA, located between P and HA. Both mutant RCs showed significant slowing down of this back electron transfer reaction with decreasing temperature, similar to that observed with the wild type RC, but contrasting with a number of single point mutant RCs studied previously. The observed similarities and differences are explained in the framework of a (P+BA− ↔ P+HA−) equilibrium model with an important role played by protein relaxation. The major cause of the observed temperature dependence, both in the wild type RC and in the mutant proteins, is a limitation in access to the thermally activated pathway of charge recombination via the state P+BA− at low temperatures. The data indicate that in all RCs both charge recombination pathways, the thermally activated one and a direct one without involvement of the P+BA− state, are controlled by the protein dynamics. It is concluded that the modifications of the protein environment affect the overall back electron transfer kinetics primarily by changing the redox potential of BA and not by changing the protein relaxation dynamics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Photosynthetic proteins offer a unique opportunity to study the influence of the protein environment on intra-protein electron transfer (ET) reactions. This is because ET can be easily followed by transient absorption measurements due to strong light absorption by the ET cofactors (pigments) that are embedded inside these proteins and which interact with nearby amino acid residues. There is increasing evidence from theoretical predictions [1,2,3,4,5] and experimentation [6,7,8,9] that the local protein dynamics affect the rate constants of intra-protein ET. Such protein dynamics are spread over very broad range of time scales, from picoseconds to seconds [10], matching the time range of ET reactions in photosynthetic proteins [11,12,13,14]. In this contribution we search for a relationship between the protein dynamics and ET reactions occurring on a nanosecond time scale at different temperatures and with different protein-ET cofactor interactions. The model system is the photosynthetic reaction center (RC) from the purple bacterium Rhodobacter (Rba.) sphaeroides, whose structure and function are very well characterized [11, 12, 15].
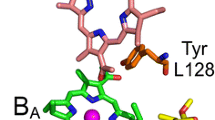
The bacteriochlorin and ubiquinone cofactors in the Rba. sphaeroides RC form two quasi-symmetric branches, only one of which is fully engaged in membrane-spanning ET (Fig. 1). Light-triggered ET usually starts from the excited electronic state of the primary electron donor, P*, a dimer of bacteriochlorophyll (BChl) a molecules, and then proceeds via a monomeric BChl a, BA, to a bacteriopheophytin, HA (Fig. 1). These reactions occur on the time scale of a few picoseconds (ps) [16, 17]. Next, within ~ 200 ps, an electron is transferred from HA to a ubiquinone molecule, QA, and after ~ 200 µs reaches the final electron acceptor, a dissociable QB ubiquinone on the second cofactor branch [18]. Weak background illumination of RCs in the presence of ascorbate, an artificial electron donor to the oxidized P (P+) leads to the quasi-permanent reduction of QA to QA−, particularly in the presence of ortho-phenanthroline which can substitute for the quinone in the QB pocket [19, 20]. Under these conditions, an electron which was transferred from P* to HA may only be transferred back to P+. In such a case, the sequence of reactions may be written in a simplified way as follows:

Arrangement of electron transfer cofactors in Rba. sphaeroides RCs and the location of the phenylalanine L146 residue that is substituted in the mutant RCs. Arrows depict the two competitive charge recombination pathways
The last of these reactions, charge recombination, occurs on a nanosecond time scale and is kinetically complex [21, 22]. It is usually modelled as a three-exponential decay process with lifetimes ranging from ~ 0.5 to ~ 20 ns [23, 24]. Since first identified the complexity of this reaction was ascribed to protein relaxation [23, 24]. Detailed studies of charge recombination revealed a significant but rather weak temperature dependence for this ET reaction [9, 25]. Significant effects on the rate of this reaction were found to be caused by mutations of single amino acids in the vicinity of the ET cofactors [26, 27]. To quantify the influence of the dynamics of protein relaxation on the rate of the charge recombination reaction, a mathematical model was proposed which separated the ET events from the protein relaxation steps (Fig. 2) [9]. Two phases of this relaxation were reported, occurring within ~ 1 and ~ 10 ns in detergent-purified wild type (WT) RCs, that were essentially independent of temperature between 78 and 298 K [9]. In membrane-embedded RCs these two phases were slower (lifetime of ~ 2 and ~ 15 ns) and also rather temperature-independent [25]. Studies on a few RCs with single point mutations near the ET cofactors, characterized by a particularly weak temperature dependence of the charge recombination rate, indicated a modest modulation of the protein relaxation rate in the membrane-bound RCs by the mutations. Estimated values for this rate, in a one-step relaxation model, ranged between ~ 1 and ~ 9 ns, depending on the RC and model, compared to ~ 3–6 ns in the WT RC [28].

Kinetic-energetic diagram used for modelling experimental transient absorption kinetics arising from charge recombination
In this contribution we investigated two single point mutant RCs characterized by particularly fast charge recombination, caused by modification of the interaction between BA and the surrounding protein [27], and by a pronounced temperature dependence. We conclude that the protein dynamics are an important factor that strongly influence the overall charge recombination and its temperature dependence.
2 Materials and methods
2.1 Preparation of samples
Antenna-deficient strains of Rba. sphaeroides assembling WT or mutant RCs were grown under dark/semiaerobic conditions as described previously [29]. Intracytoplasmic membranes were isolated by the breakage of harvested cells in a French pressure cell, followed by purification on 15%/40% (w/v) sucrose density step gradients [30]. These membrane fragments lack the native LH1 and LH2 antenna proteins and are enriched in either WT or mutant RCs.
2.2 Transient absorption experiment
For transient absorption experiments, concentrated membrane-bound RCs were diluted in 1.5 mM Tris buffer (pH 8.2), containing ∼0.0001% β-dodecyl maltoside (β-DM), 0.1 mM EDTA, and 60% glycerol (v/v). To the samples, 20 mM sodium ascorbate and 12 mM o-phenanthroline were added, the latter to block forward transfer from QA to QB [19, 20, 31, 32]. During the measurements the samples were constantly illuminated with a background white light (∼1 mW/cm2). Under such conditions, the quinone QA is permanently reduced [27, 33, 34].
Solutions of membrane-bound RCs were placed in a plastic cuvette formed by two round windows separated with a 1.5 mm thick O-ring. The cuvette was set at a 45° angle relative to the directions of the excitation, probe and background beams. The excitation and background beams illuminated the samples from two opposite sides, whereas the probe beam entered the sample at a right angle to the excitation beam. The optical density of the samples at 800 nm was OD1.5 mm = ~ 0.5. The cuvette was placed in a cryostat (Janis VPF-100) cooled with liquid nitrogen. For each sample, a kinetic curve was collected at each temperature between 78 and 298 K, as an average of 1024 individual laser-shot traces.
The transient absorption instrument used was as described previously [27, 35]. Vertically polarized saturating 532-nm excitation flashes of ∼ 2 mJ energy and of ~ 100 ps duration were provided by a Nd:YAG laser (Continuum Leopard SS-10) at a repetition rate of ∼ 2 Hz. A diode laser system (EOSI 2010) was used as a probe light source at 690 nm. The probe light was chopped to minimize its excitation effect. The monitoring beam was detected by a fast photodiode (rise time 200 ps; model UPD-200-UP from Alphalas) plugged into a digitizing oscilloscope (Agilent Infinium 81004B; 10 GHz; sampling rate, 40 Gsamples/s). The time resolution of the measurements was about 300 ps. The experiments were performed in a 6 μs time window.
2.3 Multiexponential analysis and further modelling
For each of the samples, transient absorption kinetics collected at different temperatures were fitted globally in a 200-ns time window using a three-exponential function with shared preexponential factors (amplitudes) over the temperatures. Independent fitting of individual kinetic traces collected at different temperatures was also performed but resulted in very irregular dependencies of both amplitudes and lifetimes as a function of temperature, caused by limited signal to noise ratio of experimental data (data not shown); in particular, the amplitudes did not show any clear systematic dependence on temperature in such an approach. The fit parameters resulting from the global analysis, three preexponential factors, three lifetimes and an offset, were used as input data for further modelling as presented graphically in Fig. 2. A detailed description of the mathematical modelling and the relationship between the three-exponential fit parameters, and the model parameters—the molecular lifetimes and free energy gaps shown in Fig. 2—is presented in the Supplementary Information.
3 Results and discussion
3.1 Mutant RCs with decreased free energy gaps between P+ B A − and P + H A −
Mutant RCs were investigated in which a phenylalanine residue at the 146 position of the PufL polypeptide (Fig. 1) was replaced with either a tyrosine (“FLY” mutant) or an alanine (“FLA” mutant). A tyrosine residue in L146 position introduces an additional hydroxyl group near the 31-acetyl group of BA. The alanine residue, which is much smaller than phenylalanine, leaves an empty space in the vicinity of the 31-acetyl group of BA, which could easily accommodate one or more water molecules. Both of these mutations affect the interaction between BA and the nearby L146 residue. In the case of the FLY mutant, it was shown that the mutation alters the electronic coupling between the RC pigments and thereby influences the rate of the charge separation between P and BA [36]. In both FLY and FLA membrane-bound RCs with QA permanently reduced, the mutation caused a significant acceleration of the averaged charge recombination lifetime from ~ 8 ns in the WT RC to ~ 3 ns in either mutant [27]. (The average charge recombination lifetime reported in the literature for WT RCs is even larger than 8 ns and ranges from 12 to14 ns [21 and literature referenced there, 37]. The difference was shown to originate from different methods of blocking forward electron transfer: the charge recombination in RCs with reduced QA is significantly faster than that in RCs without QA [31].) This effect is likely caused by an increase of the redox midpoint potential of BA and, consequently, stabilization (decrease) of the free energy level of the state P+BA− [27]. The likely origin of these changes is the formation of a hydrogen bond between the tyrosine at L146 in the FLY RC or a water molecule in the cavity in the FLA RC and the 31-acetyl group of BA. A similar effect of additional hydrogen bonds to the primary donor P in mutant RCs was reported previously [38]. The expected consequence of the stabilization (decrease) of the free energy level of the state P+BA− is a decreased free energy gap between P+BA− and P+HA−. The observed acceleration of the overall charge separation, from ~ 8 ns in WT to ~ 3 ns in the mutants with a decreased (P+BA−–P+HA−) free energy gap, is therefore consistent with the idea of thermally-activated P+HA− charge recombination via the rapidly recombining state P+BA−. However, to fully explain the multiexponential charge recombination kinetics and their temperature dependence presented below, as well as to answer the question of how the mutations affect the free energy gap in the sequential protein relaxation states, it is necessary to employ the more complex model presented in Fig. 2.
3.2 Model
The model shown in Fig. 2. explains the three-exponential nature of the kinetics of P+HA− charge recombination. Its essential feature is relaxation of the energy level of the state P+HA− relative to that of P+BA−. This relaxation has been assigned to protein dynamics [9, 23, 24]. From each of the three relaxation states, (P+HA−)i (where i = 1, 2, 3), charge recombination may occur directly and in an energetically downhill fashion with a molecular rate constant kdiri (or its reciprocal, a molecular lifetime τdiri), or indirectly through a thermally activated pathway involving P+BA− with a molecular rate constant kTi (or a molecular lifetime τTi). The protein relaxation rate constants, k12 and k23, co-determine the overall charge recombination kinetics, since they limit the lifetime of each of the relaxation states. Thus, protein relaxation makes the state P+BA− progressively less and less accessible.
3.3 Transient absorption kinetics
Figure 3 shows the kinetics of charge recombination in WT, FLY and FLA membrane-bound RCs, measured as transient absorption signals at 690 nm. At this wavelength there is strong positive absorption by the HA− anion and a smaller positive absorption by P+, both in WT RCs [32, 39] and in the mutant RCs (our unpublished results). At a longer time scale after excitation there is also a small component, non-decaying on a 200-ns time scale, which for the WT RC varies from 4% of the initial signal at room temperature (RT) to 7.5% at 78 K. In the FLY and FLA RCs this component varies, respectively, from 0 to 4% or oscillates at about 0%. This small component is assigned to the triplet state of the primary electron donor, 3P, which subsequently is transferred to the nearby carotenoid molecule [40] and then decays within ~ 4 µs [41]. The low signal from triplet state correlates with the results of our previous room temperature broad-band transient absorption studies on WT, FLA and FLA RCs showing the triplet yield of only 14% for WT and 5–10% for the two mutants under study [41].

Transient absorption kinetics of charge recombination in WT (A) and mutant, FLY (B) and FLA (C) membrane-bound RCs measured at 690 nm at a range of temperatures between 78 and 298 K. Excitation was at 532 nm. Insets: zoom of first 10 ns of transient absorption traces and fitted three-exponential decay curves
The charge recombination kinetics in all the three samples, at temperatures between 78 and 298 K, were fitted with a three-exponential function plus an offset (Eq. S1). To obtain a consistent picture of the temperature dependence of the three lifetimes, the preexponential factors associated with these lifetimes were assumed to be temperature-independent and therefore, in the fitting procedure, they were shared between the kinetics measured at different temperatures. The lifetimes resulting from the three-exponential fitting (in the form of their reciprocals, rate constants k1–k3) are presented as black squares in Fig. 4.

Temperature dependence of the experimental (in black) and model (in red) rate constants associated with three phases of charge recombination in the three RCs under study. A1–A3 are preexponential factors of the respective exponential phases (see Eq. S3) at room temperature (Panels A-C – WT; panels D-F – FLY; panels G-J – FLA)
Due to the limitation of the temporal resolution of the instrument, for all the RCs the fastest rate constant (k1) of the values greater than (1 ns)−1, was characterized by the most irregular dependence on temperature (Fig. 4A, D, G). In the case of the WT RC it was difficult to conclude that there was any systematic dependence on temperature (Fig. 4A). This is the reason why, in a previous paper, the initial detected P+HA− state, (P+HA−)1, was assumed to be isoenergetic with the state P+BA− and, consequently, the fastest charge recombination was approximated by a constant, temperature-independent value [9]. However, for the two mutant RCs the rate constant k1, although irregularly, seemed to show a general increase with increasing temperature (Fig. 4D and G). For all three RCs the dependence on temperature was very clear for the two slower charge recombination rate constants, k2 and k3 (Fig. 4). Depending on the sample, k2 decreased with decreasing temperature from ~ (2 ns)−1 (FLA at RT) to ~ (12 ns)−1 (WT and FLY at 78 K). Similarly, the rate constant k3 decreased from ~ (9 ns)−1 (FLA at RT) to ~ (40 ns)−1 (WT and FLY at 78 K).
The overall charge recombination dynamics depend not only on the lifetimes but also on the associated amplitudes (pre-exponential factors), Ai. In WT RCs the values of A1-A3 at RT were 0.19, 0.41 and 0.36, respectively (Fig. 4A–C), and were not significantly different at lower temperatures (see Eqs. S1–S3). The values of these amplitudes were significantly different in the mutants: A1–A3 were, respectively, 0.45, 0.43 and 0.12 for FLY, and 0.51, 0.31 and 0.18 for FLA (Fig. 4D–J). A dominating contribution of the fastest, sub-nanosecond phase was the major reason for overall faster charge recombination in the mutant RCs compared to the WT protein.
3.4 Protein dynamics and triplet formation parameters
The parameters of the three-exponential fits, the lifetimes τi (or rate constants ki) and amplitudes Ai, were sufficient to estimate the rate constants depicting the two protein relaxation steps, k12 and k23, and that for triplet formation, kt. The mathematical formulae for these rate constants are given in the Supplementary Information (Eqs. S27-S29).
Variations of the estimated rate constants k12, k23 and kt with temperature are presented in Fig. 5D–F, respectively. The values of k12 oscillated between ~ (2 ns)−1 and ~ (1 ns)−1 for all the samples with no clear systematic temperature dependence (Fig. 5D). The relatively noisy character of these curves comes from the similar character of the k1(T) curves shown in Fig. 4A, D, G. Similar temperature independence of this fast protein relaxation step was reported previously [9, 25]. The temperature dependence of the second protein relaxation step, k23, was more systematic. In the case of WT RCs, it was weakly temperature-dependent and decreased from (20 ns)−1 at RT to (30 ns)−1 at 78 K (compare Fig. 6A), consistent with a previous report [25]. In the case of the mutant RCs, the k23 rate constant was more strongly dependent on temperature. This step of protein relaxation slowed down ~ threefold for both RCs (Fig. 5E, compare also Fig. 6B, C). In the FLY mutant, k23 decreased from (17 ns)−1 at RT to (53 ns)−1 at 78 K. In the FLA mutant, the protein relaxed ~ 3 times faster than in WT at RT (6.2 ns), and ~ 2 times faster at 78 K (17 ns). We speculate that the faster dynamics in the FLA RC may be related to the empty space in the vicinity of BA due to replacement of bulky tyrosine with the small alanine residue in this mutant. Alternatively, the space in the vicinity of BA may be filled with water molecule(s) which may also affect the local dynamics of the protein.

Temperature dependences of the nine molecular lifetime parameters estimated for the model shown in Fig. 2 (Panels A-C – kT1(T)-kT3(T); panels D-F – k12(T), k23(T), kt(T); panels G-J – kdir1(T)-kdir3(T), respectively)

Values of parameters of the model shown in Fig. 2 estimated for the RCs (A – WT, B – FLY, C – FLA)
The low yield of triplet formation and its poor detection at 690 nm in the mutants did not allow solid conclusions to be drawn on the obtained values of the kt rate constant, which exceeded (100 ns)−1 at all temperatures even for WT RCs that show a higher yield of triplet formation (Figs. 5F and 6) [41]. We may speculate that the small value of kt may be related with the reduced state of QA and specific mutation effects.
3.5 Free energy gaps, direct P+ H A − recombination and indirect P + H A − recombination
To estimate the free energy gaps and direct P+HA− recombination rate constants shown in Fig. 2, the experimentally measured temperature dependencies of the rate constants k1–k3 were fitted with the following functions originating from the model presented in Fig. 2 (see SI for details):
The rates kPB, \(k_{{{\text{dir1}}}}\), \(k_{{12}}\), \(k_{{23}}\), and \(k_{t}\), as well as the free energy gaps \(\Delta G_{i}\), are the parameters of the fits—four parameters for each of the curves (2)–(4). To simplify the analyses and make their results from different samples more comparable, in the first approach, the molecular rate constant for P+BA− → PBA charge recombination, kPB, was kept constant for all the temperatures and all the samples, and equal to (0.2 ns)−1 in line with earlier reports [42, 43]. Moreover, the protein dynamics and the triplet formation rate constants (\(k_{{12}}\), \(k_{{23}}\), and \(k_{t}\)) were estimated independently, as described above. Thus, only two parameters were optimized to obtain good fit for each experimental curve, \(\Delta G_{i}\) and \(k_{{{\text{dir}}i}}\). It turned out that good fits (Fig. 4) could have been obtained either with a single value of \(k_{{{\text{dir}}i}}\) for all the temperatures for a particular i-th state of an individual sample (WT, FLY) or with a value of \(k_{{{\text{dir}}i}}\) slightly changing with temperature in a linear way (FLY) (Fig. 5G–J). Thus, the shapes of all of the temperature dependencies shown in Fig. 4 were properly modelled using essentially only one parameter (\(\Delta G_{i}\)) for each of the curves. On the other hand, the rate constant \(k_{{{\text{dir}}i}}\) was mostly constrained by the low temperature limits of the ki(T) dependencies.
Fixing the value of kPB = (0.2 ns)−1 for all the samples was somewhat arbitrary. Moreover, the amino acid substitutions in the FLY and FLA mutants, modifying interactions between BA and the protein, could have changed the recombination rate described by kPB. Therefore, we also performed fits of the data on the mutant RCs with other values of kPB. This parameter turned out to be strongly coupled with the \(\Delta G_{i}\) parameter, such that an increase of one of them had to be compensated by an increase of the other one to get a good fit. Some fits obtained for different kPB values, ranging from (0.5 ns)−1 to (0.05 ns)−1, were of comparable quality to those presented in Fig. 4. In the case of the FLY mutant, acceleration of the P+BA− → PBA charge recombination from 0.2 to 0.05 ns even slightly improved the quality of the fits (Fig. S1 B, C).
Among the \(\Delta G_{i}\) parameters shown in Fig. 4 fitted with kPB = (0.2 ns)−1, the least reliable were the values for \(\Delta G_{1}\) (panels A, D, G). This is because of the large errors of the k1 parameter caused by the limited temporal resolution of the transient absorption measurement. Thus, the obtained \(\Delta G_{1}\) values should be regarded as approximate ones, with a ~ ± 10 meV error. On the other hand, the error associated with \(\Delta G_{2}\) and \(\Delta G_{3}\), for the fixed kPB value, was not larger than ~ ± 2 meV. The fitted \(\Delta G_{i}\) values are presented in each of the panels and, in a graphical way, in Fig. 6. One can see that in line with the predictions about the effect of mutations, the free energy levels of all the three states (P+HA−)i in the mutant RCs are shifted up, closer to P+BA−, compared to the WT RC (100–125–133 meV for the WT compared to 75–91–130 meV for FLY and 58–89–105 meV for FLA).
According to Eq. (5), the free energy gaps \(\Delta G_{i}\) are unambiguously related with the indirect, thermally activated rate constants kTi in an exponential way (Fig. 5A–C).
3.6 Competition between indirect recombination, direct recombination and protein relaxation
According to Eqs. (2)–(5), each of the three charge recombination lifetimes resolved in the transient absorption experiment is a result of three competing processes: indirect recombination (kTi), direct recombination (kdiri) and protein relaxation/triplet formation (k12, k23, kt). These three processes contribute differently for each relaxation state, (P+HA−)i, of a given sample, and for each individual temperature. Figure 6 presents the lifetimes associated with the three competing processes for each relaxation state at two extreme temperatures: RT (red) and 78 K (blue). With this figure, one can analyze which process predominates for a particular state and temperature. A more comprehensive picture showing this competition at all temperatures is presented in Fig. 7. This figure presents the same data as in Fig. 5 but in a different manner. In each panel, there are collected rate constants of the three competing processes related to one relaxation state of one sample. Thus, the sum of the three curves in an individual panel is equal to the fit curve shown in the corresponding panel in Fig. 4.

Contributions from activated recombination (kTi), direct recombination (kdiri) and protein relaxation/triplet formation (k12, k23, kt) components in each of the charge recombination phase (kPHi comp.) in the three RCs. The sum of the three contributions from each panel gives the total fit curve in the corresponding panel in Fig. 4 (Panels A-C – WT; panels D-F – FLY; panels G-J – FLA)
Analysis of Fig. 7 leads to the conclusion that in the state i = 1, protein relaxation clearly predominates at all temperatures in the WT RC, while in the mutants direct recombination is roughly as efficient as the protein relaxation. In the state i = 2, direct charge recombination predominates at most temperatures in the WT and FLA RCs, whereas in the FLY RCs at higher temperatures the depopulation of this state is dominated by the thermally activated pathway. In the state i = 3, for all the samples the thermally activated recombination predominates at higher temperatures, but at lower temperatures direct recombination prevails.
It is also interesting to note that the temperature dependence of the three experimentally resolved phases (Fig. 4) is dominated in all samples by the temperature dependence of the rate constant of thermally activated pathway (Fig. 7), although, particularly in the FLY RCs, the contributions of the temperature dependence of protein relaxation and even direct recombination are substantial.
Finally, it is worth noting that that to observe protein relaxation (protein dynamics) while measuring P+HA− recombination, the characteristic time constant of this relaxation has to be comparable with the time constants of the two charge recombination routes for P+HA− (see Fig. 6). If the protein relaxation was much slower than this the recombination would occur from the unrelaxed P+HA− state only, and no conversion of one P+HA− relaxation state to another one (caused by protein relaxation) would be observed. Conversely, if the protein relaxation was much faster than the recombination from the unrelaxed P+HA− state, the recombination from this unrelaxed state would not be observed.
4 Conclusions
Our modelling shows that the hypothetical introduction of a hydrogen bond to BChl BA decreases the free energy gap between the states P+BA− and P+HA− in all the three protein relaxation states modelled on the basis of the transient absorption experiment. The two-step protein dynamics were modelled with the faster phase, of 1–2 ns lifetime, being independent of temperature, and the slower one, of 6–50 ns lifetime, showing 1.5–threefold slowdown at 78 K relative to RT. This temperature-dependence was more pronounced in the mutant RCs than in the WT RCs. The slower step of RT protein relaxation was ~ 3 times faster in the FLA mutant (6.2 ns) than in the WT RC (20 ns). This effect was tentatively assigned to appearance of the empty space, possibly filled with water molecules, in the vicinity of BChl BA caused by the mutation. Our modelling indicates significant differences in the values of direct charge recombination rate constants in the three relaxation states of the RCs, ranging from ~ (1–5) ns in the first relaxation state to ~ (30–60) ns in the third relaxation state of the protein. We conclude that protein relaxation controls the charge recombination lifetime via both thermally-activated and direct charge recombination pathways. Finally, the acceleration of the charge recombination in the mutants is assigned to a modification of the interaction between BA and the protein and not to modification of the protein dynamics.
References
Warshel, A., Chu, Z. T., & Parson, W. W. (1989). Dispersed polaron simulations of electron transfer in photosynthetic reaction centers. Science, 246, 112–116. https://doi.org/10.1126/science.2675313
Gehlen, J. N., Marchi, M., & Chandler, D. (1994). Dynamics affecting the primary charge transfer in photosynthesis. Science, 263, 499–502. https://doi.org/10.1126/science.263.5146.499
Graige, M. S., Feher, G., & Okamura, M. Y. (1998). Conformational gating of the electron transfer reaction QA-QB/QAQB-. in bacterial reaction centers of Rhodobacter sphaeroides determined by a driving force assay. Proceedings of the National Academy of Sciences of the United States of America, 95, 11679–11684. https://doi.org/10.1073/pnas.95.20.11679
McMahon, B. H., Muller, J. D., & Nienhaus, G. U. (1998). Electron transfer and protein dynamics in the photosynthetic reaction center. Biophysical Journal, 74, 2567–2587. https://doi.org/10.1016/S0006-3495(98)77964-0
Parson, W. W., & Warshel, A. (2004). Dependence of photosynthetic electron-transfer kinetics on temperature and energy in a density-matrix model. The Journal of Physical Chemistry B, 108, 10474–10483. https://doi.org/10.1021/jp0495904
Wang, H., Lin, S., Allen, J. P., Williams, J. A. C., Blankert, S., Laser, C., & Woodbury, N. W. (2007). Protein dynamics control the kinetics of initial electron transfer in photosynthesis. Science, 316(5825), 747–750. https://doi.org/10.1126/science.1140030
Pawlowicz, N. P., van Grondelle, R., van Stokkum, I. H. M., Jones, M. R., Breton, J., & Groot, M. L. (2008). Identification of the first steps in charge separation in bacterial photosynthetic reaction centers of rhodobacter sphaeroides by ultrafast mid-infrared spectroscopy: electron transfer and protein dynamics. Biophysical Journal, 95, 1268–1284. https://doi.org/10.1529/biophysj.108.130880
Guo, Z., Woodbury, N. W., Pan, J., & Lin, S. (2012). Protein dielectric environment modulates the electron-transfer pathway in photosynthetic reaction centers. Biophysical Journal, 103, 1979–1988. https://doi.org/10.1016/j.bpj.2012.09.027
Gibasiewicz, K., Pajzderska, M., Dobek, A., Karolczak, J., Burdziński, G., Brettel, K., & Jones, M. R. (2013). Analysis of the temperature dependence of P+HA- charge recombination in the Rhodobacter sphaeroides reaction center suggests nanosecond temperature independent protein relaxation. Physical Chemistry Chemical Physics: PCCP, 15, 16321–16333. https://doi.org/10.1039/C3CP44187C
Frauenfelder, H. (2011). The physics of proteins: An introduction to biological physics and molecular biophysics. Springer-Verlag.
Blankenship, R. E., Madigan, M. T., & Bauer, C. E. (Eds.). (1995). Anoxygenic photosynthetic bacteria. Kluwer.
Hunter, C. N., Daldal, F., Thurnauer, M. C., & Beatty, J. T. (Eds.). (2009). The purple phototrophic bacteria. Springer.
Wydrzynski, T. J., & Satoh, K. (Eds.). (2005). Photosystem II: The light-driven water:Plastoquinone oxidoreductase. Springer.
Golbeck, J., & van der Est, A. (Eds.). (2014). Photosynthetic reaction centers in the biophysics of photosynthesis. Springer.
Allen, J. P., Feher, G., Yeates, T. O., Komiya, H., & Rees, D. C. (1987). Structure of the reaction center from Rhodobacter sphaeroides R-26: The cofactors. Proceedings of the National Academy of Sciences of the United States of America, 84(16), 5730–5734. https://doi.org/10.1073/pnas.84.16.5730
Zinth, W., & Wachtveitl, J. (2005). The first picoseconds in bacterial photosynthesis—ultrafast electron transfer for the efficient conversion of light energy. ChemPhysChem, 6, 871–880. https://doi.org/10.1002/cphc.200400458
Parson, W. W., & Warshel, A. (2009). Mechanism of charge separation in purple bacterial reaction centers. In C. N. Hunter, F. Daldal, M. C. Thurnauer, & J. T. Beatty (Eds.), The purple phototropic bacteria. Springer Netherlands.
Woodbury, N. W. T., & Allen, J. P. (1995). Electron transfer in purple nonsulfur bacteria. In R. E. Blankenship, M. T. Madigan, & C. E. Bauer (Eds.), Anoxygenic photosynthetic bacteria (p. 527). Kluwer Academic Publishers.
Okamura, M. Y., Isaacson, R. A., & Feher, G. (1975). Primary acceptor in bacterial photosynthesis: obligatory role of ubiquinone in photoactive reaction centers of Rhodopseudomonas spheroides. Proceedings of the National Academy of Sciences of the United States of America, 79, 3491–3495. https://doi.org/10.1073/pnas.72.9.3491
Michel, H., Epp, O., & Deisenhofer, J. (1986). Pigment-protein interactions in the photosynthetic reaction centre from Rhodopseudomonas viridis. EMBO Journal, 5, 2445–2451.
Volk, M., Ogrodnik, A., & Michel-Beyerle, M. E. (1995). The recombination dynamics of the radical pair P+H- in external magnetic and electric fields. In R. E. Blankenship, M. T. Madigan, & C. E. Bauer (Eds.), Anoxygenic photosynthetic bacteria. Kluwer Academic Publishers.
Hartwich, G., Lossau, H., Michel-Beyerle, M. E., & Ogrodnik, A. (1998). Nonexponential fluorescence decay in reaction centers of Rhodobacter sphaeroides reflecting dispersive charge separation up to 1 ns. The Journal of Physical Chemistry B, 102, 3815–3820. https://doi.org/10.1021/jp973472h
Woodbury, N. W. T., & Parson, W. W. (1984). Nanosecond fluorescence from islolated photosynthetic reaction centers of Rhodopseudomonas sphaeroides. Biochimica et Biophysica Acta, 767(2), 345–361. https://doi.org/10.1016/0005-2728(84)90205-6
Woodbury, N. W., Parson, W. W., Gunner, M. R., Prince, R. C., & Dutton, P. L. (1986). Radical-pair energetics and decay mechanisms in reaction centers containing anthraquinones, naphthoquinones or benzoquinones in place of ubiquinone. Biochimica et Biophysica Acta, 851, 6–22. https://doi.org/10.1016/0005-2728(86)90243-4
Gibasiewicz, K., Dobek, A., Brettel, K., & Jones, M. R. (2013). Analysis of the kinetics of P+HA− recombination in membrane-embedded wild-type and mutant Rhodobacter sphaeroides reaction centers between 298 and 77 K indicates that the adjacent negatively charged QA ubiquinone modulates the free energy of P+HA− and may influence the rate of the protein dielectric response. The Journal of Physical Chemistry B, 117, 11112–11123. https://doi.org/10.1021/jp4011235
Tang, C. K., Williams, J. C., Taguchi, A. K. W., Allen, J. P., & Woodbury, N. W. (1999). P+HA− charge recombination reaction rate constant in rhodobacter sphaeroides reaction centers is independent of the P/P+ midpoint potential. Biochemistry, 38, 8794–8799. https://doi.org/10.1021/bi990346q
Gibasiewicz, K., Pajzderska, M., Potter, J. A., Fyfe, P. K., Dobek, A., Brettel, K., & Jones, M. R. (2011). Mechanism of recombination of the P+HA− radical pair in mutant Rhodobacter sphaeroides reaction centers with modified free energy gaps between P+BA− and P+HA−. The Journal of Physical Chemistry B, 115, 13037–13050. https://doi.org/10.1021/jp206462g
Gibasiewicz, K., et al. (2016). Weak temperature dependence of P+HA− recombination in mutant Rhodobacter sphaeroides reaction centers. Photosynthesis Research, 128, 243–258. https://doi.org/10.1007/s11120-016-0239-9
Jones, M. R., Fowler, G. J. S., Gibson, L. C. D., Grief, G. G., Olsen, J. D., Crielaard, W., & Hunter, C. N. (1992). Mutants of Rhodobacter sphaeroides lacking one or more pigment-protein complexes and complementation with reaction-centre, LH1, and LH2 genes. Molecular Microbiology, 6, 1173–1184. https://doi.org/10.1111/j.1365-2958.1992.tb01556.x
Jones, M. R., Heer-Dawson, M., Mattioli, T. A., Hunter, C. N., & Robert, B. (1994). Site-specific mutagenesis of the reaction centre from Rhodobacter sphaeroides studied by Fourier transform Raman spectroscopy: mutations at tyrosine M210 do not affect the electronic structure of the primary donor. FEBS Letters, 339, 18–24. https://doi.org/10.1016/0014-5793(94)80376-5
Gibasiewicz, K., & Pajzderska, M. (2008). Primary radical pair P+H− lifetime in Rhodobacter sphaeroides with blocked electron transfer to QA effect of o-phenanthroline. Journal of Physical Chemistry B, 112(6), 1858–1865. https://doi.org/10.1021/jp075184j
Gibasiewicz, K., Pajzderska, M., Ziółek, M., Karolczak, J., & Dobek, A. (2009). Internal electrostatic control of the primary charge separation and recombination in reaction centers from Rhodobacter sphaeroides revealed by femtosecond transient absorption. Journal of Physical Chemistry B, 113(31), 11023–11031. https://doi.org/10.1021/jp811234q
Shuvalov, V. A., & Parson, W. W. (1981). Energies and kinetics of radical pairs involving bacteriochlorophyll and bacteriopheophytin in bacterial reaction centers. Proceedings of the National Academy of Sciences of the United States of America, 78(2), 957–961. https://doi.org/10.1073/pnas.78.2.957
Schenck, C. C., Blankenship, R. E., & Parson, W. W. (1982). Radical-pair decay kinetics, triplet yields, and delayed fl uorescence from bacterial reaction centers. Biochimica et Biophysica Acta, 680, 44–59. https://doi.org/10.1016/0005-2728(82)90315-2
Byrdin, M., Thiagarajan, V., Villette, S., Espagne, A., & Brettel, K. (2009). Use of ruthenium dyes for subnanosecond detector fidelity testing in real time transient absorption. Review of Scientific Instruments, 80, 043102. https://doi.org/10.1063/1.3117208
Schenkl, S., Spörlein, S., Müh, F., Witt, H., Lubitz, W., Zinth, W., & Wachtveitl, J. (2002). Selective perturbation of the second electron transfer step in mutant bacterial reaction centers. Biochimica et Biophysica Acta, 1554, 36–47. https://doi.org/10.1016/S0005-2728(02)00211-6
Mandal, S., Espiritu, E., Akram, N., Lin, S., Williams, J. C., & Woodbury, N. W. (2018). Influence of the electrochemical properties of the bacteriochlorophyll dimer on triplet energy-transfer dynamics in bacterial reaction centers. The Journal of Physical Chemistry B, 122(44), 10097–10107. https://doi.org/10.1021/acs.jpcb.8b07985
Lin, X., Murchison, H. A., Nagarajan, V., Parson, W. W., Allen, J. P., & Williams, J. C. (1994). Specific alteration of the oxidation potential of the electron donor in reaction centers from Rhodobacter sphaeroides. Proceedings of the National Academy of Sciences of the United States of America, 91, 10265–10269. https://doi.org/10.1073/pnas.91.22.10265
Fajer, J., Brune, D. C., Davis, M. S., Forman, M., & Spaulding, L. D. (1975). Primary charge separation in bacterial photosynthesis: Oxidized chlorophylls and reduced pheophytin. Proceedings of the National academy of Sciences of the United States of America, 72(12), 4956–4960. https://doi.org/10.1073/pnas.72.12.4956
Mandal, S., et al. (2017). Mechanism of triplet energy transfer in photosynthetic bacterial reaction centers. The Journal of Physical Chemistry B, 121, 6499–6510. https://doi.org/10.1021/acs.jpcb.7b03373
Białek, R., Burdzinski, G., Jones, M. R., & Gibasiewicz, K. (2016). Bacteriopheophytin triplet state in Rhodobacter sphaeroides reaction centers. Photosynthesis Research, 129, 205–216. https://doi.org/10.1007/s11120-016-0290-6
Heller, B. A., Holten, D., & Kirmaier, C. (1996). Effects of Asp residues near the L-side pigments in bacterial reaction centers. Biochemistry, 35, 15418–15427. https://doi.org/10.1021/bi961362f
Katilius, E., Turanchik, T., Lin, S., Taguchi, A. K. W., & Woodbury, N. W. (1996). B-side electron transfer in a rhodobacter sphaeroides reaction center mutant in which the B-side monomer bacteriochlorophyll is replaced with bacteriopheophytin. The Journal of Physical Chemistry B, 1999(103), 7386–7389. https://doi.org/10.1021/jp991670y
Acknowledgements
K.G. acknowledges financial support from the Polish government (project entitled “Electrostatic control of electron transfer in purple bacteria reaction center” no N N202 127 437). The authors are grateful to Klauss Brettel for the access to the nanosecond transient absorption instrumentation and his support in conducting the experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Pushing the limits of flash photolysis to unravel the secrets of biological electron and proton transfer—a topical issue in honour of Klaus Brettel.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gibasiewicz, K., Pajzderska, M., Białek, R. et al. Temperature dependence of nanosecond charge recombination in mutant Rhodobacter sphaeroides reaction centers: modelling of the protein dynamics. Photochem Photobiol Sci 20, 913–922 (2021). https://doi.org/10.1007/s43630-021-00069-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43630-021-00069-z