
Abstract
Below 5 °C, Portland cement (PC) experiences delayed hydration, slowing strength development, making it unsuitable for winter. Alkali-activated slag (AAS) emerges as a viable alternative with continuous hydration in low-temperature conditions. The effect of the activator nature on the performance of AAS cured at normal temperatures is well known, but further studies are required for low-temperature conditions. This study investigates the synergistic impact of activator modulus (1.2 and 1.5), alkali dosage (5, 7, and 9%), and PC substitution rates (0, 10, and 20%), on low-temperature cured AAS properties. Eighteen mixtures were prepared and cured at 2 °C. Compression and ultrasonic pulse velocity tests were conducted after 7, 28, and 90 days. Fourier transform infrared spectroscopy (FTIR) and scanning electron microscopy analyses were performed to examine the microstructure of the samples. Elevating alkali dosage enhanced early age strength but resulted in a drop in 90-day strength. Simultaneous increases in modulus and PC substitution rate reduced strength due to shrinkage-induced crack formation. Optimal mixture design options included using 10% PC in the 1.2 modulus and omitting PC when the 1.5 modulus was selected. Despite low temperatures, the use of PC significantly accelerated the setting time. Altering modulus and alkali dosage caused a considerable change in the intensity of the peaks in the FTIR spectrum. The findings indicate that AAS shows promise when adjusting the mixture design for temperatures below 5 °C, which are unfavorable for the hydration of PC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Although concreting in cold weather presents its own set of challenges, waiting for suitable weather conditions may not always be a good option due to project deadlines, scheduling constraints, or other logistical considerations [1]. Advancements in concrete technology allow for concrete construction activities even in cold weather conditions, resulting in a substantial reduction in the overall construction period [2]. Moreover, the importance of cold weather concreting technology becomes evident in its capacity to enable prompt repairs of critical infrastructure damaged by natural disasters and severe accidents [3]. The engineering properties of Portland cement (PC) composites can be adversely affected by low curing temperatures [4]. The recommended temperature range for curing PC is between 10 and 20 °C. This range is considered optimal for the hydration process, which is essential for the development of concrete strength. The lowest temperature of 5 °C is specified to ensure that the standard process of PC hydration is maintained [3]. Below this temperature, the hydration reaction is delayed, leading to a slower strength improvement in the concrete [2]. PC is not considered a suitable material for winter conditions without implementing specific precautions [5]. Implementing protective measures like constructing a warm shed and incorporating isolation materials can be efficient and solid ways to ensure normal hydration [6]. However, these complex operations significantly raise project costs [5]. Currently, measures implemented during winter construction can be classified into two distinct groups [7]. The safest approach involves maintaining the concrete at an ideal curing temperature. To ensure optimal resistance to cold weather, it is necessary to heat the raw materials before stirring and casting during winter construction [8]. Methods such as constructing a warm shed and utilizing electric blankets are commonly employed to maintain a suitable ambient temperature until the concrete achieves the desired properties [9]. However, these temperature maintenance methods consume significant energy and result in a substantial amount of CO2 emissions [8]. The alternative approach involves changing binders or adding chemical admixtures to quickly enhance concrete strength. Binders like sulfoaluminate and magnesium cement show promise at low temperatures but are mostly limited to repairs due to cost and scarce raw materials [10]. On the other hand, using antifreeze agents and accelerators is a cheaper and more convenient option. However, these chemical admixtures may lead to strength reduction and durability issues later on [11].
The cement industry generates approximately 4.5 bn tons of CO2 each year, representing 8% of total carbon emissions [12]. Developing a cold climate construction material with self-curing properties would be advantageous for reducing carbon emissions and saving energy [3]. Researchers are increasingly focusing on the development of alkali-activated materials (AAMs) due to their potential for reducing carbon emissions [13]. Commonly used precursors for AAMs include fly ash [14], ground granulated blast-furnace slag (GGBS) [15], metakaolin [16], and natural pozzolans [17]. Alkali-activated slag (AAS) stands out as it can achieve the demanded mechanical properties without heat procedure [18]. AAS concrete can reach compressive strengths of 60 MPa–150 MPa at ambient conditions [19]. Additionally, the alkali activator solution contains ample free ions, significantly lowering its freezing point. In general, it is possible to prepare AAS pastes at low temperatures [3].
Several studies have been conducted to examine the properties of AAS at low temperatures. Xiao et al. [20] performed research to examine the impact of PC clinker on the strength of AAS concrete within the temperature range of 0–20 °C. The findings indicated that the addition of clinker enhanced the initial strength of AAS mortar. However, it had a detrimental impact on the later strength. Zhang et al. [8] carried out a study to explore the impact of PC substitution on the strength of AAS up to day 28. The curing temperatures ranged from − 20 to 20 °C. The findings demonstrated that PC-incorporated AAS binders outperformed PC, especially in terms of strength. Furthermore, the binder containing both GGBS and PC showed significantly superior performance at low temperatures compared to the binder with GGBS alone. In a study by Ju et al. [21], calcium oxide was added to GGBS to enhance the performance of AAS. The substitution of 3% calcium oxide resulted in a 40% increment in strength after a day of curing, and a 5% increase was observed at 28-d. According to Zhang et al.’s [3] report, AAS samples cured at low temperatures exhibited lower strength in comparison to specimens cured at ambient condition. However, they showed a higher rate of strength development.
Earlier research has indicated that the dissolution of precursor particles is primarily influenced by the alkali dosage in the activator [22]. A higher alkali dosage results in an increased presence of OH– ions in the system which leads to faster dissolution of precursor particles and an elevated Si concentration in the pore solution. Furthermore, utilizing a higher activator modulus enhances the availability of Si species in the aqueous phase, resulting in more extensively polymerized rings and planes that can accommodate additional Si–O bridging within the formed gel [23]. Several studies have explored how activator modulus and alkali dosage influence the hydration and strength of AAS cured at ambient temperature. Abubakr et al. [24] stated that an alkali dosage of 6% serves as a critical threshold, indicating a dissolution rate sufficient to achieve adequate strength levels. A further increase in alkali dosage can potentially cause a reduction in strength [25]. The impact of activator modulus on strength is intricate [26]. At a constant alkali dosage, a higher modulus contributes more to silica gel, resulting in increased compressive strength within a specific range. However, beyond this range, elevating the modulus can cause a decrease in strength [27].
Utilizing the alkali dosage around 6% and adjusting the activator modulus between 0.6 and 1.2 might not be suitable for AAS samples cured under low-temperature conditions. Insufficient alkali dosage fails to activate GGBS effectively, which leads to low early strength and prolonged hardening time [28]. Abubakr et al. [24] used a 6% alkali dosage and a 1.2 activator modulus for AAS samples cured under ambient conditions, and they reported that the 3-d compressive strength exceeded 40 MPa. In the preliminary experiment, when an alkali dosage of 6% and a modulus of 1.2 were employed for AAS cured at 2 °C, the samples were unable to be extracted from the mold due to insufficient hardening after 3-d. Increasing the amount of C-(A)-S-H by introducing more Ca2+ into the system is a reasonable approach to improve the strength of AAS cured under low temperatures [29]. The introduction of calcium sources like PC can enhance compressive strength [30]. PC is advantageous not only for elevating calcium content but also for raising the temperature through an exothermic chemical reaction between CaO and water [21]. The current research addresses a significant gap in the literature regarding the production of AAS at low temperatures (2 °C). While previous studies have explored the alkali dosage for AAS cured at normal temperatures, there is limited understanding of the optimal alkali dosage and alkali modulus for AAS production under low-temperature conditions. This study investigates the impact of activator dosage and modulus on AAS performance at low temperatures, considering both partial substitution of PC and the use of blast-furnace slag completely. Unlike previous studies that kept alkali dosage and modulus constant, this research explores how these variables, along with PC substitution, influence AAS properties. By simultaneously investigating the effects of activator dosage, modulus, and PC substitution, this study aims to contribute valuable insights to the field. PC substitution rates were set at 0%, 10%, and 20%. The chosen alkali dosages were 5%, 7%, and 9%, while the activator modulus was established at 1.2 and 1.5. The results indicate that with a well-designed mixture recipe, AAS exhibits satisfactory mechanical performance at a curing temperature of 2 °C without the need for any chemical additives.
2 Experimental
2.1 Raw materials and mixing proportions
42.5 PC and GGBS were used as raw materials. The Blaine surface area of GGBS and PC is 4400 cm2/g and 3600 cm2/g, respectively, and their specific gravities are 2.8 and 3.13. The chemical compositions of GGBS and PC are provided in Table 1. This information was obtained from the corporate suppliers providing the materials. The X-ray diffraction analysis of the GGBS, conducted using the RIGAKU ULTIMA IV device, is depicted in Fig. 1, revealing the presence of an amorphous hump primarily around 30°, indicative of substantial quantities of glass within the sample. An activator blend of Na2SiO3 and NaOH was employed. The Na2SiO3 solution comprises 28% SiO2, 13.5% Na2O, and 58.4% H2O by wt., while the NaOH pellets utilized possess a purity of 99%.

X-ray diffraction pattern of GGBS
The parameters that are kept variable when designing mixtures are as follows: alkali dosage (5, 7, and 9%), activator modulus (1.2 and 1.5), and PC replacement rate (0, 10, and 20%). Table 2 shows the mixture combinations prepared. The alkali dosage was determined by calculating the sodium oxide content within the activator solution. This was accomplished by blending NaOH and Na2SiO3 to achieve the desired alkali dosage for each mixture [24]. The activator modulus indicates to the ratio of SiO2 to Na2O of the activator mixture [28]. Considering the flowability and cohesiveness of the paste, the total water-to-binder ratio was set at 0.34. Each AAS mixture is denoted by a three-number name, where the first indicates the modulus, the second indicates alkali dosage, and the last indicates the PC replacement rate.
2.2 Test methods
Samples were manufactured at room temperature by blending the raw materials with an alkali activator. The preparation of the activator involved the combination of Na2SiO3, NaOH, and water, followed by cooling to room temperature before incorporation. Subsequently, the mixtures were poured into cubic molds measuring 50 × 50 × 50 mm3 and placed in a refrigerator set at 2 °C. The compressive strength testing involved measuring a set of three samples for each mixture using a hydraulic universal testing machine at 7, 28, and 90 days, respectively. The compression test was conducted at a loading rate of 0.9 kN/s using a 200-ton capacity testing device following ASTM C109 [31].
The UPV tests were conducted with a portable non-destructive digital indicating tester (Proceq Pundit Lab), following the ASTM C 597–16 standard procedure [32]. The test employed a set of transducers capable of transmitting and receiving 54 kHz ultrasonic pulses. To aid signal reception, a gel was applied to opposing sides of the sample. The calculation involved determining the transit time of an ultrasonic pulse as it traversed through the sample. Three samples were tested for each mixture, and measurements were taken from two different side surfaces of each sample. The determination of both the initial and final setting times of the cement paste was carried out employing Vicat's needle technique, as prescribed by ASTM C191 [33]. The initial setting time was operationally defined as the interval commencing from the incorporation of the alkali activator until the penetration depth of the 1 mm needle reached a reduction to 25 mm. Subsequently, the final setting time was gauged from the introduction of the activator until the needle left an impression without being severed by the edge, positioned at a 0.5 mm distance behind the tip. Analysis of microstructure was performed through scanning electron microscopy (SEM) and Fourier transform infrared spectroscopy (FTIR) examinations. FTIR spectra of the finely ground samples were taken using a Perkin Elmer Spektrum 100 FTIR spectrometer, with a single reflectance silicon ATR attachment. In preparation for observation under the SEM, the samples underwent a gold coating application to optimize imaging clarity, utilizing the JEOL JSM 6510 instrument for analysis. The standards adhered to during the experiments and the samples utilized in the experiments are delineated in Table 3.
3 Results and discussion
3.1 Compressive strength
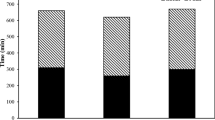
The compressive strength results for samples with activator modulus of 1.2 and 1.5 at 7, 28, and 90 days are depicted in Fig. 2a and b, respectively. At a modulus of 1.2, samples without PC exhibited low compressive strength after 7-d. Although raising the alkali dosage from 5 to 9% resulted in a minor improvement, the highest achieved compressive strength remained relatively low at 4.9 MPa. The modulus had a pronounced impact on the early age strength of AAS when exposed to low-temperature curing conditions. Elevating the modulus to 1.5 resulted in a notable increase in early strength. Furthermore, at the modulus of 1.5, increasing the alkali dosage from 5 to 9% significantly enhanced the early compressive strength. The 7-d compressive strengths were recorded as 10.8 MPa, 14.1 MPa, and 18.6 MPa for alkali dosage of 5%, 7%, and 9%, respectively. The rise in strength observed in PC-free samples with increasing alkali dosage can be attributed to the elevation of the pH value of the activator and the augmentation of the slag's hydration capacity [34]. Abubakr et al. [24] stated that, in the case of AAS cured under ambient conditions, elevating the modulus from 1 to 1.25 and 1.5 resulted in a notable decline in early compressive strength. The results obtained from the current study indicate that a higher modulus was necessary for the geopolymerization process to progress effectively at lower curing temperatures [35]. Substituting both 10% and 20% PC resulted in development of early age compressive strength at a modulus of 1.2. The chemical reaction between PC and water exhibits rapid and intense behavior, emphasizing that the beneficial impact of PC on the strength of AAS mortar is primarily concentrated in the early stages of development [21]. Upon increasing the modulus to 1.5, it was observed that 10% PC substitution enhanced the early strength of samples with 5% and 7% alkali dosage. However, it had a detrimental effect on the samples with 9% alkali dosage. Ju et al. [21] noticed that the early age strength of AAS cured at sub-zero temperatures increased with CaO substitution. In this study, PC substitution in cases of a high activator modulus and alkali dosage showed a negative effect on compressive strength by inducing shrinkage cracks in AAS samples cured at 2 °C.

Compressive strength results of the samples: a modulus of 1.2 and b modulus of 1.5
A substantial rise in strength was obtained with the extension of the curing period to 28-d. In the context of a modulus of 1.2, it was observed that raising the alkali dosage from 5 to 7% resulted in an increase in compressive strength. However, when the alkali dosage was further increased to 9%, the compressive strength declined in comparison to the 7% alkali dosage. Regarding a modulus of 1.5, it was noted that increasing the alkali dosage had an unfavorable impact on the 28-d compressive strength of the PC-containing samples. The compressive strengths of samples containing 10% PC at alkali dosages of 5%, 7%, and 9% were measured as 39.6 MPa, 32.1 MPa, and 21.8 MPa, respectively. At 20% PC replacement rate, the compressive strengths were 45.8, 33.5, and 23.1 MPa, respectively. The incorporation of PC resulted in an enhancement of the 28-d compressive strength of samples with a modulus of 1.2. However, when the modulus was increased to 1.5, it was more advantageous for the samples to be without PC, as it led to better compressive strength. The decline in the 28-d compressive strength of samples containing PC as the alkali dosage increased can be primarily attributed to the formation of cracks resulting from shrinkage [36]. In addition, increasing the alkali dosage inhibits the solubility of Ca from dissolved slag and diminishes the formation of binding phases [37].
Regardless of the variations in alkali dosage, modulus, and PC substitution rate, the compressive strength of all samples exhibited improvement on the 90th day. The most substantial increase in compressive strength was noted in samples with a modulus of 1.2, an alkali dosage of 5%, and the absence of PC. Although this combination delayed the early age hydration of low-temperature cured AAS and caused low compressive strength (4.2 MPa), it resulted in a significant strength improvement (55.5 MPa) at the end of 90-d. Samples incorporating PC at a modulus of 1.2 exhibited higher compressive strength when compared with their counterparts lacking PC. The optimal PC substitution rate for this modulus was identified as 10%. The substitution of PC at a modulus of 1.5 resulted in a reduction of compressive strength across all alkali dosages. The compressive strength of the sample with a 5% alkali dosage and no PC was measured at 69.5 MPa. However, for samples with the same alkali dosage but with 10% and 20% PC replacement, the compressive strengths decreased to 58.4 MPa and 50.7 MPa, respectively. At the end of 90-d, regardless of PC substitution rate and modulus, the best compressive strengths were obtained with a 5% alkali dosage. After 90-d, the highest compressive strength among samples cured at 2 °C was achieved by samples M1.2/N5/P10 (79.2 MPa) and M1.5/N5/P0 (69.5 MPa), respectively. While a 20% PC substitution provided very high early compressive strength for the 1.2 modulus sample, the mixture with 10% PC proved more advantageous at the 90-d mark. In the case of a 1.5 modulus, the use of PC at any rate significantly reduced the 90-d compressive strength. Consequently, the alkali-activated slag/PC binder exhibited a faster reaction rate at low temperatures, which facilitated strength enhancement and established its suitability as a material for cold weather conditions [8].
3.2 Ultrasonic pulse velocity
The UPV measurement results for samples with activator modulus of 1.2 and 1.5 at 7, 28, and 90 days are depicted in Fig. 3a and b, respectively. The samples with a modulus of 1.2 and no PC had the lowest UPV values after 7-d. Even though increasing the alkali dosage from 5 to 9% in this modulus led to an increment in the UPV value, it did not result in a substantial increase in the UPV value, as was obtained in the strength results. The substitution of PC, without altering the modulus, notably improved the 7-d UPV value. The UPV value of the 7-day-old samples with a 10% PC content ranged from 2545 to 2884 m/s. Furthermore, when the PC ratio was increased to 20%, UPV values exceeding 3247 m/s became achievable. The UPV results suggest that PC substitution was crucial for the early mechanical development of samples when cured at low temperatures, with a modulus of 1.2. With an increase in modulus to 1.5, it was observed that increasing the alkali dosage led to an increment in the 7-d UPV of samples without PC substitution. However, in cases where the mixtures contained PC, the increase in dosage had adverse effects on the UPV value.

UPV measurement results of the samples: a modulus of 1.2 and b modulus of 1.5
The correlations between compressive strength and UPV value for different curing periods are depicted in Fig. 4. The UPV values obtained at the end of the 28-d exhibit a strong correlation with the compressive strength results. After this curing period, the coefficient of determination (R2) between UPV and compressive strength was calculated as 0.89. Even though the sample with a modulus of 1.2, an alkali dosage of 5%, and no PC demonstrates a substantial proportional improvement, similar to the compressive strength, its UPV value remains the lowest among all mixtures. The sample with a modulus of 1.2 and a dosage of 5% demonstrated a notable increase in UPV after 90-d, attributed to the sample's increasing compactness resulting from ongoing reactions [38] Conversely, at the 90-d mark, UPV values for all samples an alkali modulus of 1.5 containing 20% PC exhibited a decline compared to the 28-d results. This stark difference sets the UPV results apart from the strength results and contributes to the relatively low correlation between compressive strength and UPV after 90-d [39]. In this study, the incorporation of PC, coupled with high activator modulus and alkali dosage in the paste samples, led to shrinkage-related crack formation as the curing time extended. The divergent sensitivities of compressive strength and UPV measurements to crack development played an important role in the calculated low coefficient of determination [28].

The correlations between compressive strength and UPV for different curing periods
3.3 Setting time
M1.2/N5/P10 and M1.5/5/P0 mixtures, which had the most favorable results in compressive strength and combinations of these mixtures, were chosen for the setting time experiment. The results are given in Fig. 5. Cementitious materials should demonstrate a setting time adequate for thorough mixing and casting without losing plasticity [28]. In accordance with ASTM C595 [40], Portland blast-furnace slag cement is required to have an initial setting time of no less than 45 min, and the final setting time should not surpass 7 h. M1.2/N5/P0 and M1.5/N5/P0 blends with the modulus of 1.2 and 1.5, respectively, without PC, demonstrated ideal hardening times at a curing temperature of 2 °C. However, the mixture with an activator modulus of 1.2, while exhibiting good compressive strength after 90-d, displayed a relatively slow rate of strength gain. Even though increasing PC substitution rate to 10% in the 1.2 modulus yielded good performance in terms of compressive strength, the rapid setting time emerged as a limitation for the M1.2/N5/P10 mixture. For the PC-free mixture, the impact of the increment in modulus on the hardening time was quite limited. Raising the modulus from 1.2 to 1.5 dropped the final setting time from 285 to 280 min. In a study by Chen et al. [41], it was stated that the setting time was prolonged with the increment in silicate modulus, but with a further increase in the modulus, excessive silicate in the activator caused a retarding impact on AAS paste. In other research [35], it was reported that in the case of high alkali dosage, the effect of the increase in silicates in the activator on the setting time was limited. The introduction of PC led to a reduction in the setting time of pastes, a characteristic outcome when PC is incorporated into AAM [42]. When the modulus was 1.2, and 10% and 20% PC were substituted into the mixture, respectively, the final setting time decreased from 285 to 47 min and 18 min, respectively. When the modulus was 1.5, it decreased from 280 to 32 min and 14 min, respectively. When PC reacts with water, it produces colloidal particles of Ca(OH)2, elevating Ca2+ ions and pH due to their high solubility. The increased pH expedites the dissolution of GGBS, releasing Ca2+, Al3+, Si(OH)4, and Al(OH)−4. Saturation of these ions in the solution promotes bond nucleation and growth. Simultaneously, heat facilitates aluminosilicate particle dissolution, collectively reducing the setting time in the mixture [21].

Setting time of the mixtures
3.4 FTIR analysis
Figure 6a presents the FTIR spectra of the unreacted GGBS and Fig. 6b presents the FTIR spectra of the samples that were tested after 28-d of curing, with variations in activator modulus and alkali dosage. Figure 7a and b displays the FTIR spectra acquired at the end of 28 and 90 days for samples coded as M1.2/N5/P10 and M1.5/N5/P0. The rationale for selecting these samples is that the first one demonstrates the highest 90-d compressive strength among those containing PC, while the second one exhibits the highest 90-d compressive strength among mixtures without PC. It is noted that there are similar band locations in the spectra, but variations in intensity can be observed. The bands at 1645 cm−1 correspond to the stretching and bending vibrations of the fundamental O–H bond in water [43]. This observation indicates the presence of hydrated phases in the samples [44]. Traces of carbonates were identified in the spectra within the range of 1405–1424 cm−1. These bands are related to the asymmetric stretching mode of the O–C–O bonds [45]. It suggests that CO2 from the air was adsorbed, leading to the formation of carbonates during the mixing and casting processes of the samples [46]. In the case of the M1.2/N5/P10 sample, the presence of carbonates can also be attributed to the carbonate content within the cement blend fraction [47]. The most prominent vibration associated with the Si–O bond, occurring near 960 cm−1 in the spectrum, corresponds to the formation of C–S–H or C–A–S–H gel. This is typically attributed to the characteristics of AAS or hybrid binders [8]. The presence of a sharp shoulder around 870–875 cm−1 is associated with the carbonation of soluble alkali species in the samples [48]. Intensification of the band between 1405 and 1424 cm−1 and the appearance of the band between 870 and 875 cm−1 in the FTIR spectra indicate the presence of more carbonate in the samples [49]. Regarding unreacted GGBS, the Si–O bond is situated around 900 cm−1; an elevation in wavenumber signifies the establishment of a more highly polymerized Si–O network. It can be deduced that the overall location of the peak representing the hydration products remains consistent regardless of the activator modulus and alkali dosage. However, there is a notable alteration in the intensity of these peaks. The compressive strength of AAS is intimately linked to their micro-structural properties, particularly the type and concentration of gel formed during the activation process. The intensity of the Si–O bond band in FTIR spectra serves as a key indicator of the gel formation within these materials. Comparing samples with different moduli, such as 1.2 and 1.5, reveals significant insights into the micro-structural variations. A higher intensity of the Si–O bond band in samples with a modulus of 1.5 indicates a greater concentration of C–S–H or C–(A)–S–H gel compared to those with a modulus of 1.2. This suggests that the AAS with a modulus of 1.5 possesses a more abundant and densely polymerized gel structure. This enhanced gel structure is associated with superior strength development, particularly evident at 28 days. This underscores the critical role of gel formation in dictating the mechanical properties of AAS [50]. This is significant when these materials are cured at low temperatures. It is observed that the absorption peak, typically occurring around 675 cm−1 when cured at normal temperatures, isn’t present at 2 °C. This suggests that low temperatures are not conducive to the formation of the bridge position in C–S–H [8]. FTIR spectra of the M1.5/N5/P0 sample show limited variation between 28 and 90 days. The Si–O bond at 952 cm−1 did not change significantly in intensity but shifted to a higher wavenumber (963 cm−1). On the other hand, the 90-day-old M1.2/N5/P10 sample exhibited a sharper and stronger Si–O bond compared to the 28-day-old sample, which is consistent with its higher strength at the end of 90-d.

a FTIR spectra of GGBS. b FTIR spectra of the samples with variations in activator modulus and alkali dosage

FTIR spectra of the samples at the end of 28 and 90 days: a M1.2/N5/P10, and b M1.5/N5/P0
3.5 SEM analysis
Figure 8 presents SEM images of M1.5/N5/P0 and M1.2/N5/P10 samples obtained after 28 and 90 days. The selection criteria for these samples are based on the first one demonstrating the highest 90-d strength among those containing PC, while the second one exhibits the highest 90-d strength among mixtures without PC. The 28-day-old sample lacking PC and with an activator modulus of 1.5 exhibits unhydrated GGBS particles with clearly visible sharp edges. In the SEM image of the M1.2/N5/P0 sample containing 10% PC, unhydrated GGBS particles were also observed; however, the quantity of GGBS particles appears to be less. This is related to the increased release of hydration heat and the enhanced concentration of calcium ions, facilitated by the combination of calcium oxide and H2O [21]. As depicted in Fig. 8b–d, the structure becomes denser as the curing time increases. In samples cured at lower temperatures, unreacted particles seem to be more abundant, especially when a CaO source is not utilized. With prolonged curing time and advancing reactions, unhydrated GGBS particles integrate into the reaction, contributing to a more compact structure. It is noteworthy that these samples showed an approximately 50% increment in strength after 90-d compared to the 28-d period [51]. Micro-cracks in the 90-d samples are distinctly observable in both cases, likely arising from drying shrinkage [52]. The substantial temperature difference in the early reaction of the AAS sample with PC caused greater thermal stress, leading to more intense and wider cracks [53]. Figure 9 depicts the change in the microstructure of samples without PC at a constant activator modulus. Micro-cracks, commonly observed in AAS, are evident at both alkali dosages [54]. The escalation of the alkali dosage from 5 to 9% markedly intensified the extent of cracks, primarily attributed to high shrinkage deformation [55]. As evident in the figure, unhydrated GGBS particles with sharp edges are also observed in the M1.5/N9/P0 sample with a 9% alkali dosage. An increase in alkali dosage provides more network-breaking cations, influencing the formation of the Al–O–Al framework. This ultimately accommodates the Si species. Consequently, the reaction products formed at this alkalinity level are expected to envelop the slag particles, impeding further reactions sufficiently [24].

SEM images of the samples at the end of 28 and 90 days: a M1.5/N5/P0 and b M1.2/N5/P10

Change in the microstructure of samples at a constant activator modulus: a M1.5/N5/P0 and b M1.5/N9/P0
4 Conclusion
This study provides valuable insights into the nuanced interactions among key parameters, offering a roadmap for designing alkali-activated slag binders with enhanced performance and versatility at low curing temperature. The findings underscore the intricate balance required for achieving optimal strength development in AAS. Tailoring formulations based on activator modulus, alkali dosage, and PC substitution is crucial for adapting to low curing temperature. The conclusions drawn are as follows:
-
Modifying the activator modulus and alkali dosage significantly influences early age compressive strength. While PC substitution enhanced strength at modulus 1.2, it had varying effects at 1.5, emphasizing the need for tailored formulations.
-
Compressive strength at 28-d benefited from 10% PC substitution at modulus 1.2, but without PC, modulus 1.5 yielded superior results. A 5% alkali dosage was the most ideal option in terms of ultimate compressive strength in both modules. Higher alkali dosage and PC incorporation led to strength reduction, primarily due to crack formation.
-
UPV values demonstrated a strong correlation with strength at 28-d but diverged at later ages due to crack development.
-
Activator modulus variations had limited impact on setting time, emphasizing the role of PC in influencing this crucial property.
-
FTIR spectra highlighted consistent hydration product formation, with a denser gel structure observed at higher modulus (1.5). SEM microstructure analysis showcased the reduction of unhydrated particles with time, contributing to a more compact structure, albeit with observed micro-cracks.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Mei Y, Zhao L, Nong X, Yang T, Zhang X, Wang R, Wang X. Field test study on early strain development law of mass concrete in cold weather. Case Stud Constr Mater. 2022. https://doi.org/10.1016/j.cscm.2022.e01455.
Yu K, Jia M, Yang Y, Liu Y. A clean strategy of concrete curing in cold climate: solar thermal energy storage based on phase change material. Appl Energy. 2023. https://doi.org/10.1016/j.apenergy.2022.120375.
Zhang H, Ai J, Ren Q, Zhu X, He B, Jiang Z. Understanding the strength evolution of alkali-activated slag pastes cured at subzero temperature. Cem Concr Compos. 2023;138: 104993. https://doi.org/10.1016/j.cemconcomp.2023.104993.
Alzaza A, Ohenoja K, Illikainen M. Enhancing the mechanical and durability properties of subzero-cured one-part alkali-activated blast furnace slag mortar by using submicron metallurgical residue as an additive. Cem Concr Compos. 2021;122: 104128. https://doi.org/10.1016/j.cemconcomp.2021.104128.
Ju C, Ye R, Wu Y, Sun P, Liu Y, Yang Y. Effect of alkali cation on performance of alkali-activated slag mortar in cold environments. Separations. 2022. https://doi.org/10.3390/separations9120450.
Liu Y, Sun F, Yu K, Yang Y. Experimental and numerical research on development of synthetic heat storage form incorporating phase change materials to protect concrete in cold weather. Renew Energy. 2020. https://doi.org/10.1016/j.renene.2019.10.142.
Demirboǧa R, Karagöl F, Polat R, Kaygusuz MA. The effects of urea on strength gaining of fresh concrete under the cold weather conditions. Constr Build Mater. 2014. https://doi.org/10.1016/j.conbuildmat.2014.04.008.
Zhang G, Yang H, Ju C, Yang Y. Novel selection of environment-friendly cementitious materials for winter construction: Alkali-activated slag/Portland cement. J Clean Prod. 2020. https://doi.org/10.1016/j.jclepro.2020.120592.
American Concrete Institute (ACI), ACI 306R-10, Am Concr Inst. 2016.
Haque MA, Chen B. Research progresses on magnesium phosphate cement: a review. Constr Build Mater. 2019. https://doi.org/10.1016/j.conbuildmat.2019.03.304.
Liu J, Li Y, Yang Y, Cui Y. Effect of low temperature on hydration performance of the complex binder of silica fume-portland cement. J Wuhan Univ Technol Mater Sci Ed. 2014. https://doi.org/10.1007/s11595-014-0870-2.
Miller SA, Horvath A, Monteiro PJM. Impacts of booming concrete production on water resources worldwide. Nat Sustain. 2018. https://doi.org/10.1038/s41893-017-0009-5.
Dener M. Effect of Ferrochrome slag substitution on high temperature resistance and setting time of alkali-activated slag mortars. Iran J Sci Technol Trans Civ Eng. 2023. https://doi.org/10.1007/s40996-023-01087-w.
Bodur B, Bayraktar OY, Benli A, Kaplan G, Tobbala DE, Tayeh B. Effect of using wastewater from the ready-mixed concrete plant on the performance of one-part alkali-activated GBFS/FA composites: Fresh, mechanical and durability properties, J Build Eng. 2023;107167.
Bayraktar OY, Yakupoglu U, Benli A. Slag/diatomite-based alkali-activated lightweight composites containing waste andesite sand: mechanical, insulating, microstructural and durability properties. Arch Civ Mech Eng. 2023. https://doi.org/10.1007/s43452-023-00774-9.
Karatas M, Dener M, Mohabbi M, Benli A. A study on the compressive strength and microstructure characteristic of alkali-activated metakaolin cement, Matéria (Rio Janeiro). 2019;24.
Balun B, Karataş M. Influence of curing conditions on pumice-based alkali activated composites incorporating Portland cement. J Build Eng. 2021. https://doi.org/10.1016/j.jobe.2021.102605.
Zhang H, Shi X, Wang Q. Effect of curing condition on compressive strength of fly ash geopolymer concrete. ACI Mater J. 2018. https://doi.org/10.14359/51701124.
Bakharev T, Sanjayan JG, Cheng YB. Alkali activation of Australian slag cements. Cem Concr Res. 1999;29:113–20. https://doi.org/10.1016/S0008-8846(98)00170-7.
Xiao L, Mingi X, Guang X, Jingting X. The effect of different conditions on the strength of alkali slag concrete in low temperature, 2017;1007–9467.
Ju C, Liu Y, Jia M, Yu K, Yu Z, Yang Y. Effect of calcium oxide on mechanical properties and microstructure of alkali-activated slag composites at sub-zero temperature. J Build Eng. 2020;32: 101561. https://doi.org/10.1016/j.jobe.2020.101561.
Neupane K. High-strength geopolymer concrete- properties, advantages and challenges. Adv Mater. 2018;7:15. https://doi.org/10.11648/j.am.20180702.11.
Duxson P, Mallicoat SW, Lukey GC, Kriven WM, van Deventer JSJ. The effect of alkali and Si/Al ratio on the development of mechanical properties of metakaolin-based geopolymers. Colloids Surf A Physicochem Eng Asp. 2007. https://doi.org/10.1016/j.colsurfa.2006.05.044.
Abubakr AE, Soliman AM, Diab SH. Effect of activator nature on the impact behaviour of alkali-activated slag mortar. Constr Build Mater. 2020;257: 119531. https://doi.org/10.1016/j.conbuildmat.2020.119531.
Wang SD, Scrivener KL, Pratt PL. Factors affecting the strength of alkali-activated slag. Cem Concr Res. 1994;24:1033–43. https://doi.org/10.1016/0008-8846(94)90026-4.
Duran Atiş C, Bilim C, Çelik Ö, Karahan O. Influence of activator on the strength and drying shrinkage of alkali-activated slag mortar. Constr Build Mater. 2009;23:548–55. https://doi.org/10.1016/j.conbuildmat.2007.10.011.
Shi Z, Shi C, Wan S, Zhang Z. Effects of alkali dosage and silicate modulus on alkali-silica reaction in alkali-activated slag mortars. Cem Concr Res. 2018. https://doi.org/10.1016/j.cemconres.2018.06.005.
Fang S, Lam ESS, Li B, Wu B. Effect of alkali contents, moduli and curing time on engineering properties of alkali activated slag. Constr Build Mater. 2020. https://doi.org/10.1016/j.conbuildmat.2020.118799.
Wongsa A, Boonserm K, Waisurasingha C, Sata V, Chindaprasirt P. Use of municipal solid waste incinerator (MSWI) bottom ash in high calcium fly ash geopolymer matrix. J Clean Prod. 2017. https://doi.org/10.1016/j.jclepro.2017.01.147.
Yang KH, Cho AR, Song JK, Nam SH. Hydration products and strength development of calcium hydroxide-based alkali-activated slag mortars. Constr Build Mater. 2012;29:410–9. https://doi.org/10.1016/j.conbuildmat.2011.10.063.
A. ASTM C109/C109M, Compressive Strength of Hydraulic Cement Mortars ( Using 2-in . or [ 50-mm ] Cube Specimens ) 1, Am Soc Test Mater. 2007.
ASTM C597, Standard Test Method for Pulse Velocity Through Concrete, Am. Soc. Test. Mater. West Conshohocken, PA, USA, 2016. pp. 1–4. https://doi.org/10.1520/C0597-09.
ASTM Standard C191, Standard test methods for Time of Setting of Hydraulic Cement by Vicat Needle, 2019. www.astm.org
Fernández-Jiménez A, Puertas F. Effect of activator mix on the hydration and strength behaviour of alkali-activated slag cements. Adv Cem Res. 2003. https://doi.org/10.1680/adcr.2003.15.3.129.
Al Makhadmeh W, Soliman A. Effect of activator nature on property development of alkali-activated slag binders. J Sustain Cem Mater. 2020;10:240–56. https://doi.org/10.1080/21650373.2020.1833256.
Ye H, Cartwright C, Rajabipour F, Radlińska A. Understanding the drying shrinkage performance of alkali-activated slag mortars. Cem Concr Compos. 2017. https://doi.org/10.1016/j.cemconcomp.2016.11.010.
Bernal SA. Effect of the activator dose on the compressive strength and accelerated carbonation resistance of alkali silicate-activated slag/metakaolin blended materials. Constr Build Mater. 2015. https://doi.org/10.1016/j.conbuildmat.2015.08.013.
Nasr D, Pakshir AH, Ghayour H. The influence of curing conditions and alkaline activator concentration on elevated temperature behavior of alkali activated slag (AAS) mortars. Constr Build Mater. 2018;190:108–19. https://doi.org/10.1016/j.conbuildmat.2018.09.099.
Živica V. Effects of type and dosage of alkaline activator and temperature on the properties of alkali-activated slag mixtures. Constr Build Mater. 2007. https://doi.org/10.1016/j.conbuildmat.2006.07.002.
ASTM International, ASTM C595-17, Standard Specification for Blended Hydraulic Cements, ASTM Int. 2017.
Chen W, Li B, Wang J, Thom N. Effects of alkali dosage and silicate modulus on autogenous shrinkage of alkali-activated slag cement paste. Cem Concr Res. 2021. https://doi.org/10.1016/j.cemconres.2020.106322.
Balun B, Karataş M. Factors affecting the setting times of pumice based alkali-activated hybrid cements. Iran J Sci Technol Trans Civ Eng. 2023. https://doi.org/10.1007/s40996-023-01184-w.
Ibrahim M, Megat Johari MA, Rahman MK, Maslehuddin M. Effect of alkaline activators and binder content on the properties of natural pozzolan-based alkali activated concrete. Constr Build Mater. 2017. https://doi.org/10.1016/j.conbuildmat.2017.04.163.
Bernal SA, Rodríguez ED, De Gutiérrez RM, Provis JL. Performance at high temperature of alkali-activated slag pastes produced with silica fume and rice husk ash based activators. Mater Constr. 2015. https://doi.org/10.3989/mc.2015.03114.
Sakulich AR, Anderson E, Schauer C, Barsoum MW. Mechanical and microstructural characterization of an alkali-activated slag/limestone fine aggregate concrete. Constr Build Mater. 2009;23:2951–7. https://doi.org/10.1016/j.conbuildmat.2009.02.022.
Liu L, Xu Y, Liu H, Xie M, He Y, Shi C. Drying shrinkage and microstructure of alkali-activated slag with different mixing time at low temperatures (−5 to 5 °C). Constr Build Mater. 2022. https://doi.org/10.1016/j.conbuildmat.2022.129529.
Sánchez R, Palacios M, Puertas F. Characteristics and properties of oil-well cements additioned with blast furnace slag. Mater Constr. 2011. https://doi.org/10.3989/mc.2010.54110.
Rovnaník P, Bayer P, Rovnaníková P. Characterization of alkali activated slag paste after exposure to high temperatures. Constr Build Mater. 2013;47:1479–87. https://doi.org/10.1016/j.conbuildmat.2013.06.070.
Ya-min G, Yong-hao F, Duo Y, Yong-fan G, Chen-hui Z. Properties and microstructure of alkali-activated slag cement cured at below- And about-normal temperature. Constr Build Mater. 2015;79:1–8. https://doi.org/10.1016/j.conbuildmat.2014.12.068.
Chen K, Lin WT, Liu W. Effect of NaOH concentration on properties and microstructure of a novel reactive ultra-fine fly ash geopolymer. Adv Powder Technol. 2021;32:2929–39. https://doi.org/10.1016/j.apt.2021.06.008.
El-Hassan H, Shehab E, Al-Sallamin A. Influence of different curing regimes on the performance and microstructure of alkali-activated slag concrete. J Mater Civ Eng. 2018. https://doi.org/10.1061/(asce)mt.1943-5533.0002436.
Puertas F, Torres-Carrasco M. Use of glass waste as an activator in the preparation of alkali-activated slag. Mechanical strength and paste characterisation. Cem Concr Res. 2014. https://doi.org/10.1016/j.cemconres.2013.12.005.
Yang K, Yang Y, Deng J, Xiong D, Zhu X, Li Q, Yang C, Basheer M. Using calcium-rich precursors to improve the early-compressive strength of alkali-activated slag cement at low temperature. Struct Concr. 2022. https://doi.org/10.1002/suco.202100021.
Lima VME, Basto PA, Henrique MA, Almeida YMB, de Melo Neto AA. Optimizing the concentration of Na2O in alkaline activators to improve mechanical properties and reduce costs and CO2 emissions in alkali-activated mixtures. Constr Build Mater. 2022. https://doi.org/10.1016/j.conbuildmat.2022.128185.
Tran TT, Kwon HM. Influence of activator Na2O concentration on residual strengths of alkali-activated slag mortar upon exposure to elevated temperatures. Materials (Basel). 2018. https://doi.org/10.3390/ma11081296.
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK). This research received no external funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dener, M., Altunhan, U. & Benli, A. A green binder for cold weather applications: enhancing mechanical performance of alkali-activated slag through modulus, alkali dosage, and Portland cement substitution. Arch. Civ. Mech. Eng. 24, 176 (2024). https://doi.org/10.1007/s43452-024-00991-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s43452-024-00991-w