
Abstract
Background
Irritable bowel syndrome (IBS) is a chronic condition with recurring gastrointestinal (GI) symptoms: altered motility and abdominal pain. As endogenous opioid system participates in pain perception and in the control of GI peristalsis, opioids have been proposed as a promising therapy in IBS. In a previous study, we observed that morphiceptin derivative, P-317 (Dmt-cyclo-(d-Lys-Phe-d-Pro-Asp)-NH2), presents promising features to be applied in IBS. In this project, we tested whether modifications in cyclic morphiceptin-based structure: fluorination (compound 1) or peptide bond reduction (compound 2) improve pharmacological effect.
Methods
We evaluated tested derivatives in the mouse GI system under physiological (GI transit) and pathophysiological (castor oil diarrhea, stress-induced hypermotility, visceral pain) conditions.
Results
Both compounds prolonged GI transit. Compound 1 and P-317 inhibited upper GI transit and motility of the colon; compound 2 remained inactive. Compound 1 and P-317 inhibited hypermotility in stressed mice and delayed the acute diarrhea in comparison to control. Only P-317 exerted antinociceptive effect. None of tested derivatives, similar to P-317, affected locomotor activity.
Conclusions
Compound 1 is equally effective as P-317 in the mouse GI tract. The peptide bond reduction decreased the activity of compound 2. Fluorination appears to be an efficient way to increase the effects of morphiceptin analogs in the GI tract.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Irritable bowel syndrome (IBS), a result of brain–gut axis disturbance with persistent and recurring gastrointestinal (GI) symptoms, affects even up to 11% of the worldwide population [1]. IBS is characterized by disturbed bowel motility, abdominal pain, bowel distension and thus abdominal discomfort. Based on the stool consistency (assessed using the Bristol stool scale) several subtypes of IBS can be distinguished: diarrhea predominant, IBS-D; constipation predominant, IBS-C; IBS with mixed bowel habits, IBS-M; IBS with no significant abnormalities in patient’s stool, IBS-U [2].
Due to the involvement of the endogenous opioid system (EOS) in the proper functioning of the GI system, opioids have been considered an interesting treatment option in the therapy of GI diseases. In the family of ORs, three types are distinguished: µ-(MOP), δ-(DOP) and ĸ-(KOP) receptors. In the GI system, each of these receptors maintains different functions, e.g., MOP receptors participate in the control of motility, DOP receptors—secretion and absorption and KOP receptors—mainly immunomodulation [3, 4]. One of the agonists of MOP receptors, loperamide, is a commonly used drug to treat acute diarrhea. However, as loperamide inhibits the transit along the intestines and induces constipation instead, it cannot be applied in patients with chronic diarrhea, for example, functional diarrhea (FD) or IBS-D [5].
The new generation of opioids with mixed activity at ORs presents high therapeutic potency. In general, mixed agonists are characterized as more potent in antinociception and safer in comparison to ‘classic opioids’ (as the risk of adverse effects such as tolerance and physical dependence development or occurrence of constipation is lower). One example is a mixed MOP/DOP agonist, biphalin, which possesses stronger analgesic activity than morphine in preclinical studies and during chronic treatment its ability to develop tolerance is significantly lower in comparison to morphine [6]. Another opioid with mixed MOP/DOP receptor affinity is chimeric peptide MCRT (heptapeptide based on morphiceptin and a neuropeptide FF). It was assessed that the dose of MCRT required to induce analgesia in tail-flick test in mice is lower than that which evokes alterations in the GI peristalsis [7]. Biphalin and MCRT never entered the clinical studies. However, there is currently a mixed OR ligand that has already been accepted for the treatment of IBS-D: eluxadoline (named originally MuDelta). Eluxadoline, an agonist of MOP and antagonist of DOP receptors, improves the functioning of the GI tract in the course of IBS; however, its efficacy is not satisfactory. The antagonistic component of eluxadoline at DOP receptors has a positive effect on the GI peristalsis, as it counteracts the inhibitory action of the MOP receptor activation [8].
Morphiceptin (Tyr-Pro-Phe-Pro-NH2) is a tetrapeptide opioid isolated from the enzymatic digest of bovine β-casein that induces a potent antinociceptive effect after intra-cerebroventricular (icv) injection, but not after peripheral administration, as it is prone to enzymatic degradation [9]. To find an alternative to currently used opioids in the therapy of IBS, our group has previously evaluated the action of morphiceptin derivative, cyclic pentapeptide P-317 (Dmt-cyclo-(d-Lys-Phe-d-Pro-Asp)-NH2; Dmt = 2,6-dimethyl-l-tyrosine) on the GI functions [10]. P-317 is an orally available OR agonist with high affinity for MOP receptor and moderate for KOP receptor that turned out to be effective in the mouse models mimicking the symptoms of IBS-D. P-317 modulated GI motility under physiological and pathophysiological conditions and attenuated abdominal pain after intraperitoneal (ip) and intragastric (ig) administration. In contrast to morphiceptin, P-317 was not enzymatically degraded out of the CNS [10].
The goal of this study was to continue the research on cyclic derivatives of morphiceptin in the GI tract to evaluate how modifications of the P-317 chemical structure: incorporation of fluorine in the Pro (compound 1) and the reduction of peptide bond between Pro and Asp (compound 2; Table 1) will change its pharmacological activity in the GI tract to develop the most promising drug candidate for the therapy of IBS.
Materials and methods
Animals
In this study, male Balb/C mice (University of Lodz, Lodz, Poland), weighing from 22 to 26 g, were used. Mice were maintained under a 12-h light/dark cycle and at 22–23 °C temperature. Animals were housed in sawdust-lined plastic transparent cages with a free access to tap water and laboratory chow. Experiments in this project were performed in accordance with respective national guidelines and animal use was approved by the Local Ethical Committee (48/ŁB700/DLZ/2015).
Whole GI transit
Whole GI transit test was performed as described earlier [11]. Tested derivatives, P-317 or vehicle were injected ip 15 min before ig administration of 0.15 ml of blue marker.
Upper GI transit
The transit of the upper part of the GI tract was assessed in mice fasted overnight with free access to top water according to the protocol described by Zielińska et al. [10]. On the experimental day, animals received tested derivatives, P-317 or vehicle (ip) 15 min before the gavage of 0.2 ml of a red marker solution (50 mg phenol red in 100 ml 1.5% methylcellulose). Upper GI transit, geometric center of the GI tract, and gastric emptying were evaluated.
Colonic bead expulsion test
Colonic expulsion test was performed as described earlier by our group [12]. Tested compounds (compound 1, compound 2), P-317 or vehicle were injected ip 15, 30, 45, 60, 90 or 120 min before colon bead insertion.
Castor oil-induced diarrhea
The anti-diarrheal action was assessed in the mouse model of castor oil-induced diarrhea [12]. Compounds or vehicle was injected ip 15 min before castor oil administration.
Fecal pellet output
The exposition to novel environment was used to mimic stress conditions and induce hypermotility. To induce hypermotility mice were not separated for habituation before the experiment. Fecal pellet output was assessed in habituated and non-habituated mice according to the protocol described earlier [10].
Mustard oil-induced pain
Behavioral pain test with mustard oil (MO, administered intracolonically, ic) was performed as described by Sobczak et al. [11].
Locomotor activity
The measurement of locomotor activity was made automatically in a computerized actimeter (PanLab, LE8825, Witko, Lodz, Poland) according to the protocol [10].
Drugs
All reagents and drugs, unless otherwise stated, were purchased from Sigma-Aldrich (Poznan, Poland). Tested derivatives (compound 1, compound 2) and P-317 were synthesized by TriMen Chemicals, Lodz, Poland. Peptides were synthesized on the Rink resin using Fmoc-group strategy with allyl-based protecting groups for side chains of d-Lys and Asp residues (alloc- and allyl esters, respectively). Allyl-based group removal and cyclization (amide bond formation) of the side chains were performed on-resin. The reduced peptide bond between d-Pro and Asp residues in compound 2 was synthesized by on-resin reductive alkylation of the amino group of Asp with Fmoc-d-prolinal obtained in two synthetic steps from commercially available Fmoc-d-Pro-OH. After cleavage from the resin, peptides were purified by preparative reversed-phase HPLC and freeze-dried [13].
In the in vitro experiments, all drugs were dissolved in dimethyl sulfoxide (DMSO). In the in vivo tests, drugs were dissolved in 5% DMSO in 0.9% NaCl, which was used as vehicle in control group. The vehicle given alone had no effects on the observed parameters.
Statistics
Statistical analysis was performed using Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA). The data are expressed as mean ± SEM. In the in vitro experiments, n indicates the number of individual tissue samples from ≥ 3 different animals, while in the in vivo studies n stands for the number of animals. Student’s t test was used to compare single treatment means with control means. Analysis of one-way variance (ANOVA) followed by Newman–Keuls post hoc test was used for the analysis of multiple treatment means. P values < 0.05 were considered as statistically significant.
Results
Physiological conditions
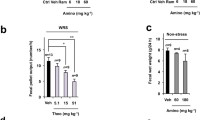
We observed that compound 1, compound 2 and P-317 were resistant to enzymatic degradation, as after systemic administration (ip), as they prolonged whole GI transit in comparison to control (Fig. 1). To explore in more detail the effect of P-317 and its derivatives in the GI tract we examined the influence of compound 1, compound 2 and P-317 on the motility of particular parts of the GI tract. First, we observed that compound 1 and P-317 significantly reduced the transit through the upper GI tract in comparison to control, while compound 2 remained inactive (Fig. 2a). In further analysis of the upper GI transit, the position of geometric center of the small intestine was assessed. None of the tested derivatives or P-317 altered this parameter in a statistically significant manner (Fig. 2b). Notably, tested derivatives and P-317 affected the emptying of the stomach: compound 1 and P-317 inhibited gastric emptying. The opposite was reported for compound 2: it accelerated the emptying of the stomach (Fig. 2c). Second, we evaluated the effect of morphiceptin derivatives on the motility of the distal colon (Fig. 3). Compound 1 and P-317 significantly inhibited colonic motility in the glass expulsion test. Compound 1 remained active up to 30 min following its administration, while the action of P-317 was pronounced up to 60 min. Compound 2 did not affect the colonic motility.

P-317, compound 1 and compound 2 significantly prolonged transit along GI tract. Data represent mean ± SEM for n = 6–8 mice per group. ***P < 0.001 as compared to control

This figure shows the effect of P-317, compound 1 and compound 2 on the motility of the upper part of the mouse GI tract (a), the position of geometric center in the small intestine (b) and gastric emptying (c). Data represent mean ± SEM for n = 6–8 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001 as compared to control

The effect of P-317, compound 1 and compound 2 (0.01 mg/kg, ip) on motility of the mouse distal colon. The experiments varied by the time between drug administration and insertion of the glass bead: 15, 30, 45, 60, 90, 120 min, respectively. Data represent mean ± SEM for n = 6–8 mice per group. *P < 0.05, **P < 0.01, ***P < 0.001 as compared to control
Pathophysiological conditions
Compound 1 and P-317 evoked a strong antidiarrheal action: the time to the occurrence of watery stools was 2.5-fold longer in comparison to control. There was no difference between group treated with compound 2 and the controls (Fig. 4a).

The action of P-317, compound 1 and compound 2 under pathophysiological conditions in the GI tract: the antidiarrheal effect of tested compounds in the castor oil-induced acute diarrhea (a) and stress-induced hypermotility (b). Data represent mean ± SEM for n = 6–8 mice per group. ***P < 0.001 as compared to control
In our study, compound 1, compound 2 and P-317 did not affect the number of fecal pellets excreted within a 1-h period when compared to control (Fig. 4b). Interestingly, when the intestinal motility was accelerated by stress, compound 1 and P-317 decreased the fecal pellet output, while compound 2 remained inactive.
In the MO pain test, compounds 1 and 2 slightly decreased the number of behavioral pain reactions; however, the effect was not statistically significant. In contrast, P-317 induced pronounced antinociceptive action when compared to control group (Fig. 5).

The antinociceptive action of P-317, compound 1 and compound 2 in the mustard oil induced pain test in mice. Data represent mean ± SEM for n = 6–8 mice per group. *P < 0.05 as compared to control
Locomotor activity
Interestingly, most of the adverse effects during therapy with opioids are related to the CNS (i.e., respiratory depression, mood changes, sedation). In our study, tested compounds did not penetrate blood–brain barrier as they did not affect horizontal locomotor activity up to 60 min following administration in comparison to control (Fig. 6).

The influence of P-317, compound 1 and compound 2 on the locomotor activity in mice. Data represent mean ± SEM for n = 3–5 mice per group
Discussion
In the present study, we assessed if chemical modification, including fluorination (compound 1) and reduction of peptide bond (compound 2), affects pharmacological activity of cyclic morphiceptin derivatives. We observed that the activity of tested derivatives varied in the GI tract: compound 1, in contrast to compound 2 significantly inhibited the transit along GI tract (in the upper and lower part) similar to P-317. Neither compound 1 nor compound 2 entailed antinociceptive effect, while P-317 alleviated abdominal pain.
Compound 1 contains in its chemical structure fluorine in Pro (4-fluoroproline). Literature shows that the effect of fluorine incorporation is hard to predict and strongly depends on its position in the derivative; the major effect is the alteration of acidity or basicity of the parent compound that impacts its pharmacological properties, bioavailability and receptor affinity [14]. Previously, fluorinated derivatives of morphiceptin were synthesized and evaluated by Adamska et al. [15]. They assessed how the incorporation of fluorine in positions 2 or/and 4 by introduction of 4,4-difluoroproline (F2Pro) affects the receptor binding affinity of four fluorinated morphiceptin analogs. Tested compounds varied by the affinity at MOP receptors: ligand 4 (Tyr-F2Pro-Phe-d-F2Pro-NH2) presented the highest activity at MOP and relative low affinity at KOP receptors. It was concluded that the substitution of more hydrophobic derivative with F2Pro2 instead of Pro2 results in the formation of stronger ligand–receptor interactions [15].
The amino acid chain of the compound 2 contains reduced peptide bond between d-Pro and Asp. In the study of Delaet et al. [16], it was observed that the reduction of the peptide bond in morphiceptin improved an enzymatic resistance in the human plasma. Moreover, as assessed in the guinea pig brain, the incorporation of reduced peptide in the structure of morphiceptin resulted in a significant increase in the affinity and selectivity to MOP receptors. Compound 5 (H-Tyr-Pro-Phe-(CH2-NH)-Pro-NH2) from the study of Delaet et al. [16] inhibited electrically evoked contractions in the guinea pig ileum and this effect was dependent on MOP receptors. In contrast, compound 4 (H-Tyr-Pro-(CH2-NH)-Phe-Pro-NH2) presented very weak response. It was concluded that reduction of peptide bond between Pro and Phe amino acids did not alter the binding to ORs receptors in comparison to morphiceptin. Concurrently, the same modification between Phe and Pro2 amino acids increased the affinity at MOP receptors in the guinea pig brain but it entailed lack of effect in the functional assays on peripheral MOP receptors [16].
Our results indicate that P-317 and derivatives are superior to morphiceptin because they are enzymatically stable after systemic administration (i.e., they exert inhibitory effect in the whole GI transit test after ip administration). In the previous studies on morphiceptin derivatives conducted by Fichna et al. [17], it was shown that cyclization improves the stability of morphiceptin-based compounds against enzymatic degradation. Noteworthy, morphiceptin (0.3 and 1 mg/kg)-injected ip was inactive in the mouse GI tract [9]. However, when administered directly into the central nervous system (morphiceptin (10–30 nmol/mouse) significantly inhibited the transit along the small intestine [18].
Noteworthy, all tested compounds prolonged whole GI transit. However, this inhibitory action was observed in more detailed studies on the upper and lower parts of the GI tract only in case of compound 1 and P-317. Similarly, morphiceptin administered centrally (icv) significantly prolonged gastric emptying and inhibited upper GI transit in a dose-dependent manner (10–30 nmol/mouse) [18]. Moreover, He et al. [18] evaluated the effect of MCRT (heptapeptide based on morphiceptin and a neuropeptide FF) at the upper GI transit. MCRT inhibited the emptying of the stomach and prolonged the transit along the small intestine in a dose-dependent manner (10–30 nmol/mouse, icv). This effect was reversed by non-selective OR antagonist, naloxone and selective DOP antagonist (naltrindole), but not cyprodime (MOP antagonist) [18].
As both, P-317 and compound 1, prolonged the process of gastric emptying it is worth mentioning that delayed gastric emptying is not an eligible feature for potential therapeutics in GI diseases, because it may cause nausea and emesis [19]. However, MuDelta, which is currently applied in clinics as eluxadoline reduced upper GI transit by 50% already at the lowest dose tested (25 mg/kg, po) in mice. Nevertheless, nausea/emesis was not a limitation of the therapy with eluxadoline in clinical trial: 8.1% of patients treated with eluxadoline at the dose of 75 mg and 7.5% eluxadoline at 100 mg vs. 5.1% in the placebo group experienced nausea [20].
Interestingly, compound 2 prolonged whole GI transit time, while it did not affect upper GI transit and colonic motility. We presume that the lack of action of compound 2 in the upper GI transit test may occur due to the acceleration of gastric emptying combined with the inhibitory effect on the motility of the proximal part of small intestine.
In further experiments, we assessed the action of tested derivatives and P-317 under pathophysiological conditions: castor oil-induced acute diarrhea, stress-induced hypermotility, and MO-induced pain test. These mouse models allowed us to mimic IBS symptoms and to assess potential application of tested derivatives in clinics.
In our study, compound 1 and P-317, but not compound 2, evoked a strong antidiarrheal action. Gach et al. [21] synthesized and tested several morphiceptin derivatives in which morphiceptin was modified at position 1 by replacing Tyr by Dmt (2′,6′-dimethyltyrosine) and at positions 2 or 3 by incorporation of β2- or β3-amino acids (β-Ala and β-1-3-(1-naphthyl)-alanine). Two among tested peptides (peptide 2 (Dmt-(R)-β2-Ala-d-1-Nal-Pro-NH2) and peptide 4 (Dmt-d-Ala-(R)-β2-1-Nal-Pro-NH2)) were characterized by high affinity at MOP and DOP receptors; peptide 4 was also active at KOP receptor. Peptide 2 and peptide 4 were tested in the model of acute diarrhea. Both compounds at the dose of 0.1 mg/kg exerted a strong antidiarrheal activity, comparable to loperamide (0.1 mg/kg, ip). Peptide 4 was also active after oral administration (1 mg/kg); however, the effect was not as strong as after loperamide (1 mg/kg).
The important feature of potential therapeutic for IBS is the ability to control accelerated peristalsis (for example being a result of stress) without affecting the regular peristalsis. Noteworthy, compound 1 and P-317, but not compound 2, reduced the number of fecal pellets in mouse model of hypermotility induced by stress, while it did not impact the excretion in non-stressed animals. The similar action was reported for MuDelta: at the doses range 10–25 mg/kg, po, it did not affect fecal pellet output in acclimated mice although in stressed mice MuDelta was effective already at the lowest dose tested (10 mg/kg, po) [22]. Chimeric peptide, MRCT, administered centrally significantly reduced fecal pellet output in mice (1.5–4 nmol/mouse, icv) in naloxone-reversible manner [18]. The results on mixed OR agonists and their influence on GI peristalsis are comparable: it appears that mixed OR agonists normalize accelerated GI peristalsis without constipating effect.
In our study, P-317 induced pronounced antinociceptive action, while compound 1 and compound 2 did not decrease the number of behavioral pain reactions. Despite the fact that the results of MO-induced pain test of compound 1 were not statistically significant, there is a positive tendency in its action (29 pain-related behaviors in mice treated with compound 1 vs. 41.2 in control group, during 20 min of observation). Nevertheless, the efficacy of compound 1 is still about 50% weaker than P-317. Therefore, we assume that the dose we used is not enough to induce a potent antinociceptive action in MO assay. The separation of the analgesic action and the influence on the GI tract is a critical feature that differs between the OR agonists. The occurrence of opioid-induced bowel dysfunction is an important among adverse effects related to pain therapy with opioids. Thus, in the process of development new analgesics, the goal is to minimize the action in the GI tract at doses that produce analgesia. For example, MCRT administrated icv produced significant analgesic effect (in tail flick test, ED50 = 0.03 nmol/mouse) and a required dose was far lower than those needed for its action in the GI tract (over 1 nmol/mouse). Therefore, MCRT presents a separation of analgesic action and the effects in the GI tract [18]. Similar results were reported for morphine-6-O-sulphate in several tests: the tail flick test, chronic constriction nerve injury hyperalgesia and allodynia, and formalin test [23]. On the contrary, the doses of morphine-6-O-sulphate required to alleviate visceral pain in acetic acid-induced writhing in mice were equal to doses that affect GI peristalsis [24]. Morphiceptin derivatives with β2- or β3-amino acids synthesized by Gach et al. [21]: peptide 2 and peptide 4 exerted strong antinociceptive action in acetic acid writhing test in mice, the effect was comparable to morphiceptin (0.1 mg/kg, ip). Both compounds at the dose of 0.1 mg/kg exerted a strong antidiarrheal activity. In case of compound 1 and compound 2, tested in our experiments, the dose required to induce antinociceptive effect in MO model of visceral pain may be a higher than that required to influence the GI peristalsis.
In summary, compounds 1 and 2, despite their similar effect in whole GI transit test, differ in more detailed study on particular GI segments. Fluorinated analog (compound 1) appears to be equally effective to P-317 in the mouse GI tract, while compound 2 did not alter the upper GI transit and remained inactive in the colon. These cyclic derivatives of morphiceptin are a promising template for further studies as they are superior to the parent compound due to their resistance to enzymatic degradation and lack of the activity in the CNS. However, the modification of a peptide bond between Pro and Asp resulted in decreased activity of tested compound in the GI tract in comparison to P-317. Therefore, fluorination appears to be an efficient way to modify and increase the effects of morphiceptin analogs in the GI tract.
References
Enck P, Aziz Q, Barbara G, Farmer AD, Fukudo S, Mayer EA, et al. Irritable bowel syndrome. Nat Rev Dis Prim. 2016;2:16014. https://www.ncbi.nlm.nih.gov/pubmed/27159638.
Zeevenhooven J, Koppen IJN, Benninga MA. The new Rome IV criteria for functional gastrointestinal disorders in infants and toddlers. Pediatr Gastroenterol Hepatol Nutr. 2017;20(1):1.
Sobczak M, Sałaga M, Storr MA, Fichna J. Physiology, signaling, and pharmacology of opioid receptors and their ligands in the gastrointestinal tract: current concepts and future perspectives. J Gastroenterol. 2014;49(1):24–45. https://www.ncbi.nlm.nih.gov/pubmed/23397116.
Jarmuż A, Banaszek M, Leń K, Storr M, Zielińska M, Fichna J. The role of MOP and DOP receptors in treatment of diarrhea-predominant irritable bowel syndrome. Mini Rev Med Chem. 2016. https://www.ncbi.nlm.nih.gov/pubmed/27494159.
Awouters F, Megens A, Verlinden M, Schuurkes J, Niemegeers C, Janssen PAJ. Loperamide. Dig Dis Sci. 1993;38(6):977–95. https://link.springer.com/10.1007/BF01295711.
Shen K-F, Crain SM. Biphalin, an enkephalin analog with unexpectedly high antinociceptive potency and low dependence liability in vivo, selectively antagonizes excitatory opioid receptor functions of sensory neurons in culture. Brain Res. 1995;701(1–2):158–66. https://linkinghub.elsevier.com/retrieve/pii/0006899395009991.
Li M, Zhou L, Ma G, Dong S. Analgesic properties of chimeric peptide based on morphiceptin and PFRTic-amide. Regul Pept. 2012;179(1–3):23–8. https://www.ncbi.nlm.nih.gov/pubmed/22960407.
Lembo AJ, Lacy BE, Zuckerman MJ, Schey R, Dove LS, Andrae DA, et al. Eluxadoline for irritable bowel syndrome with diarrhea. N Engl J Med. 2016;374(3):242–53.
Gach K, do-Rego JC, Fichna J, Storr M, Delbro D, Toth G, et al. Synthesis and biological evaluation of novel peripherally active morphiceptin analogs. Peptides. 2010;31(8):1617–24. https://www.sciencedirect.com/science/article/abs/pii/S0196978110001725?via%3Dihub.
Zielińska M, Chen C, Mokrowiecka A, Cygankiewicz AI, Zakrzewski PK, Sałaga M, et al. Orally administered novel cyclic pentapeptide P-317 alleviates symptoms of diarrhoea-predominant irritable bowel syndrome. J Pharm Pharmacol. 2015;67(2):244–54. https://www.ncbi.nlm.nih.gov/pubmed/25515402.
Sobczak M, Cami-Kobeci G, Sałaga M, Husbands SM, Fichna J. Novel mixed NOP/MOP agonist BU08070 alleviates pain and inhibits gastrointestinal motility in mouse models mimicking diarrhea-predominant irritable bowel syndrome symptoms. Eur J Pharmacol. 2014;736:63–9.
Zielińska M, Jarmuż A, Sałaga M, Lipkowski AW, Fichna J. Mixed MOP/DOP agonist biphalin elicits anti-transit effect in mouse models mimicking diarrhea-predominant irritable bowel syndrome symptoms. Pharmacol Rep. 2016;68(1):32–6. https://www.ncbi.nlm.nih.gov/pubmed/26721348.
Pels K, Kodadek T. Solid-phase synthesis of diverse peptide tertiary amides by reductive amination. ACS Comb Sci. 2015;17(3):152–5. https://www.ncbi.nlm.nih.gov/pubmed/25695359.
Wang J, Sa M, Acen L, del Pozo C, Sorochinsky AE, Fustero S, et al. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011). 2013. https://pubs.acs.org/sharingguidelines.
Adamska A, Kluczyk A, Cerlesi MC, Calo G, Janecka A, Borics A. Synthesis, biological evaluation and structural analysis of novel peripherally active morphiceptin analogs. Bioorg Med Chem. 2016;24(7):1582–8. https://www.sciencedirect.com/science/article/pii/S0968089616301237?via%3Dihub.
Delaet NGJ, Verheyden PMF, Tourwe D, Van Binst G, Davis P, Burks TF. Morphiceptin and β-casomorphin-5 analogues containing a reduced peptide bond: selective μ-receptor agonists and a novel μ antagonist, H-Tyr-Proψ(CH2-NH)Phe-Pro-Gly-OH. Biopolymers. 1992;32(8):957–69. https://www.ncbi.nlm.nih.gov/pubmed/1330047.
Fichna J, Perlikowska R, Wyrębska A, Gach K, Piekielna J, do-Rego JC, et al. Effect of 2,6-dimethyl-l-tyrosine (Dmt) on pharmacological activity of cyclic endomorphin-2 and morphiceptin analogs. Bioorg Med Chem. 2011;19(23):6977–81. https://www.sciencedirect.com/science/article/pii/S0968089611008492?via%3Dihub.
He C, Li H, Zhang J, Kang Y, Jia F, Dong S, et al. Supraspinal inhibitory effects of chimeric peptide MCRT on gastrointestinal motility in mice. J Pharm Pharmacol. 2017;69(9):1244–51. https://doi.org/10.1111/jphp.12761.
Camilleri M, Lembo A, Katzka DA. Opioids in gastroenterology: treating adverse effects and creating therapeutic benefits. Clin Gastroenterol Hepatol. 2017;15(9):1338. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5565678/.
Lembo AJ, Lacy BE, Zuckerman MJ, Schey R, Dove LS, Andrae DA, et al. Eluxadoline for irritable bowel syndrome with diarrhea. N Engl J Med. 2016;374(3):242–53. https://www.ncbi.nlm.nih.gov/pubmed/26789872.
Gach-Janczak K, Piekielna-Ciesielska J, Adamska-Bartłomiejczyk A, Perlikowska R, Kruszyński R, Kluczyk A, et al. Synthesis and activity of opioid peptidomimetics with β2- and β3-amino acids. Peptides. 2017;95:116–23. https://www.sciencedirect.com/science/article/abs/pii/S0196978117302504?via%3Dihub.
Wade PR, Palmer JM, Mckenney S, Kenigs V, Chevalier K, Moore BA, et al. Modulation of gastrointestinal function by MuDelta, a mixed m opioid receptor agonist/ m opioid receptor antagonist. Br J Pharmacol. 2012;167:1111. https://www.brjpharmacol.org.
Holtman JR, Crooks PA, Johnson-Hardy J, Wala EP. Antinociceptive effects and toxicity of morphine-6-O-sulfate sodium salt in rat models of pain. Eur J Pharmacol. 2010;648(1–3):87–94.
Lackó E, Riba P, Giricz Z, Váradi A, Cornic L, Balogh M, et al. New morphine analogs produce peripheral antinociception within a certain dose range of their systemic administration. J Pharmacol Exp Ther. 2016;359(1):171–81.
Acknowledgements
Support by grants from the National Science Centre #UMO-2013/11/N/NZ7/00724 (to MZ) is acknowledged.
Author information
Authors and Affiliations
Contributions
MZ and JF designed the research study; AS, JW, and MZ performed the research; AS and MZ analyzed the data; AS and MZ wrote the paper; JO and MM provided morphiceptin derivatives. All authors critically revised the paper and approved the final version of manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Authors have no conflicts of interest to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szymaszkiewicz, A., Włodarczyk, J., Mazur, M. et al. Cyclic derivatives of morphiceptin possess anti-transit effect in the gastrointestinal tract and alleviate abdominal pain in mice. Pharmacol. Rep 72, 314–321 (2020). https://doi.org/10.1007/s43440-020-00084-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43440-020-00084-4