
Abstract
Breast cancer is the most common female malignancy and the second leading cause of cancer related deaths. It is estimated that about 40% of all cancer in women is hormonally mediated. Both estrogens and androgens play critical roles in the initiation and development of breast cancer. Estrogens influence normal physiological growth, proliferation, and differentiation of breast tissues, as well as the development and progression of breast malignancy. Breast cancer is caused by numerous endo- and exogenous risk factors. The paper presents estrogen metabolism, in particular 17β-estradiol and related hormones. The mechanisms of estrogen carcinogenesis include the participation of estrogen receptors, the genotoxic effect of the estrogen metabolites, and epigenetic processes that are also presented. The role of reactive oxygen species in breast cancer has been described. It called attention to a role of numerous signaling pathways in neoplastic transformation. Chemoprotective agents, besides other phytoestrogens, classical antioxidants, synthetic compounds, and their mechanisms of action have been shown.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common malignancy in women and the second leading cause of cancer-related deaths in developed industrialized countries. In 2020, there were an estimated 2.261 million new cases (11.7% of all sites) and 0.685 million new deaths (6.9% of all sites) from breast cancer affecting women worldwide [1]. In Poland breast cancer is responsible for about a quarter of all cancer cases (in 2010 over 15,700 cases). It was assessed that within the next 15 years the number of new cases will exceed 21, 000 and the risk of breast cancer will be comparable to that observed in Europe (roughly 90/10). In Poland, the mortality level from breast cancer is alarming. The countries with 1.5–2 times higher incidence of breast cancer than Poland have identical levels of mortality [2].
Breast cancer is a complex disease. It is one of most malignant cancers with an increased risk of relapse and metastasis. No single factor is likely to be the cause of it. Many endogenous and exogenous factors have been identified with relevance to breast cancer etiology. Age is the strongest risk factor for this disease. More than 2/3 of all new cases occur after the age of 55 and women older than 65 have a relatively greater risk than 4.0 in comparison with those younger than 65. Many additional risk factors for breast cancer have been identified. Some risk factors are invariable, such as age, BRCA1and BRCA2 gene mutations, family history, and reproductive history. Others are potentially variable, such as high endogenous estrogens, hormone therapy, obesity and alcohol consumption. These endogenous factors include increasing age, menarche age (the younger the age during the first menarche, the higher the risk), and menopause age (the older the age at menopause, the higher the risk) [3, 4].
Moreover, there are exogenous risk factors known as external and environmental factors. Among others these comprise: alcohol consumption, poor diet, contraceptive pill and hormone replacement therapy, overweight and obesity, sleep deprivation, and physical inactivity [5,6,7].
Prolonged lactation and physical activity can decrease the number of ovulatory cycles. A meta-analysis revealed that women could significantly reduce the risk of breast cancer by approximately 25% if they exercise regularly. The interdependence was most evident in post-menopausal women who performed moderate to vigorous exercises throughout their lifetime [8]. Physical activity leads to lower circulating steroid hormone levels, mainly estrogens, and other key hormones, such as leptin and insulin. Physical activity reduces levels of chronic inflammation factors, e.g., C-reactive protein (CRP), which can contribute to breast cancer risk. Physical activity also activates immune system surveillance and decreases oxidative stress [9].
It has been suggested that dietary and environmental factors may be responsible for up to 50% of breast cancer incidence [10]. A low-fat diet and a vegetarian diet [11] decrease levels of sex-steroid hormones. These data suggest that reduction of serum sex-steroid hormone concentrations may diminish the risk of breast cancer.
The normal physiological growth, proliferation and differentiation of breast tissues as well as the development and progression of breast malignancy [12] are mediated by 17β-estradiol (E2) and related compounds upon binding to two members of the nuclear receptor superfamily—estrogen receptor (ER) ERα and ERβ [13].
Upon ligand binding, ERs undergo a conformational change which allows chromatin interaction and the regulation of target genes transcription [14].
Both estrogens and androgens play critical roles in the development of breast cancer. Higher levels of serum estradiol and estrone (E1) were found in post-menopausal women with breast cancer compared to controls [15]. In a case–control study which included postmenopausal women, a positive association with serum E1, androstenedione, and inverse association with sex hormone-binding globulin (SHBG) after adjustment for hormonal variables and body mass index (BMI) were observed [16]. In another study, free E2, albumin-bound estradiol and estrone were all associated with an increased risk of breast cancer after adjustments of BMI in a prospective study [17]. There are data that post-menopausal women with breast cancer also show high serum levels of other sex hormones, i.e. testosterone, dehydro-epiandrosterone sulfate (DHEAS) and androstendione. These hormones are precursors of estrogenes [18]. Estradiol is biosynthesized from androstendione via testosterone or E1 [19]. In a case–control study which included white women with breast cancer and proper controls, the age-adjusted relative risk (RR) of breast cancer in women with the highest level of E2 and free testosterone was 3.6 (95% confidence interval, CI 1.3–10.0) and 3.3 (95% CI 1.1–10.3), respectively, in comparison with those with the lowest concentrations of both hormones [20].
In a prospective case–control study conducted on women with invasive breast cancer and control subjects aged 55–74 years, the serum concentration of unconjugated E2 was strongly associated with the risk of breast cancer (hazard ratio, HR = 2.07; 95% CI 1.19–3.62) [21]. In addition, an enhanced 2-hydroxilation of estrogens, E1 and E2, was associated with reduced risk of postmenopausal breast cancer [22]. There is a suggestion that more extensive 2-hydroxylation of estrogens is associated with lower risk, whereas less extensive methylation of 4-hydroxylated catechols is associated with higher risk of postmenopausal breast cancer [21].
Relatively few studies have reported estrogen and breast cancer risk in pre-menopausal women. One research suggested that mean plasma estradiol concentrations may be higher in pre-menopausal women than in controls but were found not statistically significant in either of these studies [23]. Another study conducted on premenopausal breast cancer women and suitable controls demonstrated that higher urinary estrone (E1) and E2 levels were significantly associated with lower risk of breast cancer. Also, inverse association with parent estrogen metabolites and the parent estrogen metabolite/non-parent estrogen metabolite ratio were observed and it has been suggested that these women are at lower risk [24].
Metabolism of endogenous estrogens
Estrogens are synthesized by the ovaries and partly by the adrenal gland in the woman’s body. Cholesterol is a precursor of all steroid hormones (Fig. 1). The main source of this compound is LDL-cholesterol [25]. Cholesterol is metabolized to the 21-, 19-, and 18-carbon steroid hormones, respectively [26].

Synthesis of sex hormones. CYP11A1 mitochondrial cholesterol side-chain cleavage enzyme, P450scc, 3β-HSD 3β-hydroxysteroid dehydrogenase, CYP17 17α-hydroxylase/17,20-lyase, P450c17, CYP19 aromatase, P450aro, 17β-HSD 17β-hydroxysteroid dehydrogenases
The first step in ovarian steroidogenesis is the passage of cholesterol into the mitochondria. The next step that the biotransformation of cholesterol to pregnenolone, catalyzed by the mitochondrial enzymes. Pregnenolone is a final precursor for everything sex steroid hormones. This compound is biotransformed to progesterone or androstenedione. Androstenedione is transformed to other androgens or estrogens, i.e., testosterone, E1, and E2 [26].
Testosterone is rapidly demethylated at the C-19 position and converted to E2 by the action of steroid aromatase (CYP19) in the peripheral tissues. CYP19 is the rate-limiting enzyme in the conversion of androgens to estrogens. An aromatase mediates in the metabolism of C19 androgen steroids to estrogens. This enzyme and its mRNA have been demonstrated in both the epithelial cells of the terminal ductal lobular units and stromal cells of normal human breast [27]. The high activity of CYP19 may increase breast cancer risk [28] by providing more estrogen for activation to genotoxic metabolites and by stimulating breast epithelial cell mitosis. The blocking CYP19 activity is an important pharmacological method for the treatment of estrogen-dependent breast cancer. The parent estrogens, E1 and E2, are reversibly inter-converted by the 17β-hydroxysteroid dehydrogenase enzyme. In premenopausal women, E2 synthesized in the ovaries is the most important estrogen, whereas in postmenopausal women, E1 present in peripheral tissues is predominant [26]. An important source of estrogens is the peripheral conversion of androgens in the adipose tissue to free estrogens.
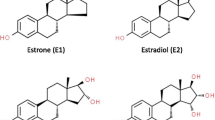
Estradiol and estrone are metabolized by three competitive pathways involving irreversible hydroxylations catalyzed by the NADPH-dependent cytochrome P450 (CYP) enzymes including CYP1A1, CYP1B1, and CYP1A2 (Fig. 2). E1 and E2 are hydroxylated at positions C2, C4 and C16. Therefore, both E1 and E2 are biotransformed to catechol estrogens including 2-hydroxyestrone (2-OHE1), 4-hydroxyestrone (4-OHE1), 2-hydroxyestradiol (2-OHE2), 4-hydroxyestradiol (4-OHE2), 16α-hydroxyestrone (16α-OHE1), or estriol. Estriol is produced by the hydroxylation of E2 or reduction of 16α-OHE1 [26]. Estriol is considered to be a peripheral metabolite and inactivation product of E1 and E2 because it is biologically less active than estrogens.

Metabolic pathway of estrogens. 2-OHE1 2-hydroxyestrone, 4-OHE1 4-hydroxyestrone, 16α-OHE1 16α-hydroxyestrone, 2-OHE2 2-hydroxyestradiol, 4-OHE2 4-hydroxyestradiol, E3 estriol, 2-MeOE1 2-methoxyestrone, 4-MeOE1 4-methoxyestrone, 2-MeOE2 2-methoxyestradiol, 2-OH-3-MeOE2 2-hydroxy-3-methoxyestradiol, 4-MeOE2 4-methoxyestradiol, 17β-HSD 17β-hydroxysteroid dehydrogenases, COMT catechol–O–methyltransferase, GST glutathione S-transferase, CYP cytochrome P450, 1N3-Ade 1–N3–Adenine, 1-N7-Gua 1–N7–Guanine
Catechol estrogens are methylated by the catechol–O–methyltransferase (COMT) to 2-, 3- and 4-methoxyestrogens. From hydroxyestradiols COMT generates two products, 2-methoxyestradiol (2-MeOE2) and 2–hydroxy–3–methoxyestradiol (2–OH–3–MeOE2), from 2-OHE2, but only one product, 4-methoxyestradiol (4-MeOE2), from 4-OHE2 [29]. The O-methylation of catechol estrogen further blocks their two-electron oxidation to semiquinones (SQ) and quinones (Q). Parent estrogens, catechol estrogens, and methoxyestrogens can be also conjugated with glucuronic acid, sulfate, and glutathione by UDP-glucuronosyltransferases, sulfotransferases (SULTs), and glutathione S-transferase P1 (GSTP1), respectively. These enzymes are expressed in breast tissue. SULTs inactivate estrogens because the introduction of the sulfate group prevents the binding of the steroid to its receptor and reduces its mitogenic effects. GSTP1 yields two conjugates, 2–OHE2–1–SG and 2–OHE2–4–SG, from the corresponding quinone 2–hydroxyestradiol–quinone and one conjugate, 4–OHE2–2–SG, from 4–hydroxyestradiol–quinone [29]. Conjugation is a detoxication process by which estrogens either become water soluble and are excreted in the urine or feces, or turn into a more lipophilic moiety with longer half-lives [30].
When catechol estrogen biosynthesis is increased and/or COMT and other conjugated enzymes activity is decreased, the catechol estrogens are easily oxidized to reactive SQ and Q. These electrophilic products, E1/E2–3,4–quinones (E1/E2–3,4–Q), react with DNA to form the depurinating adducts 4–OHE1/E2–1–N3Adenine (4–OHE1/E2–1–N3Ade) and 4–OHE1/E2–1–N7Guanine (4–OHE1/E2–1–N7Gua), which constitute over 99% of the total DNA adducts formed [31, 32]. These adducts are rapidly lost from DNA by cleavage of the glycosyl bond. The apurinic sites, as a result of oxygenation reaction, generate the mutations that may lead to the initiation of cancer [33, 34].
The 2-hydroxylation process is a major metabolic pathway compared to the 4- and 16-hydroxylation pathways. The CYP1A1/2 and CYP3A4 catalyze C2 hydroxylation of parent hormones to their respective catechol products. The catechol estrogens possess a low binding affinity for the ER [35]. These metabolites show lower hormonal potency when compared to E2, and both non-estrogenic and anti-estrogenic activities. There is some evidence that 2-OHE1 and 2-OHE2 inhibit cell growth and proliferation [36]. In addition, 2-hydroxyestrogens have been associated with normal cell differentiation and apoptosis [37].
The lack of carcinogenic activity of 2-hydroxyestrogens has been attributed to a high rate of clearance, more rapid rate of O-methylation and lower hormonal potency in estrogen target tissues, plus produces 2-methoxyestradiol, which suppress tumor cell proliferation and angiogenesis [19]. On the other hand, it has been shown that 2-hydroxyestrogen can damage DNA and generate free radicals when they go through redox cycling or when COMT is inhibited [38]. Methoxyestrogens, including 2-methoxyestradiol (2-MeOE2) and 4-methoxyestradiol (4-MeOE2), have been shown to inhibit both CYP1A1 and CYP1B1 activities. For both enzymes, the order of inhibition by methoxyestrogens was 2–OH–3–MeOE2 > or = 2-MeOE2 > 4-MeOE2. In the in vitro study it was demonstrated that CYP1A1 and CYP1B1 O-demethylated the methoxyestrogens to catechol derivatives [39]. Thus, both the CYP-mediated oxidation of estrogens and the O-demethylation of methoxyestrogens yield identical products, catechol estrogens.
2-Methoxyestradiol exhibits potent apoptotic activity against rapidly growing tumor cells and possesses antiangiogenic action. This metabolite has a lower binding affinity for both ERα and ERβ compared with that of E2 [40]. 2-MeOE2 has a 500- and 3200-fold lower affinity than that of E2 for α and β estrogen receptors, respectively [41]. While 2-OHE2 enhanced growth of MCF-7 breast cancer cells, 2-MeOE2 inhibited DNA synthesis and mitosis. It was suggested that 2-MeOE2 acts as a cytostatic, whereas, 2-OHE2 may be the potent mitogen [42].
There are polymorphisms in the genes encoding CYP1A1, CYP1A2, CYP3A4, and CYP1B1. Some products of this gene exhibit altered kinetics with distinctly lower or higher Km values for both 2- and 4-hydroxylation of 17β-estradiol. For example, in the CYP1B1*3 product a lower Km value for both 2- and 4-hydroxylation has been found [43]. In a case–control study, the carriers of the CYP1B1*3 allele were significantly more frequent among breast cancer women, than those in the controls [44].
Enzymes of the CYP3A family are most important in hepatic drug metabolism because of their wide substrate specificity. The CYP3A4 hydroxylates E1 to 16α-hydroxyestrone, a metabolite involved in breast cancer induction [45]. CYP3A4 and CYP3A5 mRNAs have been detected in mammary tissues [46].
A genetic polymorphism of COMT, creating a COMT(Val) → COMT(Met) substitution at codon 158, is associated with low enzyme activity. It was suggested that its carriers may be at increased breast cancer risk. In a study of pre-menopausal women, a significantly higher risk for breast cancer with COMT(Met/Met) genotypes, compared with the homozygotes COMT(Val/Val) genotype women, was revealed. Women with high BMI and the COMT (Met/Met) genotype demonstrated high breast cancer risk (odds ratio (OR) = 5.7; 95% CI 1.1–30.1) [47]. In Taiwanese women it was observed that of the three gens, i.e., CYP17, CYP1A1, and COMT, low activity of the COMT genotype was associated with the highest relative risk of breast cancer (RR = 4.0; 95% CI 1.12–19.8) [48].
The mechanisms of mammary gland carcinogenesis
Breast cancer is mainly a disease caused by DNA mutations and epigenetic disorders that make an abnormal cell multiply uncontrollably with the potential to invade other parts of the organism, which leads to its death. Carcinogenesis is usually considered as a process that starts with genotoxic effects—initiation followed by enhanced cell proliferation—promotion. In the breast the main estrogen, E2, is both a substrate for the metabolizing enzymes and a ligand for the ER (Fig. 3).

Mechanisms of the carcinogenic effect of estrogens in breast cancer. NQO-1 NADPH-quinone oxidoreductase 1, ROS-reactive oxygen species, 2-OHE2 2-hydroxyestradiol, 4-OHE2 4-hydroxyestradiol, E2–2,3–Q estradiol–2,3–quinone, E2–3,4–Q estradiol–3,4–quinone, 2–OHE2–6–N3Ade 2–hydroxyestradiol–6–N3 Adenine, 4–OHE2–1N7Gua 4–hydroxyestradiol–1N7Guanine, 4–OHE2–1–N3Ade 4–hydroxyestradiol–1–N3 Adenine
Estrogen carcinogenesis can be connected with ER mediated growth and proliferation as a result of the hormone’s ability to stimulate the expression of genes encoding different growth factors [49]. DNA depurinating adducts formation derived from activated metabolites, e.g., catechol estrogens, semiquinones, quinones and free radicals, generated during estrogen metabolism, and also production of reactive oxygen species (ROS) may play a critical role in breast cancer initiation [49]. Excess ROS not only exerts genotoxicity, but also increases progression of mammmary carcinogenicity by inducing a redox-associated signaling pathway [50].
In experimental conditions, it was demonstrated that MCF-7 breast cancer cells and normal breast tissue in aromatase transfected mice contain the enzymes necessary for the metabolism of E2 to the estradiol DNA adducts. After administration of E2 to animals the genotoxic products in breast tissue were observed. In ERα knockout (ERKO/Wnt) transgenic animals a reduction in incidence of tumor formation and a delay in the occurrence of those that formed were found. Oophorectomy reduced the incidence of tumors and delayed their onset, whereas E2 add-back returned the incidence rate to that observed before oopherectomy. The aromatase inhibitor, letrazole, delayed the onset of tumor formation. These data support both ER-dependent and genotoxic ER-independent effects of E2 mediated breast cancer development [51, 52].
The metabolism of estrogens can lead to DNA damage in two ways. The first way is exemplified by the formation of estrogen-adenine/guanine adducts which are released from the DNA by rapid depurination. This phenomenon is a consequence of oxidation of the catechol estrogen metabolites, 4-OHE1/E2 and 2-OHE1/E2, to the quinones, i.e., E1/E2–3,4–Q and E1/E2–2,3–Q.
The reaction of E2–3,4–Q with DNA mainly affords the depurinating adducts 4–OHE2–1–N3Ade and 4–OHE2–1N7Gua, whereas the reaction of E2–2,3–Q with DNA leads to synthesis of the depurinating adduct 2–OHE2–6–N3Ade. The E2–3,4–Q is much more reactive with DNA than E2–2,3–Q. The levels of depurinating adducts formed from E2–3,4–Q are much higher than that of the depurinating adduct from E2–2,3–Q [52].
Increased levels of estrogen quinones and depurinating adducts occur when estrogen metabolism is unbalanced. This unbalanced metabolism is the result of overexpression of estrogen activating enzymes and/or deficiency of the deactivating enzymes. Treatment of MCF-10F cells, which are ERα-negative and ERβ-positive human breast epithelial cells, with E2, 4-OHE2 or 2-OHE2 induces their neoplastic transformation in vitro, even in the presence of the antiestrogen. This suggests that the transformation process is independent of the estrogen receptor and is more probably due to the genotoxic effect of the estrogen metabolites [53].
The second reason for DNA damage is because of the oxidation of the catechol estrogen metabolites 4-OHE2 and 2-OHE2 to the quinones, i.e., E2–3,4–Q and E2–2,3–Q. The quinones, as well as catechol estrogens activated by lactoperoxidase, tyrosinase or prostaglandin H synthase in vitro react with DNA to depurinating and stable adducts which are endogenous initiators of breast cancer. The E2–3,4–Q is much more reactive with DNA than E2–2,3–Q [54]. Moreover, generation of oxygen-free radicals, e.g., the hydroxyl radical and then hydrogen peroxide, resulting from redox cycling of 4-OHE2 to the E2–3,4–Q and back conversion to 4-OHE2, lead to oxidative modification of DNA. If these processes are sufficient, a number of mutations accumulate over a long period to induce neoplastic transformation.
The catechol estrogens, including 2-OHE2, 4-OHE2, 2-OHE1 and 4-OHE1, undergo redox cycling, which results in the production of ROS. These catechol estrogens were found to generate H2O2 and hydroxyl radicals in lysates of MCF-7, MDA-MB-231 and MCF-10A breast epithelial cells. Intracellular ROS accumulation leads to oxidative DNA modification expressed by 8–oxo–7,8–dihydroxy–2′–deoxyguanosine (8–oxo–dG) level [55]. Dicoumarol, the inhibitor of NAD(P)H:quinone 1 (NQO1) activity, enhances 4-OHE2-induced ROS production, whereas, the antioxidant N-acetylcysteine inhibites ROS accumulation and the neoplastic transformation induced by 4-OHE2 [50]. Redox cycling of the quinones and semiquinones and subsequent generation ROS is an important mechanism of DNA damage and cell death.
In cancerogenesis induced by estrogen and their metabolites activation of the numerous signaling pathways occurs.
A high level of ROS increased the translocation of the nuclear factor—kappa B (NF-kB) to the nucleus and its DNA binding through induction of Ikappa B kinase alpha (IκK alpha) and IκK beta activities. The inhibition of the IκK activities significantly reduced the independent growth colony formation in MCF-10A cells treated with 4-OHE2 [50]. The redox-sensitive transcription factor in the nucleus, factor ĸB (NF-ĸB), was transiently activated by 4-OHE2 exposure. Co-exposure of MCF-10A cells to the NF-ĸB inhibitor increased cell death induced by 4-OHE2. In addition, 4-OHE2 caused transient activation of extracellular signal-regulated protein kinases (ERK). An inhibition of ERK aggravated the 4-OHE2-induced cytotoxicity. ERK plays the main role in protection against catechol estrogen-induced cell death [56].
ROS, not only leads to oxidative DNA damage, but also promotes neoplastic transformation of initiated cells, which is a critical process in the induction of carcinogenesis. There is a suggestion that 4-OHE2-induced cell transformation is independent of ER binding [53]. When MCF-10F cells were exposed to benz[a]pyrene, E2, 2-OHE2, 4-OHE2, or 16α-OHE2, 4-OHE2 effectively induced larger colonies at low doses [57]. It was observed that the effect of 4-OHE2 on neoplastic transformation in MCF-10A cells take places via activation of IκK-NF-ĸB signaling pathways [50].
During the malignant transformation process in MCF-10A cells treated with 4-OHE2 the increased AKT phosphorylation through phosphatidylinositol 3-kinase (PI3K) activation was observed. These data indicate that 4-OHE2-induced cell transformation may be mediated through redox-sensitive serine/threonine protein kinase (Akt) signal transduction pathways by up-regulating the expression of cell cycle genes CDC2, PRC1, and PCNA, and also the transcription of nuclear respiratory factor-1 (NRF-1). The study has demonstrated that 4-OHE2 is the main estrogen metabolite responsible for induction of malignant phenotype cells. Moreover, ROS are crucial species in the development of estrogen-induced breast cancer [57].
4-OHE2 not only induces cell transformation but also promotes the growth of cancer cells. There are a lot of studies that 4-OHE2 stimulated specific intracellular signaling pathways, which supports the role of 4-OHE2 in oncogenesis. In particular, the effect of E2 metabolites on the cell cycle has been investigated [58]. Among these metabolites, 4-OHE2 and 16-OHE1 showed a proliferative effect on MCF-7 human ER-positive metastatic breast cancer cells, which was accompanied by a downregulation of apoptosis [59]. On the other hand, the expression of cell cycle-related genes such as CDC2, the protein regulator of cytokinesis 1 (PRC1), the proliferation of the cell nuclear antigen (PCNA), and the transcription factor NRF-1 were up-regulated by 4-OHE2 [57].
It has been demonstrated that 4-OHE2 induces hypoxia-inducible factor-1 alpha (HIF-1) and vascular endothelial growth factor A (VEGF-A) expression in two human ovarian cancer cell lines, OVCAR-3 and A2780-CP70 cells, in dose- and time-dependent manners. HIF-1 is a transcription factor which is induced by hypoxia, growth factors, and activation of oncogens. HIF-1 activates downstream target genes such as VEGF-A, which plays a significant role in tumor progression and angiogenesis. Moreover, it was observed that PI3K inhibitors blocked HIF-1 alpha and VEGF-A expression, whereas the mitogen-activating protein kinase (MAPK) inhibitor did not alter the expression of both indices induced by 4-OHE2. 4-OHE2 also induced AKT phosphorylation at Ser473. These data suggest that the PI3K/AKT/FRAP signaling pathway is required for HIF-1 alpha and VEGF-A expression induced by 4-OHE2, although the MAPK pathway is not required. The findings that induction of HIF-1 alpha and VEGF-A expression occurs via the activation of the PI3K/AKT/FRAP signaling pathway could be an important mechanism of 4-OHE2-induced tumorogenesis [60].
In MCF-7 and MCF-10A CYP1B1 promoted cell proliferation, migration, and invasion. CYP1B1 induced epithelial–mesenchymal transition (EMT) and activated Wnt/β-catenin signaling pathway via upregulation of a key oncogenic proteins. Specific protein 1 (Sp 1), a transcription factor involved in cell growth and metastasis is upregulated by CYP1B1. The suppression of Sp 1 expression by mithramycin A blocked oncogenic transformation induced by CYP1B1.This indicates that Sp 1 acts as a key mediator for CYP1B1 action. Treatment of MCF-7 and MCF-10A cells with 4-OHE2 showed similar effects to the CYP1B1 overexpression. These data suggest that both CYP1B1 and 4-OHE2 promote cell proliferation and metastasis by inducing EMT and Wnt/β-catenin signaling via Sp 1 induction [61]. In addition, CYP1B1 promotes cancer cell survival through involvement of DNA hypermethylation-mediated death receptor 4 (DR4) inhibition via Sp 1, which may act as a key mediator for oncogenic effect [62].
The invasion and metastases of cancer follow the degradation of the extracellular matrix, which acts as a barrier to cancer cells. The proteins of the extracellular matrix are degradated by proteases, mainly the matrix metalloproteinases (MMPs), which increase the risk of invasive and metastatic processes. It was demonstrated that 4-OHE2 led to the conversion of pro MMP-2 and pro MMP-9 to activate both MMPs, which resulted in dissemination of cancer cells [63].
Chemoprevention
Chemoprevention alone and chemoprevention in combination with anticancer treatment is an important way to reduce morbidity and mortality caused by breast cancer [64].
Numerous chemical agents, natural and synthetic, exert a protective effect on the breast cancer cells. These agents include, among others, phytoestrogens, aromatase inhibitors, marijuana smoke condensate, classical antioxidants, and synthetic compounds.
There is a theory which postulates that estrogens increase the rate of cell proliferation by stimulating ER-mediating transcription, thereby increasing the number of errors occuring during DNA replication. The other theory suggests that E2 is metabolized to Q-derivatives, which directly remove base pairs from DNA through a depurination process. Error-prone DNA repair leads to point mutations. It was postulated that both processes interact in an additive or synergistic manner. Thus, antiestrogens would only block receptor-mediated effects, whereas aromatase inhibitors would inhibit both processes. Therefore, aromatase inhibitors would be more powerful in prevention of breast cancer than antiestrogens. In in vitro studies it was observed that catechol-estrogen metabolites are produced in MCF-7 human breast cancer cells. The animal study obtained the dissociation of ER-mediated function from the effects of E2 metabolites. The absence of E2 markedly reduced the incidence of tumors and delayed their onset. The metabolites of E2 may induce breast cancer simultaneously with the ER-mediated mechanism. The study supported the possibility that aromatase inhibitors might be more effective than tamoxifen and other antiestrogens in prevention of breast cancer [65].
In breast cancer epithelial cells, the mitogenic activity of E2 is transduced through binding to two receptors, ERα and ERβ, as transcription factors. Anti-estrogens and aromatase inhibitors are applied in clinic to arrest the estrogen-dependent growth of breast cancer cells. Estrogen effects are mediated through nuclear ERs and cytoplasmic/membrane ERs and G-protein-coupled ERs. These estrogen-binding systems connected with different proteins that lead to cell cycle signaling, proliferation and survival. The partners of nuclear ER include steroid receptor coactivator 1–3 (SRC1-3), histone deacetylases (HDACs), ERβ, and also proteins such as LKB1, PELP1, PAX2 and TOXA1. The extra-nuclear ERα factors include PI3K, and the sarcoma virus tyrosine kinase (Src). These factors are all potential targets for therapeutic agents. In addition, proliferation of breast cancer is enhanced by insulin-like growth factor 1 (IGF-1) and epidermal growth factor (EGF), which stimulate signaling through the MAPK and PI3K/AKT pathways by activation of IGF-1R and EGFR receptors, respectively. These pathways are linked with ER-activated signaling, and the membrane ERα forms complexes with Src and PI3K. Inhibiting these pathways with specific inhibitors or activators may be therapeutically beneficial for arresting breast cancer cell cycle progression and for inducing apoptosis [66]
Phytoestrogens are a biologically active organic compounds derived from plants and have structures like to the main mammalian estrogen, E2. There are five major classes of phytoestrogens, namely: flovonoids, isoflavonoids, lignants, coumestans, and stilbenes. The data about association between breast cancer and phytoestrogens issue from epidemiological studies showing that the incidence of breast cancer is low in Asian women who consume high dietary levels of soy products which have a high isoflavone content [67, 68].
Resveratrol, quercetin, daidzein, and genistein represent a group of the most commonly consumed phytoestrogens. In East Asia, the average daily consumption of phytoestrogens is evaluated to be 20–50 mg. In Europe the average daily phytoestrogen consumption is assessed to be 0.49–1.0 mg. Plasma isoflavone concentrations range from 2 µM to 5 nM. However, local tissue phytoestrogen concentrations are probable to be 2–3 times higher than plasma concentrations [67, 68].
Phytoestrogens are thought to exert anticancer actions through different molecular pathways. They act within target cells on estrogen receptors, cell signaling pathways, the cell cycle, apoptosis, steroid synthesis and induction of DNA damage in cancer cells [68].
Numerous studies in humans have examined the effects of phytoestrogens on estrogen biosynthesis and their biosynthetic enzyme activities. It has been observed a decrease of plasma/serum levels of the follicle-stimulating hormone (FSH), luteinizing hormone (LH), E2, E1, end progesterone following phytoestrogen consumption. In addition, the isoflavone diet increased the ratio of 2-hydroxyestrone to 16α-hydroxyestrone and decreased in the ratio of genotoxic estrogens to total estrogens. In women consuming 40 mg isoflavones every day for 3 months, the average menstruation length and follicular phase of the menstrual cycle were elongated. Flavones and flavonones, including 7-hydroxyflovone, apigenin, chrisin and hesperetin, are the most potent inhibitors of aromatase. Also, quercetin, genistein, and daidzein inhibited the transcription of CYP19 mRNA in human granulosa luteal cells. A mixture of genistein, daidzein, and biochain significantly reduced aromatase activity. Resveratrol exerted inhibitory effects on aromatase activity in MCF-7 cells in vitro and suppressed transcription of CYP19 mRNA in SK-BR-3 breast cancer cells [67].
In addition, phytoestrogens have been shown to inhibit 17β-hydroxyestradiol dehydrogenase; e.g., genistein decreased this enzyme activity in human placental microsomes, granulosa luteal cells, MCF-7, and T47D human breast cancer cells [67]. Phytoestrogens can bind weakly to ERs and some have a preferential affinity for ERβ, which can inhibit the transcriptional growth-promoting activity of ERα. Instead, only higher doses of phytoestrogens exert their growth inhibitory effects through the ERα and Erβ. It has also been shown to inhibit oncogenic cyclin D1 expression, but increase the level of cyclin-dependent kinase inhibitors, p21 and p27, and tumor suppressor genes p53, APC, AMP, and mammary serpin peptidase inhibitor (SERPINB5). These effects were only observed at high doses (> 10 µmol/l) of phytoestrogens. The effects of phytoestrogens on breast cancer may be also caused by their ability to inhibit local estrogen synthesis, induce epigenetic changes, and suppression of ERα signaling pathways [68, 69].
The majority of breast cancer patients have ER-positive breast cancer. E2 is the most potent endogenous estrogen, which expresses its activity through binding to ERs. Most of the growth-inducing effects of estrogens are exerted by their link to ERα. ERα is considered the first target for inhibition of tumor proliferation. In contrast, the proliferative activity of estrogens can be opposed by ERβ. In breast cancer cells E2 induces proliferation and inhibits apoptosis via ERα. ERβ inhibits the growth effects of ERα. ERβ-mediated inhibitory effect against ERα depends on the participation of transcription factors able to repress most of the genes activated by ERα and on the degradation of this receptor [70].
Furanodienone
The ERα is critical in the development and progression of breast cancer. The mitigation of ERα activities is a promising strategy in pharmacotherapy. Furanodienone is the main bioactive compound of Rhizoma Curcumae. It is commonly used in Chinese medicine for cancer therapy. This compound in vitro inhibited MCF-7, T47D, and MDA-MB-231 breast cancer cells’ proliferation in a dose-dependent manner. ERα-negative MDA-MB-231 cells were less sensitive to furanodienone than ERα-positive MCF-7 and T47D cells. Furanodienone effectively blocked E2-stimulated MCF-7 cell proliferation and cell cycle progression, and induced apoptosis. Furanodienone down-regulated ERα protein and mRNA expression levels without effect to ERβ expression. Exposure to furanodienone inhibited E2-stimulation of the estrogen response element (ERE) activity and oblated E2-targeted gene, e.g., oncogenic transcription factor (c-Myc), B-cell lymphoma-2 (Bcl-2), and cyclin D1 expression, which manifested by the inhibition of the cell cycle and cell proliferation and also induction apoptosis. Diminished the cell growth inhibitory effect of furanodienone in MCF-7 cells with knockdown of ERα suggest that these effects are mediated by inhibition of ERα signaling pathways [71].
Basal phenotype breast cancer is one of the most aggressive tumors that frequently metastasizes to the brain.
Current antihormonal therapies are frequently applied for the treatment of hormone receptor positive breast cancer, i.e., ERα and/or nuclear progesterone receptors (PR-positive). For ERα breast cancers, antiestrogen therapies, such as tamoxifen and anastrozole, are often efficient both in primary and in metastatic stages. The status of PR expression is used with ER to indicate the potential effectiveness of antiestrogen therapies because the majority of breast cancers express ER and PR concurrently. Clinical studies revealed that ERα/PR-negative breast cancers respond less well to selective ER modulator (SERM) therapy than ERα/PR-positive tumors [72, 73]. Recent data suggest that ERα/PR-negative breast cancers may be resistant to SERM, whereas they may be less resistant to estrogen withdrawal therapy with aromatase inhibitors. Novel molecular mechanisms explaining SERM resistance in ERα/PR-negative tumors have suggested that growth factors may downregulate PR levels. The absence of PR may reflect hyperactive cross talk between ER and growth factor signaling pathways that downregulate PR even as they activate other ER functions. It was concluded that ERα/PR-negative breast cancers might be treated by blocking ER activity via estrogen withdrawal with aromatase inhibitors by targeted ER degradation, or by combined therapy that assumes both ER and growth factor signaling pathway [73].
Marijuana smoke condensate
There are data about antiestrogenic effects of marijuana smoke condensate (MSC) and cannabinoid biological active compounds. MSC, but not Δ9-tetrahydrocannabinol, cannabidiol, and cannabinol, induced the antiestrogenic effect via the ER-mediated pathway.
Moreover, MSC induced CYP1A1 activity and the E2 metabolism, but inhibited the aromatase activity, suggesting that the activity of MSC is also associated with the indirect ER-dependent pathway as a result of the depletion of the E2 level available to bind to the ER. It was suggested that pyrogenic products, including polycyclic aromatic hydrocarbons (PAHs), might be responsible for the antiestrogenic activity [74].
Genistein
An isoflavon, genistein occurs in soybeans. Cell proliferation and apoptosis inhibition in MCF-7 (high ratio of ERα/ERβ), although not in T47D (low ratio) and MDA-MB-231 (ER-negative), cells were observed after E2 and genistein treatment simultaneously. Moreover, exposure to genistein produced an up-regulation of ERβ and an increase in cytochrome c oxidase activity in T47D cells and decreased the ATP synthase/cytochrome c oxidase ratio. The beneficial effects of genistein treatment depend on the ERα/ERβ ratio in breast cancer cells. Genistein treatment leads to cell cycle arrest and an improvement of mitochondrial function in T47D cells with a low ERα/ERβ ratio, but not MCF-7 with a high ERα/ERβ ratio and MDA-MB-231 with ER-negative ones [75]. In another study genistein treatment of breast cancer cells in combination with cytotoxic agents, i.e., cisplatin, paclitaxel or tamoxifen, caused an increase in cell viability, and also in the antioxidant enzymes level, and decreased ROS production, autophagy, and apoptosis, plus enhanced the cell cycle S phase only in MCF-7 cells with a high ERα/ERβ ratio. In contrast, in the T47D (low ratio) and overexpressed ERβ cells the combination of genistein with cytotoxic compounds did not cause significant changes in the analyzed parameters. It was concluded that genistein consumption is beneficial in patients receiving anticancer therapy with a high ERα/ERβ ratio of breast cancer cells. Also, in the patients with lower ERα/ERβ ratio breast cancer cells genistein consumption could be harmless [76]. Genistein has inhibited invasion of breast cancer cells and decreased tumor growth in nude mice bearing MCF-7 and MDA-MB-231 xenografts.
Genistein, glycitein, daidzein, equol, O-desmethylangolensin and coumestrol exerted a potent inhibitory effect on cell invasion. In contrast, the lignants exerted minimal effects on invasion. Inhibition of invasion induced affects by the phytochemicals was seen without affecting cell viability [77].
Genistein induces apoptosis in breast cancer cells that are ER-positive and ER-negative. Ability of genistein to promote apoptosis requires wild-type caspase-3 activity. Moreover, it was demonstrated that genistein induced apoptosis in breast cancer cells by reducing expression of an oncogenic micro-RNA (miR-155) that occurs in breast cancer. These data indicate that genistein growth inhibitory effects proceed via activation of apoptotic pathways and also suggest that genistein can exert its antitumor actions via estrogen-independent signaling pathways. Genistein, when co-administered with adriamycin, was found to induce necrotic-like cell death in breast cancer cells by inactivating the HER-2 receptor and Akt. It was shown that genistein might be a valuable treatment option for tamoxifen-resistant breast cancer. Inhibition of invasiveness and metastatic activity of MCF-7 and MDA-MB-231 cells by genistein probably proceeds by downregulation of the transcription of various MMPs. Also, many of the anticancer properties of genistein are believed to proceed through its regulation of various molecular signaling pathways that involve numerous genes, such as Bc1-2, Bax, NF-kB, and Akt. There are data that genistein exposure results in the upregulation of BRCA1/2 genes in a dose- and time-dependent manner. Furthermore, genistein upregulates the expression p53 and TNFα genes in breast cancer cells. It was reported that genistein-mediated downregulation of miR-155 contributions to the anticancer effects of genistein in metastatic breast cancer [78].
Calycosin
The endogenous factors which the increased expression stimulates in the development of breast cancer are as follows: IGF1, PI-9, ERα, BRF2, cyclin D1, pS2, TGF-β3, monoamine oxidase A, and α-antichymotripsin. In contrast, the decreased expression of Akt, Wnt/β-catenin, miR-155, MMPs, and HSD leads to inhibition of tumor proliferation. The increased apoptosis is associated with NF-kB decrease, and p53, Caspase 3, Bax, TNF-α, and BRCA-1/2 elevated expression [78].
Calycosin is an isoflavone derived from the Chinese medical herb Radix astragoli. Calycosin at low concentrations effectively stimulated proliferation of ER-positive MCF-7 human breast cancer cells. On the other hand, this isoflavone at high concentrations significantly suppressed the proliferation of these cells and promoted cell apoptosis through the mitochondrial apoptotic pathway by upregulating RASD1. RASD1 is a Ras-family member and a regulator in MAPK-mediated cell proliferation or apoptosis. Calycosin decreased the expression of Bcl-2 and increased Bax expression, which was significantly correlated with elevated RASD1 expression [79]. The cellular effects of isoflavones, i.e., impaired proliferation and triggered apoptosis, induced by calycosin and formononetin were observed only in ER-positive breast cancer cells. No such effects were observed in ER-negative breast cancer cells, indicating the association between isoflavones’ induced inhibitory effect and ERs. After the treatment of MCF-7 cells with calycosin the ERβ expression was significantly increased, followed by a decrease of IGF-1R, activation of poly (ADP)-ribose) polymerase 1 (PARP-1) and downregulation of miR-375. Thus, calycosin exerts an inhibitory effect on breast cancer growth by ERβ-mediated regulation of IGF-1R signaling pathways [80, 81].
In addition, higher doses of calycosin (50, 100, and 150 µM) were found to inhibit migration and invasion of the two cell lines in a dose-dependent manner. Calycosin in a higher dose significantly reduced the expression levels of the forkhead box P3 (Foxp3), followed by downregulation of VEGF and MMP-9 in MCF-7 and T47D breast cancer cells. Thus, a higher dose of calycosin led to a reduction of migration and invasion of human breast cancer cells, by targeting Foxp3-mediated VEGF and MMP-9 expression [82].
The two main isoflavones, calycosin and genistein, inhibited proliferation and induced apoptosis in MCF-7 breast cancer cells. MCF-7 cells’ exposure to calycosin or genistein led to decreased phosphorylation of Akt, and reduced expression of its downstream target, HOTAIR. HOTAIR is the HOX transcript antisense RNA gene located on chromosome 12 and encodes the long non-coding RNAs molecule. HOTAIR is over-expressed in breast cancer. High expression of HATOIR in breast cancer is a predictor of metastasis and poor outcome.
Calycosin is more effective in inhibiting breast cancer growth in comparison with genistein, through its regulation of Akt signaling pathways and HOTAIR expression [83].
Formononetin
Formononetin is one of the main phytoestrogens of red clover plants. This compound inhibited the proliferation of MCF-7 cells and effectively induced cell cycle arrest. Formononetin downregulated the levels of p-IGF-1R, p-Akt, cyclin D1 protein, and cyclin D1 mRNA expression. Formononetin prevented the tumor growth of human breast cancer cells in nude mouse xenografts. It was found that this phytoestrogen causes cell arrest at the G0/G1 phase by inhibiting of IGF-1/IGF-1R-PI3K/Akt pathways and decreasing cyclin D1 mRNA and protein expression [84].
In a study of the molecular mechanisms involved in the induced apoptosis and effect of the proliferation of ER-positive only, MCF-7 and T47D cells were observed. Formononetin activated the mitogen-activated protein kinase (MAPK) signaling pathway in a dose-dependent manner, which resulted in the increased Bax/Bcl-2 ratio and induced apoptosis in MCF-7 cells. Moreover, when MCF-7 cells were pretreated with a p38MAPK inhibitor before phytoestrogen, apoptosis induced by formononetin was less pronounced. These data indicate that the apoptosis induced by formononetin in human breast cancer cells were associated with the Ras-p38MAPK pathway [85].
Baicalein
Baicalein (3,4,5-trihydroxyfurane) is the compound from the root of Scutellaria baicalensis. It has a broad spectrum of activity, including induction of tumor cell apoptosis, cell cycle arrest, inhibiting tumor proliferation and angiogenesis, and also scavenging free radicals. An in vivo study showed that baicalein inhibited metastasis in the xenograft nude mouse model. Baicalein significantly decreased the expression of AT-rich sequence-binding protein-1 (SATB1) and enhanced the expression of E-codherin. SATB1 is a speciffically expressed matrix attached to regions (MARs) binding protein.
In addition, baicalein downregulated the expression of Wnt1 and β-catenin proteins and the transcription level of Wnt/β-catenin-targeted genes. It was suggested that baicalein has the potential to suppress breast cancer metastasis, probably by inhibition of EMT, which may be attributed to downregulation of both SATB1 and the Wnt/β-catenin [86, 87].
SATB1 plays an important role in the initiation and development of cancer. It regulates the malignant progression of breast cancer. Activation of SATB1 is indispensable for tumor metastasis, for the expression of various genes involved in the growth, proliferation, angiogenesis, invasion, and metastasis.
The Wnt/β-catenin pathway is one of the most common signaling abnormalities occurring in human malignancies [88]. It is estimated that metastasis accounts for 90% of cancer-related mortality [89].
Luteolin
Luteolin (3′,4′,5,7-tetrahydroxyflavone) is a flavonoid which exhibits antioxidant, anti-inflammatory, antibacterial, antidiabetic and antiproliferative properties. It has been shown that luteolin may inhibit cell proliferation, metastasis, and angiogenesis different types of cancers, including breast cancer. These effects proceed through cell cycle arrest, and apoptosis, and by modulating cell signaling pathways. Luteolin was shown to effectively inhibit the IGF-1 stimulated ERα-positve MCF-7 cell proliferation in a dose- and time-dependent manner, and to suppress ERα-negative MDA-MB-231 cells. Supplementation by luteolin at 0.01% or 0.05% significantly reduced the tumor in nude mice inoculated with MDA-MB-231 cells. It has been shown that this flavonoid significantly inhibits the cell cycle of MCF-7 and MDA-MB-231 cells at the G2/M and S stages. Luteolin can efficiently suppress the migration and invasion of MCF-7 and MDA-MB-231 cells. Luteolin exerts its biological actions through multiple signaling pathways, such as PI3K/Akt, MAPK/ERK1/2, EGFR, and NF-kB [90, 91].
Benzanilides and dithiobenzanilides
There are synthetic estrogenic receptor modulators. Benzanilides and dithiobenzanilides are novel promising compounds which show selective antiproliferative activity against estrogen-dependent MCF-7 breast adenocarcinoma. Their estrogenic activity was confirmed in MCF-7 transfected with an estrogen receptor reporter plasmid and in HEK 239 cells over-expressing the ERα. Docking studies have shown that one of tested compounds interacts with the receptor in the same cavity as E2 [92].
Resveratrol and its analogs
Natural polyphenols are considered chemopreventive agents able to prevent tumor initiation caused by ROS targeting not only lipids and proteins, but also DNA and RNA in living cells. Free radicals react with phenolic compounds much faster than with lipids or DNA. Therefore, polyphenols protect lipids and DNA from oxidative damage induced by free radicals.
The resveratrol (3,4′,5-trans-trihydroxystilbene) present in grapes possesses chemopreventive and chemotherapeutic activities. Although resveratrol shows phytoestrogenic activity, it inhibits the growth of both hormone-dependent and hormone-independent breast cancer cells [93, 94]. This compound inhibited transformation, migration and growth of MCF-10A human breast epithelial cells treated with 4-OHE2. Resveratrol treatment suppressed the 4-OHE2 induced activation of IκKβ and phosphorylation of IκKα, and consequently NF-κB DNA binding activity and cyclooxygenase-2 (COX-2) expression. Resveratrol suppressed ROS production and phosphorylation of Akt and ERK induced by 4-OHE2 treatment. Thus, resveratrol blocks activation of IκKβ-NF-κB signaling pathway and induction of COX-2 expression in 4-OHE2-treated MCF-10A cells. These effects lead to suppression of migration and transformation of MCF-10A cells [95].
Resveratrol is extensively metabolized by hormone-dependent ZR-75-1 cells, but only marginally by hormone-independent MDA-MB-231 cells exclusively forming resveratrol–3–O–sulfate. Resveratrol–3–O-sulfate concentration in the cytoplasm and culture medium was several times higher in ZR-75-1 cells than in MDA-MB-231 cells. The expression of SULT1A1 mRNA showed a close association with resveratrol–3–sulfate formation. It was concluded that SULT1A1-based biotransformation diminishes the anticancer activity of resveratrol in breast cancer cells, which must be considered in patients following oral uptake of this polyphenol as a chemopreventive chemical [96].
It was shown that introduction of additional hydroxyl groups into the stilbene structure increases the biological activity of resveratrol. The 3,3′,4,4′,5,5′-hexahydroxystilbene caused the activation of caspase-8 in MDA-MB-231 cells, whereas, the activation of caspase-9 and caspase-3 was evident in ZR-75-1, MDA-MB-231 and T47D breast cancer cells. Activation of caspase-9 and caspase-3 was associated with a reduction of mitochondrial potential and an increase of p53, which could have been connected with the downregulation of mitochondrial superoxide dismutase (MnSOD). MnSOD is a key enzyme assuring antioxidative defense in mitochondria, the cellular center of ROS generation which can cause a significant reduction of antioxidant defense in cancer cells. An increase of oxidative stress was confirmed by loss of reduced glutathione in tested cells. These data suggest that ROS generating compounds may cause p-53-mediated MnSOD activity reduction and lead to induction p53 transcriptional functions, which subsequently lead to the activation of mitochondrial apoptotic processes. It was suggested that increased ROS generation caused by 3,3′,4,4′,5,5′-hexahydroxystilbene, as a result of the interaction of this chemical with the mitochondrial respiratory chain and a decrease in antioxidative defense, can be a promising way for selective elimination of cancer cells [97].
Higher hydroxylated resveratrol analogs (HHRA), i.e., 3,3′,4,4′–tetrahydroxy–trans–stilbene, 3,4,4′,5–tetrahydroxy–trans–stilbene and 3,3′,4,4′,5,5′–hexahydroxy–trans–stilbene, significantly demonstrate various biological activities when compared with resveratrol. These compounds possess both antioxidant and prooxidant properties. The antioxidant properties are expressed to enable these compounds to protect cells from oxidative damage, whereas prooxidant activity are shown by their cytotoxic or pro-apoptotic effects. These compounds exerted the cytotoxic effect on leukemia cells. Moreover, it was observed that there was increased activity of caspase 3 and 9. Cell death was accompanied by a decrease of mitochondrial potential, oxidative stress, loss of reduced glutathione level, and the diminishing of both mRNA expression and activity of MnSOD. Manganese superoxide dismutase is a key antioxidant enzyme that protects the mitochondrial matrix against the superoxide radical produced by respiratory chain activity. Also, MnSOD protects cancer cells from apoptosis induced by various pro-apoptotic agents like TNF and anticancer drugs. HHRA possessing ortho-hydroxy groups are stronger cytotoxic agents than compounds without such a structure. It was found that the increased cytotoxicity of ortho-hydroxystilbenes is associated with the presence of ortho-semiquinones formed during metabolism or oxidation. It was suggested that the cytotoxic activity of the tested compounds may be connected with the formation of short-living prooxidative metabolites, which cause the induction of oxidative stress in cancer cells [98, 99].
Piceatannol (3,3′,4′,5–trans–tetrahydroxystilbene is a naturally occurring analog of resveratrol. Piceatannol has been found in various plants, including grapes, passion fruit, and white tea. This compound exhibits antioxidative activity, and anticancer properties expressed by its ability to suppress proliferation among other breast cancers. The inhibition of cancer cells’ growth and apoptotic activities of piceatannol are mediated through cell cycle arrest; up-regulation of Bid, Bax, Bik, Bok, Fas, p21; down-regulation of Bcl-xL, BCL-2, cIAP, activation of caspases -3, -7, -8, -9, loss of mitochondrial potential, and release of cytochrome c. Piceatannol suppresses the activation of some transcription factors, including NF-кB, which plays a main role as a transcriptional regulator in response to oxidative stress caused by free radicals, cytokines, or microbial antigens. Moreover, piceatannol inhibits Janus kinase (JAK-1), which is a key factor of the STAT pathway that is crucial in controlling cellular activities in response to extracellular cytokines and is a COX-2-inducible enzyme involved in carcinogenesis. Piceatannol exerts apoptotic action but may also show anti-apoptotic and pro-proliferative activity. Piceatannol binds estrogen receptors and stimulates growth of estrogen-dependent cancer cells. The pharmacological properties of piceatannol, particularly its antitumor, antioxidant, and anti-inflammatory actions, indicate that this compound might be a potentially useful nutritional and therapeutic agent [100].
The protective anticancer effect of biologically active compounds may be realized by direct impact on the activity of the metabolized enzymes and E2 biotransformation.
Chrysoeriol
Chrysoeriol (luteorin–3′–methoxyether) is a natural methoxyflavonoid that selectively inhibits CYP1B1 activity and prevents the formation of carcinogenic 4-OHE2 from E2. Chrysoeriol inhibited E2 hydroxylation catalyzed by CYP1B1, but not by CYP1A1. Methylation of 4-OHE2, which is thought to be a detoxification process, was not affected by the chrysoeriol. In MCF-7 human breast cancer cells, chrysoeriol did not affect the CYP1A1 and CYP1B1 gene expression, but significantly inhibited the production of 4-MeOE2 without any effects on the formation of 2-MeOE2 [101].
A similar effect was observed in MCF-10F cells treated with resveratrol. Resveratrol has anticancer activity because it is the aryl hydrocarbon receptor (AhR) antagonist that decreases CYP1B1 expression and induces NQO1 expression in a dose- and time-dependent manner. While CYP1B1 favors qauinone formation by catalyzing estrogen 4-hydroxylation, NQO1 catalyzes the protective reduction of quinones to catechols. Resveratrol decreased estrogen metabolism and blocked formation of DNA adducts in MCF-10F cells. Thus, resveratrol can prevent breast cancer initiation by blocking multiple stages in the estrogen genotoxicity pathway [102].
The antioxidants, N-acetylcysteine, and reduced form lipoic acid showed a significant inhibition of the formation of the depurinating estrogen-DNA adducts, N3Ade and N7Gua, by E2–3,4–Q. In the reaction of lactoperoxidase-activated 4-OHE2 with DNA, resveratrol achieved the highest level of inhibition, N-acetylcysteine and reduced lipoic acid caused moderate inhibition, and melatonin had the least inhibition. These data indicate that all four antioxidants can inhibit the formation of the depurinating estrogen-DNA adducts and to prevent the indication of human breast cancer [103].
Conclusions
This review presents data on important findings concerning the risk factors, estrogen metabolism, and pathomechanism of breast cancer, as well as its chemoprevention. The initiation and development of breast cancer is caused by numerous risk factors.
The parent estrogen, mainly E2 and its metabolites, are responsible for the formation of neoplastic alterations in breast epithelial cells. Reactive oxygen species and free radicals play a significant role in the induction oncogenesis. The mechanisms of estrogen carcinogenesis include participation of E2 as an estrogen receptors’ agonist, the genotoxic effects of the estrogen metabolites, and epigenetic processes. The carcinogenic process occurs with the participation of numerous signaling pathways. Phytoestrogens, classical antioxidants, and some synthetic compounds, are considered to have a number of beneficial activities, including anticancer, anti-oxidative, anti-inflammatory and estrogenic activity. These chemicals suppress proliferation of a wide variety of tumor cells, including breast cancer cells. The growth-inhibitory and apoptotic effects of these compounds are mediated through cell-cycle arrest, upregulation and/or down-regulation of numerous biomolecules, activation of caspases, and loss of mitochondrial potential. These compounds have been shown to suppress the activation of some transcription factors, including NF-кB, which plays a central role as a transcriptional regulator in response to cellular stress caused by ROS and free radicals. The protective anticancer effect may be realized by direct impact of biologically active compounds to the metabolized enzymes’ activity and E2 biotransformation. Higher hydroxylated polyphenols possess both antioxidant and prooxidant properties. Ortho-hydroxystilbenes are a promising agent for selective elimination of cancer cells. The biological activity, especially phytoestrogens, suggests that these compounds might be potentially useful nutritional and chemopreventive agents.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataran I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;10:10. https://doi.org/10.3322/caac.21660.
Didkowska J, Wojciechowska U. Breast cancer in Poland and Europe—population and statistics. Nowotwory J Oncol. 2013;63(2):111–8.
Collaborative Group on Hormonal Factors in Breast Cancer. Menarche, menopause, and breast cancer risk: individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. The Lancet Oncol. 2012;13(11):1141–51. https://doi.org/10.1016/S1470-2045(12)70425-4.
Hulka BS, Moorman PG. Breast cancer: hormones and other risk factors. Maturitas. 2008;61(1–2):203–13. https://doi.org/10.1016/j.maturitas.2008.11.016.
Castro GD, Castro JA. Alcohol drinking and mammary cancer: Pathogenesis and potential dietary preventive alternatives. World J Clin Oncol. 2014;5(4):713–29. https://doi.org/10.5306/wjco.v5.i4.713.
Chlebowski R, Rohan T, Manson J, Aragaki A, Kaunitz A, Stefanick M, et al. Breast cancer after use of estrogen plus progestin and estrogen alone. JAMA Oncol. 2015;1(3):296–305. https://doi.org/10.1001/jamaoncol.2015.0494.
Fortner RT, Katzke V, Kuhn T, Kaaks R. Obesity and breast cancer. Recent Results Cancer Res. 2016;208:43–65. https://doi.org/10.1007/978-3-319-42542-9_3.
Lynch BM, Neilson HK, Friedeureich CM. Physical activity and breast cancer prevention. Recent Results Cancer Res. 2011;186:13–42. https://doi.org/10.1007/978-3-642-04231-7_2.
Schmidt S, Monk J, Robinson L, Mourtzakis M. The integrative role of leptin, estrogen and the insulin family in obesity-associated breast cancer: potential effects of exercise. Obes Rev. 2015;16(6):473–87. https://doi.org/10.1111/obr.12281.
Willet WC. Diet, nutrition, and avoidable cancer. Environ Health Persp. 1995;103(Suppl. 8):165–70. https://doi.org/10.1289/ehp.95103s8165.
Dorgan JF, Reichman ME, Judd JT, Brown C, Longcope C, Schatzkin A. Relation of energy, fat, and fiber intakes to plasma concentrations of estrogens and androgens in premenopausal women. Am J Clin Nutr. 1996;64(1):25–33. https://doi.org/10.1093/ajcn/64.1.25.
Yue W, Yager JD, Wang JP, Jupe ER, Santen RJ. Estrogen receptor-dependent and independent mechanisms of breast cancer carcinogenesis. Steroids. 2013;78:161–70. https://doi.org/10.1016/j.steroids.2012.11.001.
Shanle EK, Xu W. Selectively targeting estrogen receptors for cancer treatment. Drug Deliv Rev. 2010;62:1265–76. https://doi.org/10.1016/j.addr.2010.08.001.
Hervouet E, Cartron PF, Jouvenot M, Delage-Mourroux R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics. 2013;8:237–45. https://doi.org/10.4161/epi.23790.
Bernstein L, Ross RK, Pike MC, Brown JB, Henderson BE. Hormone levels in older women: a study of post-menopausal breast cancer patients and healthy population controls. Br J Cancer. 1990;61:298–302. https://doi.org/10.1038/bjc.1990.56.
Lipworth L, Adami HO, Trichopoulos D, Carlstrom K, Mantzoros C. Serum steroid hormone levels, sex hormone-binding, and body mass index in the etiology of post-menopausal breast cancer. Epidemiology. 1996;7(1):96–100.
Toniolo PG, Levitz M, Zeleniuch-Jacquotte A, Banerjee S, Koenig KL, Shore RE. A prospective study of endogenous estrogens and breast cancer risk. J Natl Cancer Inst. 1995;87(3):190–7. https://doi.org/10.1093/jnci/87.3.190.
Berrino F, Muti P, Micheli A. Serum sex hormone levels after menopause and subsequent breast cancer. J Natl Cancer Inst. 1996;88(5):291–6. https://doi.org/10.1093/jnci/88.5.291.
Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. 1998;19:1–27. https://doi.org/10.1093/carcin/19.1.1.
Cauley JA, Lucas FL, Kuller LH, Stone K, Browner W, Cummings SR. Elevated serum estradiol and testosterone concentrations are associated with a high risk for breast cancer. Ann Intern Med. 1999;130(4 Pt 1):270–7. https://doi.org/10.7326/0003-4819-130-4part1-199902160-00004.
Fuhrman BJ, Schairer C, Gail MH, Boyd-Morin J, Xu X, Sue LY, et al. Estrogen metabolism and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2012;104(4):326–39. https://doi.org/10.1093/jnci/djr531.
Ziegler RG, Fuhrman BJ, Moore SC, Mattews CE. Epidemiologic studies of estrogen metabolism and breast cancer. Steroids. 2015;99(Pt A):67–75. https://doi.org/10.1016/j.steroids.2015.02.015.
Thomas HV, Key TJ, Allen DS, Moore JW, Dowsett M, Fentiman IS, et al. A prospective study of endogenous serum hormone concentrations and breast cancer risk in premenopausal women on the island of Guernsey. Br J Cancer. 1997;75:1075–9. https://doi.org/10.1038/bjc.1997.183.
Eliassen AH, Spiegelman D, Xu X, Keefer LK, Veenstra TD, Barbieri RL, et al. Urinary estrogens and estrogen metabolites and subsequent risk of breast cancer among premenopausal women. Cancer Res. 2012;72(3):696–706. https://doi.org/10.1158/00085472.CAN-11-2507.
Carr BR, MacDonald PC, Simpson ER. The role of lipoproteins in the regulation of progesterone secretion by the human corpus luteum. Fertil Steril. 1982;38(3):303–11. https://doi.org/10.1016/S0015-0282/16/46511-8.
Samavat H, Kurzer MS. Estrogen metabolism and breast cancer. Cancer Lett. 2015;356(2PtA):231–43. https://doi.org/10.1016/j.canlet.2014.04.018.
Kristensen VN, Andersen TI, Lindblom A, Erikstein B, Magnus P, Borresen-Dale AL. A rare CYP19 (aromatase) variant may increase the risk of breast cancer. Pharmacogenetics. 1998;8:43–8.
Williams JA, Phillips DH. Mammary expression of xenobiotic metabolizing enzymes and their potential role in breast cancer. Cancer Res. 2000;60:4667–77.
Dawling S, Hachey DL, Roodi N, Parl FF. In vitro model of mammary estrogen metabolism: structural and kinetic differences between catechol estrogens 2- and 4- hydroxyestradiol. Chem Res Toxicol. 2004;17(9):1258–64. https://doi.org/10.1021/tx0498657.
Raftogianis R, Creveling C, Weinshilboum R, Weisz J. Estrogen metabolism by conjugation. J Natl Cancer Inst Monogr. 2000;27:113–24. https://doi.org/10.1093/oxfordjournals.jmcimonographs.a024234.
Li KM, Todorovic R, Devanesan P, Higginbotham S, Köfeler H, Ramanathan R, et al. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-uinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis. 2004;25(2):289–97. https://doi.org/10.1093/carcin/bgg191.
Zhao Z, Kosinska W, Khmielnitsky M, Cavalieri EL, Rogan EG, Chakravarti D, et al. Mutagenic activity of 4-hydroxyestradiol, but not 2-hydroxyestradiol, in BB rat 2 embrionic cells, and the mutational spectrum of 4-hydroxyestradiol. Chem Res Toxicol. 2006;19(3):475–9. https://doi.org/10.1021/tx0502645.
Mailander PC, Meza JL, Higginbotham S, Chakravarti D. Induction A.T to G.C mutations by erroneous repair of depurinated DNA following estrogen treatment of the mammary gland of ACI rats. J Steroid Biochem Mol Biol. 2006;101(4–5):204–15. https://doi.org/10.1016/j.jsbmb.2006.06.019.
Cavalieri EL, Rogan EG. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol. 2011;125(3–5):169–80. https://doi.org/10.1016/j.jsbmb.2011.03.008.
Van Aswegen CH, Purdy RH, Wittliff JL. Binding of 2-hydroxyestradiol and 4-hydroxyestradiol to estrogen receptors from human breast cancer. J Steroid Biochem. 1989;32(4):485–92. https://doi.org/10.1016/0022-4731(89)90380-4.
Gupta M, McDougal A, Safe S. Estrogenic and antiestrogenic activities of 16 alpha- and 2-hydroxy metabolites of 17 beta-estradiol in MCF-7 and T47D human breast cancer cells. J Steroid Biochem Mol Biol. 1998;67(5–6):413–9. https://doi.org/10.1016/s0960-07609(98)00135-6.
Vandewalle B, Lefebvre J. Opposite effects of estrogen and catecholestrogen on hormone-sensitive breast cancer cell growth and differentiation. Mol Cell Endocrinol. 1989;61(2):239–46. https://doi.org/10.1016/0303-7207(89)90135-4.
Lavigne JA, Goodman JE, Fonong T, Odwin S, He P, Roberts DW, et al. The effects of catechol-O-methyltransferase inhibition on estrogen metabolite and oxidative DNA damage levels in estradiol-treated MCF-7 cells. Cancer Res. 2001;61(20):7488–94.
Dawling S, Roddi N, Parl FF. Methoxyestrogen exert feedback inhibition on cytochrome P450 1A1 and 1B1. Cancer Res. 2003;63(12):3127–32.
Lakhani NJ, Sarkar MA, Venitz J, Figg WD. 2-Methoxyestradiol, a promising anticancer agent. Pharmacotherapy. 2003;23(2):165–72. https://doi.org/10.1592/phco.23.2.165.2088.
LaValee TM, Zhang XH, Herbstritt CJ, Kough EC, Green SJ, Pribluda VS. 2-Methoxy- estradiol inhibits proliferation and induces apoptosis independently of estrogen receptors α and β. Cancer Res. 2002;62:3691–7.
Lottering ML, Haag M, Seegers JC. Effects of 17beta-estradiol metabolites on cell cycle events in MCF-7 cells. Cancer Res. 1992;52(21):5926–32.
Li DN, Seidel A, Pritchard MP, Wolf CR, Friedberg T. Polymorphism P450 CYP1B1 affects the conversion of estradiol to the potentially carcinogenic metabolite 4-hydroxyestradiol. Pharmacogenetics. 2000;10:343–53.
Kocabas NA, Sardas S, Cholerton S, Daly AK, Karakaya AE. Cytochrome P450 CYP 1B1and catechol O-methyltransferase (COMT) genetic polymorphism and breast cancer susceptibility in a Turkish population. Arch Toxicol. 2002;76:643–9. https://doi.org/10.1007/s00204-002-0387-x.
Shou M, Korzekwa KR, Brooks EN, Krausz KW, Gonzales FJ, Gelboin HV. Role of human hepatic cytochrome P450 1A2 and 3A4 in the metabolic activation of estrone. Carcinogenesis. 1997;18(1):207–14. https://doi.org/10.1093/carcin/18.1.207.
Huang Z, Fasco MJ, Figge HL, Keyomarsi K, Kaminsky LS. Expression of cytochromes P450 in human breast tissue and tumors. Drug Metab Dispos. 1996;24(8):899–905.
Thompson PA, Shields PG, Freudenheim JL, Stone A, Vena JE, Marshall JR, et al. Genetic polymorphism in catechol-O-methyltransferase, menopausal status, and breast cancer risk. Cancer Res. 1998;58:2107–10.
Huang CS, Chern HD, Chang KJ, Cheng CW, Hsu SM, Shen CY. Breast cancer risk associated with genotype polymorphism of the estrogen-metabolising genes CYP17, CYP1A1, and COMT: a multigenetic study on cancer susceptibility. Cancer Res. 1999;59:4870–5.
Hofseth LJ, Raafat AM, Osuch JR, Pathak DR, Slomski CA, Haslam SZ. Hormone replacement therapy with estrogen or estrogen plus medroxyprogesterone acetate is associated with increased epithelial proliferation in the normal postmenopausal breast. J Clin Endocrinol Metab. 1999;84:4559–65. https://doi.org/10.1210/jcem.84.12.6194.
Park SA, Na HK, Kim EH, Cha YN, Surh YJ. 4-Hydroxyestradiol induces anchorage-independent growth of human mammary epithelial cells via activation of Ikappa B kinase: potential role of reactive oxygen species. Cancer Res. 2009;69:2416–24. https://doi.org/10.1158/0008-5472.CAN-08-2177.
Santen R, Cavalieri E, Rogan E, Russo J, Guttenplan J, Ingle J, et al. Estrogen mediation of breast tumor formation involves estrogen receptor—dependent, as well as independent, genotoxic effects. Ann N Y Acad Sci. 2009;1155:132–40. https://doi.org/10.1111/j.1749-6632.2008.03685.x.
Santen RJ, Yue W, Wang JP. Estrogen metabolites and breast cancer. Steroids. 2015;99(Pt A):61–6. https://doi.org/10.1016/j.steroids.2014.08.003.
Lareef MH, Garber J, Russo PA, Russo IH, Heulings R, Russo J. The estrogen antagonist ICI-182-780 does not inhibit the transformation phenotypes induced by 17-beta-estradiol and 4-OH estradiol in human breast epithelial cells. Int J Oncol. 2005;26(2):423–9. https://doi.org/10.3892/ijo.26.2.423.
Zahid M, Kohli E, Saeed M, Rogan E, Cavalieri E. The greater reactivity of estradiol 3,4-quinone vs estradiol-2,3-quinone with DNA in the formation of depurinating adducts: implication for tumor-initiating activity. Chem Res Toxicol. 2006;19(1):164–72. https://doi.org/10.1021/tx050229y.
Fussell KC, Udasin RG, Smith PJS, Gallo MA, Laskin JD. Catechol metabolites of endogenous estrogens induce redox cycling and generate reactive oxygen species in breast epithelial cells. Carcinogenesis. 2011;32(8):1285–93. https://doi.org/10.1093/carcin/bgr109.
Chen ZH, Na HK, Hurh YJ, Surh YJ. 4-Hydroxyestradiol induces oxidative stress and apoptosis in human mammary epithelial cells: possible protection by NF-kappa B and ERK/MAPK. Toxicol Appl Pharmacol. 2005;208(1):46–56. https://doi.org/10.1016/j.Taap.2005.01.010.
Okoh VO, Felty Q, Parkash J, Poppiti R, Roy D. Reactive oxygen species via redox signaling to PI3K/AKT pathway contribute to the malignant growth of 4-hydroxyes-tradiol-transformed mammary epithelial cells. PLoS ONE. 2013;8(2):e54206. https://doi.org/10.1371/journal.pone.0054206.
Chang M. Dual role of estrogen metabolism in mammary carcinogenesis. BMB Rep. 2011;44:423–34. https://doi.org/10.5483/BMBRep.2011.44.7.423.
Seeger H, Wallwiener D, Kraemer E, Mueck AO. Comparison of possible carcinogenic estradiol metabolites: effects on proliferation, apoptosis and metastasis of human breast cancer cells. Maturitas. 2006;54(1):72–7. https://doi.org/10.1016/j.maturitas.2005.08.010.
Gao N, Nester RA, Sarkar MA. 4-Hydroxy estradiol but not 2-hydroxy estradiol induces expression of hypoxia-inducible OVCAR-3 and A2780-CP70 human ovarian carcinoma cells. Toxicol Appl Pharmacol. 2004;196(1):124–35. https://doi.org/10.1016/j.Taap.2003.12.002.
Kwon YJ, Back HS, Ye DJ, Shin S, Kim D, Chun YJ. CYP1B1 enhances cell proliferation and metastasis through induction of EMT and activation of Wnt/β-catenin signaling via Sp1 upregulation. PLoS ONE. 2016;11(3): e0151598. https://doi.org/10.1371/journal.pone.0151598.
Kwon YJ, Cho NH, Ye DJ, Back HS, Ryu YS, Chun YJ. Cytochrome P4501B1 promotes cell survival via specificity protein 1 (Sp1)-mediated suppression of death receptor 4. J Toxicol Environ Health A. 2018;81(9):278–87. https://doi.org/10.1080/15287394.2018.1440186.
Paquette B, Bisson M, Baptiste C, Therriault H, Lemay R, Cantin AM. Invasiveness of breast cancer cells MDA-MB-231 through extracellular matrix is increased by the estradiol metabolite 4-hydroxyestradiol. Int J Cancer. 2005;113(5):706–11. https://doi.org/10.1002/ijc.20647.
Ames BN. Micronutrients prevent cancer and delay aging. Toxicol Lett. 1998;102–103:5–18.
Yue W, Wang JP, Li Y, Bocchinfuso WP, Korach KS, Devanesan PD, et al. Tamoxifen versus aromatase inhibitors for breast cancer prevention. Clin Cancer Res. 2005;11(2 Pt 2):925–30.
Renoir JM, Marsand V, Lazennec G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem Pharmacol. 2013;85(4):449–65. https://doi.org/10.1016/j.bcp.2012.10.018.
Mense SM, Hei TK, Ganju RK, Bhat HK. Phytoestrogens and breast cancer prevention: possible mechanisms of action. Environ Health Perspect. 2008;116(4):426–33. https://doi.org/10.1289/ehp.10.538.
Bilal I, Chowdhury A, Davidson J, Whitehead S. Phytoestrogens and prevention of breast cancer: the contentious debate. World J Clin Oncol. 2014;5(4):705–12. https://doi.org/10.5306/wjco.v5.i4.705.
Basu P, Maier C. Phytoestrogens and breast cancer: In vitro anticancer activities of isoflavones, lignans, coumestans, stilbens and their analogs and derivatives. Biomed Pharmacother. 2018;107:1648–66. https://doi.org/10.1016/j.biopha.2018.08.100.
Orlando L, Schiavone P, Cinieri S. Genistein: the future of prevention and treatment of breast cancer? Cancer Biol Ther. 2011;11(10):918–20. https://doi.org/10.4161/cbt.11.10.15493.
Li YW, Zhu GY, Shen XL, Chu JH, Yu ZL, Fong WF. Furanodienone inhibits cell proliferation and survival by suppressing ERα signaling in human breast cancer MCF-7 cells. J Cell Biochem. 2011;112:217–24. https://doi.org/10.1002/jeb.22922.
Cui X, Schiff R, Arpino G, Osborne CK, Lee AV. Biology of progesterone receptor loss breast cancer and its implications for endocrine therapy. J Clin Oncol. 2005;23(30):7721–35. https://doi.org/10.1200/JCO.2005.09.004.
Xie M, Zhou L, Chen X, Gainey LO, Xiao J, Nanes MS, et al. Progesterone and Src family inhibitor PP1 synergistically inhibit cell migration and invasion of human basal phenotype breast cancer cells. Biomed Res Int. 2015;2015: 426429. https://doi.org/10.1155/2015/426429.
Lee SY, Oh SM, Lee SK, Chung KH. Antiestrogenic effects of marijuana smoke condensate and cannabinoid compounds. Arch Pharmacol Res. 2005;28:1365–75. https://doi.org/10.1007/BF02977903.
Pons DG, Nadal-Serrano M, Blanquer-Rossello MM, Sastre-Serra J, Oliver J, Roca P. Genistein modulates proliferation and mitochondrial functionality in breast cancer cells depending on ERalpha/ERbeta ratio. J Cell Biochem. 2014;115:949–58. https://doi.org/10.1002/jcb.24737.
Pons DG, Nadal-Serrano M, Torrens-Mas M, Oliver J, Roca P. The phytoestrogen genistein affects breast cancer cells treatment depending on the ERα/ERβ ratio. J Cell Biochem. 2016;117:218–29. https://doi.org/10.1002/jcb.25268.
Magee PJ, McGlynn H, Rowland IR. Differential effects of isoflavones and lignans on invasiveness of MDA-MB-231 breast cancer cells in vitro. Cancer Lett. 2004;208(1):35–41. https://doi.org/10.1016/j.canlet.2003.11.12.
Ziaei S, Halaby R. Dietary isoflavones and breast cancer risk. Medicines (Basel). 2017;4(2):18–29. https://doi.org/10.3390/medicines4020018.
Tian J, Duan YX, Bei CY, Chen J. Calycosin induces apoptosis by upregulation of RASD1 in human breast cancer cells MCF-7. Horm Metab Res. 2013;45(08):593–8. https://doi.org/10.1055/s-0033-1341510.
Chen J, Zhao X, Ye Y, Wang Y, Tian J. Estrogen receptor beta-mediated proliferative inhibition and apoptosis in human breast cancer by calycosin and formononetin. Cell Physiol Biochem. 2013;32:1790–7. https://doi.org/10.1159/000356612.
Chen J, Hou R, Zhang X, Ye Y, Wang Y, Tian J. Calycosin suppresses breast cancer growth via ERβ-dependent regulation of IGF-1R, p38 MAPK and Pl3K/Akt pathways. PLoS ONE. 2014;9(3): e91245. https://doi.org/10.1371/journal.pone.0091245.
Li S, Wang Y, Feng C, Wu G, Ye Y, Tian J. Calycosin inhibits the migration and invasion of human breast cancer cells by down-regulation of Foxp3 expression. Cell Physiol Biochem. 2017;44:1775–84. https://doi.org/10.1159/000485784.
Huang CJ, Lin C, Yong W, Ye Y, Huang Z. Calycosin and genistein induce apoptosis by inactivation of HOTAIR/p-Akt signaling pathway in human breast cancer MCF-7 cells. Cell Physiol Biochem. 2015;35:722–8. https://doi.org/10.1159/000369732.
Chen J, Zeng J, Xin M, Huang W, Chen X. Formononetin induces cell arrest of human breast cancer cells via IGF1/PI3K/Akt pathways in vitro and in vivo. Horm Metab Res. 2011;43:681–6. https://doi.org/10.1055/s-0031-1286306.
Chen J, Sun L. Formononetin—induced apoptosis by activation of Ras/p38 mitogen—activated protein kinase in estrogen receptor-positive human breast cancer cells. Horm Metab Res. 2012;44:943–8. https://doi.org/10.1055/s-0032/1321818.
Ling Y, Chen Y, Chen P, Hui H, Song X, Lu Z, et al. Baicalein potentially suppresses angiogenesis induced by vascular endothelial growth factor through the p53/Rb signaling pathway leading to G1/S cell cycle arrest. Exp Biol Med (Maywood). 2011;236(7):851–8. https://doi.org/10.1258/ebm.2011.010395.
Ma X, Yan W, Dai Z, Gao X, Ma Y, Xu Q, et al. Baicalein supresses metastasis of breast cancer cells by inhibiting EMT via downregulation of SATB1 and Wnt/β-catenin pathway. Drug Des Devel Ther. 2016;10:1419–41. https://doi.org/10.2147/DDDT.S102541.
Brennan KR, Brown AMC. Wnt proteins in mammary development and cancer. J Mamm Gland Biol Neoplasia. 2004;9(2):119–31. https://doi.org/10.1023/B:JOMG.0000037157.
Christofori G. New signals from the invasive front. Nature. 2006;441(7092):444–50. https://doi.org/10.1038/nature04872.
Park SH, Ham S, Kwon TH, Kim MS, Lee DH, Kang JW, et al. Luteolin induces cell cycle arrest and apoptosis through extrinsic and intrinsic signaling pathways in MCF-7 brist cancer cells. J Environ Pathol Toxicol Oncol. 2014;33(3):219–31. https://doi.org/10.1615/JEnvironPatholToxicolOncol.2014010923.
Wang Y, Wang J, Gong X, Wen X, Gu X. Luteolin: anti-breast cancer effects and mechanisms. J Expl Res Pharmacol. 2018;3(3):85–90. https://doi.org/10.14218/JERP2018.00011.
Kucinska M, Giron MD, Piotrowska H, Lisiak N, Granik WH, Lopez-Jaramillo FJ, et al. Novel promising estrogenic receptor modulators: cytotoxic and estrogenic activity of benzanilides and dithiobenzalinides. PLoS ONE. 2016;11:1–16. https://doi.org/10.1371/journal.pone.0145615.
Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y. Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res. 2004;24:2783–840.
Perdew GH, Hollingshead BD, Di Natale BC, Morales JL, Labrecque MP, Takhar MK, et al. Estrogen receptor expression is required for low-dose resveratrol-mediated repression of aryl hydrocarbon receptor activity. J Pharmacol Exp Ther. 2010;335:273–83. https://doi.org/10.1124/jpet.110.170654.
Park SA, Na HK, Surh YJ. Resveratrol suppresses 4-hydroxyestradiol-induced transformation of human breast epithelial cells by blocking IκB kinase β-NF-κB signalling. Free Radic Res. 2012;46(8):1051–7. https://doi.org/10.3109/10715762.2012.671940.
Murias M, Miksits M, Aust S, Spatzenegger M, Thalhammer T, Szekeres T, et al. Metabolism of resveratrol in breast cancer cell lines: Impact of sulfotransferase 1A1 expression on cell growth inhibition. Cancer Lett. 2008;261:172–82. https://doi.org/10.1016/j.canlet.2007.11.008.
Murias M, Luczak MW, Niepsuj A, Krajka-Kuzniak V, Zielinska-Przyjemska M, Jagodzinski PP, et al. Cytotoxic activity of 3,3’,4,4’,5,5’-hexahydroxystilbene against Breast cancer cells is mediated by induction of p53 and downregulation of mitochondrial superoxide dysmutase. Toxicol Vitro. 2008;22:13611370. https://doi.org/10.1016/j.tiv.2008.03.002.
Kucinska M, Piotrowska H, Luczak MW, Mikula-Pietrasik J, Ksiazek K, Wozniak M, et al. Effects of hydroxylated resveratrol analogs on oxidative stress and cancer cells death in human acute T cell leukemia cell line. Prooxidative potential of hydroxylated resveratrol analogs. Chem Biol Interact. 2014;209:96–110. https://doi.org/10.1016/j.cbi.2013.12.009.
Murias M, Jager W, Handler N, Erker T, Horvath Z, Szekeres T, et al. Antioxidant, prooxidant and cytotoxic activity of hydroxylated resveratrol analogues: structure-activity relationship. Biochem Pharmacol. 2005;69:903–12. https://doi.org/10.1016/j.bcp.2004.12.001.
Piotrowska H, Kucinska M, Murias M. Biological activity of piceatannol: leaving the shadow of resveratrol. Mutat Res. 2012;750:60–82. https://doi.org/10.1016/j.mrrev.2011.11.001.
Takemura H, Uchiyama H, Ohura T, Sakakibara H, Kuruko R, Amagai T, et al. A methoxyflavonoid, chrysoeriol, selectively inhibits the formation of a carcinogenic estrogen metabolite in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol. 2010;118(1–2):70–6. https://doi.org/10.1016/j.jsbmb.2009.10.002.
Lu F, Zahid M, Wang C, Saeed M, Cavalieri EL, Rogan EG. Resveratrol prevents estrogen-DNA adduct formation and neoplastic transformation in MCF-10F cells. Cancer Prev Res. 2008;1(2):135–45. https://doi.org/10.1158/1940.6207.CAPR-08-0037.
Zahid M, Gaikwad NW, Rogan EG, Cavalieri EL. Inhibition of depurinating estrogen-DNA adduct formation by natural compounds. Chem Res Toxicol. 2007;20(12):1947–53. https://doi.org/10.1021/tx700269s.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest regarding the publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Starek-Świechowicz, B., Budziszewska, B. & Starek, A. Endogenous estrogens—breast cancer and chemoprevention. Pharmacol. Rep 73, 1497–1512 (2021). https://doi.org/10.1007/s43440-021-00317-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-021-00317-0