
Abstract
Post-approval changes (PACs) to the registered information of authorised medicinal products are introduced routinely worldwide to enhance the robustness and efficiency of the manufacturing process, ensure timely supply in case of increased demand, improve quality control techniques, respond to changes in regulatory requirements and upgrade to state-of-the-art facilities. These are critical to prevent supply disruption and continuously improve existing medicines and vaccines. Due to the complexity of current PAC systems across markets, a change can take 3 to 5 years to approval globally (Hoath et al in BioProcess Int, 2016) thus hindering innovation and increasing the risk of shortages. The key messages are as follows: 1. Industry believes that global regulatory convergence of post-approval changes to Marketing Authorisations (MAs) using science- and risk-based approaches will enable a more efficient management of quality and supply improvements and will facilitate patients’ access to innovative medicines and vaccines of the highest quality. 2. National Regulatory Authorities (NRAs) should establish national or regional guidelines in line with international standards (regarding a risk-based classification of changes and standardisation of requirements) (Guidelines on procedures and data requirements for changes to approved biotherapeutic products, in WHO Technical Report Series, 2018, Guidelines on procedures and data requirements for changes to approved vaccines, in WHO Technical Report Series, 2015), have clear procedural guidance including timelines and implement reliance pathways to accelerate the approval of changes. This paper briefly outlines the challenges for PACs and provides solutions for a more flexible and aligned global system.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Post-approval changes (PACs) to the registered information of authorised medicinal products are introduced routinely worldwide to enhance the robustness and efficiency of the manufacturing process, ensure timely supply in case of increased demand, improve quality control techniques, respond to changes in regulatory requirements and upgrade to state-of-the-art facilities. This continued effort is critical to prevent supply disruption and continuously improve existing medicines and vaccines and is, in many ways, as important as bringing new medicines and vaccines to the market. It is also important when the product development and registration process is accelerated to meet unmet medical needs, thus pushing changes that would typically be made during development into the post-approval setting.
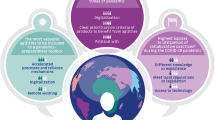
The challenges and consequences of the current global regulatory landscape for PACs are shown in Fig. 1.

Challenges of the current global regulatory landscape for PACs. This figure illustrates the challenges that are currently observed when managing PACs and the subsequent consequences that are experienced by the industry
Tools for Efficient PACs Management
Use of Reliance Practices Should Be Maximised
Collaboration amongst Regulators has been shown to be essential to make better use of available resources and to enable more efficient regulatory pathways leading to fast access of medicines and vaccines to patients [4].
Industry strongly supports the International Coalition of Medicines Regulatory Authorities (ICMRA), International Conference of Drug Regulatory Authorities (ICDRA) and World Health Organisation (WHO) positions on the use of reliance [4,5,6] and its broader application throughout the full lifecycle of a product, including for GMP inspections and lot release [7, 8]. Of note is that if a Certificate of Pharmaceutical Product (CPP) is required by the NRA, then reliance should be applied (without further data requirements), and the review shortened.
Requirements for the Submission of PACs Should Be Converged
Data requirements for PACs should be adapted to the risk level and limited to those that are scientifically justifiable. We propose that data which can be verified during inspection should be eliminated. Industry encourages the adoption of the International Council on Harmonisation (ICH)—Common Technical Document format/content for a harmonised dossier throughout the lifecycle to facilitate the preparation and submission process.
PAC Classification and Timelines Should Be Converged and Common Risk-Based Approaches Adopted
A common regulatory understanding of risk-based approaches and risk-based classification of changes is essential for post-approval changes management as highlighted in the ICH Q12 [9] and World Health Organisation (WHO) guidelines on procedures and data requirements for changes [2, 3]. To provide a further breakdown of specific changes, data requirements and timelines, we recommend alignment with the WHO guidelines [2, 3, 10].
Maximum review periods for changes should be established as recommended in the WHO guidance [3]: major changes, a maximum of 6 months review; moderate changes, a maximum of 3 months review; and minor changes, only requiring a notification to the NRA.
Finally building on the concept of reliance and risk-based approaches, the following should be implemented:
-
Changes with no impact on quality, safety or efficacy should be managed internally within companies’ Pharmaceutical Quality System (PQS) without any reporting to NRAs as per ICH Q12 [11]
-
When already reviewed and approved in a reference country, a major or moderate change should be accelerated in the relying country via a verification or abridged pathway (i.e. a risk-based approach).
The Use of Tools in ICH Q12 (Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management) Should Be Maximised [9]
The implementation of ICH Q12 introduces several important tools and related concepts which we believe can help to streamline technical change management based on process understanding and risk-based approaches [12].
To determine whether to prioritise implementation of ICH Q12 tools, we recommend to first consider the level of maturity of the regulatory framework for PACs that is in place.
For rapid benefit of ICH Q12, implementation of the post-approval change management protocol (PACMP) is recommended. This tool can allow an appropriate lower reporting category to enable the reporting of results after executing the protocol as agreed with the Health Authorities.
Flexible Market Implementation to Ensure Supply Continuity
Pre- and post-change product versions should co-exist in the market during a transition period. Flexible implementation timelines are needed to ensure smooth transition and supply continuity of the post-change product version, whilst approval processes may still be on-going in other regions of the world. This is especially important to accommodate the long lag time needed to supply global products with complex manufacturing processes to reduce potential supply chain disruption.
Emergency Preparedness Considerations
The recent Covid-19 pandemic has led to a re-think in terms of how changes should be managed especially in an emergency [13]. Regulators and the pharmaceutical Industry have made significant efforts to mitigate issues and to find innovative approaches to accelerate PACs. One example is a pilot for collaborative assessment of post-approval change mechanisms [14]. Such collaborative efforts should be continued (Fig. 2).

Solutions to the Current Global Regulatory Landscape for PACs. This figure illustrates the solutions that could help to resolve the challenges currently faced by industry when managing PACs
We believe the following elements are particularly critical for major changes applied to therapeutics or vaccines, such as addition of new manufacturing sites in the context of technology transfer to expand manufacturing capacity in a short timeframe:
-
PAC review periods for emergency applications should be accelerated, with suitable timelines given.
-
Transparent communication and coordinated dialogue amongst stakeholders are critical elements for success.
-
Rather than having multiple reference authorities in an emergency procedure, consideration should be given to limit to the first market where the relevant change package has been submitted and approved by a reference authority.
Conclusion
Regulatory oversight is critical to ensure that high-quality and effective medicines and vaccines are available in a country. Regulators and the pharmaceutical Industry have a collective responsibility to assure an uninterrupted supply of compliant, safe and efficacious medicines and vaccines to patients globally.
Regulators as well as Industry believe that the regulatory process for managing post-approval changes needs to be significantly simplified to facilitate global supply of medicines and vaccines. This can be achieved with consistent, harmonised and clear classifications and adherence to timelines, increased reliance between Regulators and the use of novel regulatory and scientific tools. International collaboration and cooperation towards regulatory convergence are a good way to address the increasing workload challenges of NRAs [15].
We acknowledge that further effort needs to be made by the Industry to reduce the complexity of managing post-approval changes. These measures include advanced planning of changes at start of the lifecycle, more strategic combination of changes as well as transparent communication of supply challenges to Regulators.
We believe action by all stakeholders is required to develop an efficient change management system that contributes to enhancing global public health by providing equitable and timely access to medicines and vaccines.
References
Hoath C, Chang L, Ramalingam Iyer K et al. Post-approval changes for biopharmaceutical drug-substance and drug-product manufacture: regulatory complexity and impact. BioProcess Int; 2016. bioprocessintl.com
Guidelines on procedures and data requirements for changes to approved biotherapeutic products. WHO technical report series no. 1011, Annex 3; 2018. pp. 181–275.
Guidelines on procedures and data requirements for changes to approved vaccines. WHO technical report series no. 993, Annex 4; 2015. pp. 175–259.
Good Reliance Practices in regulatory decision-making: high-level principles and recommendations. WHO Drug Information, vol 34, no. 2, Annex 10; 2020. pp. 201–230.
International Coalition of Medicines Regulatory Authorities (ICMRA). Statement from Global Regulators on the Value of Regulatory Reliance; 2021. Statement from Global MedicinesRegulators on the Value of Regulatory Reliance | International Coalition of Medicines RegulatoryAuthorities (ICMRA)
International Conference of Drug Regulatory Authorities (ICDRA) 20–24 September 2021, Plenary 5: Recommendations of Extraordinary Virtual International Conference of Drug Regulatory Authorities (ICDRA); 2021. WHO efforts to promote reliance
International Federation of Pharmaceutical Manufacturers and Associations (IFPMA). Position paper—Best practices for in-country testing and sample management; 2020. Position Paper - Best Practices for In-Country Testing and Sample Management - IFPMA
International Federation of Pharmaceutical Manufacturers and Associations (IFPMA). Position paper on regulatory reliance; 2019. https://www.ifpma.org/resource-centre/ifpma-position-paper-on-regulatory-reliance/.
International Council on Harmonisation. ICH Q12 (technical and regulatory considerations for pharmaceutical product lifecycle management). Q12_Guideline_Step4_2019_1119.pdf(ich.org)
General guidance on variations to multisource pharmaceutical products. WHO technical report series no. 996, Annex 10; 2016. pp. 348–358.
Ramnarine E, Vinther A, Bruhin K, et al. Effective management of post-approval changes in the pharmaceutical quality system (PQS)—through enhanced science and risk-based approaches industry One-Voice-of-Quality (1VQ) solutions. PDA J Pharm Sci Technol. 2020. https://doi.org/10.5731/pdajpst.2020.011734.
International Council on Harmonisation. ICH Q12 (technical and regulatory considerations for pharmaceutical product lifecycle management) training materials (Module 0 to module 7). https://database.ich.org/sites/default/files/ICH_Q12_IWG_TrainingMaterial_Modules0-7_2021_0611.zip.
International Coalition of Medicines Regulatory Authorities. ICMRA stakeholder workshop on enabling manufacturing capacity during the Covid-19 pandemic. covid-19_manufacturing_capacity_ws_presentation.pdf (icmra.info)
International Coalition of Medicines Regulatory Authorities. Summit and plenary meeting report, 4j) pharmaceutical quality knowledge management system (PQKMS); 2021. p. 6. ICMRA_Summit_and_Plenary_2021_Meeting_Report.pdf
Good regulatory practices in the regulation of medical products. WHO technical report series no. 1033, Annex 11; 2021. pp. 269–304.
Funding
This work is supported by European Federation of Pharmaceutical Industries and Associations (EFPIA), International Federation of Pharmaceutical Manufacturers and Associations (IFPMA) and Vaccines Europe. Whilst there was no specific funding for this work, the authors are employed by the pharmaceutical Industry or related trade associations.
Author information
Authors and Affiliations
Contributions
AD: substantial contributions to the work, drafting, critique and revision, and final approval of the version to be published. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. SA, SA, ASBN, SC, IC-P, TG, AG-M, SM, CM: substantial contributions to the work, drafting, critique and revision.
Corresponding author
Ethics declarations
Conflict of interest
Andrew Deavin is an employee of the GSK group of companies and holds shares in GSK. Sarah Adam, Susanne Ausborn, Ane Sofie Böhm Nielsen, Sonia Cappellini, Isabelle Colmagne-Poulard, Thierry Gastineau, Arturo Gonzalez-Martinez, Sylvie Meillerais, Charlie Mortazavi are employed by the pharmaceutical Industry and may hold shares in the Industry. The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Arturo Gonzalez-Martinez—Retired.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deavin, A., Adam, S., Ausborn, S. et al. Path Forward to Optimise Post-approval Change Management and Facilitate Continuous Supply of Medicines and Vaccines of High Quality Worldwide. Ther Innov Regul Sci 57, 7–11 (2023). https://doi.org/10.1007/s43441-022-00426-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43441-022-00426-9