
Abstract
Purpose of Review
Treatment of liver fibrosis benefits from hepatic stellate cell (HSC)-specific delivery. Since the description of first carrier to HSC, many developments have taken place in this area. The purpose is to give an overview of the different carriers and homing moieties that are available for HSC targeting and illustrate the opportunities and hurdles they provide.
Recent Findings
There is a growing number of homing devices to deliver drugs to HSC, and options to deliver siRNA to HSC have emerged. Other developments include controlling corona formation, development of linker technology, and design of theranostic approaches. We are on the eve of reaching the clinic with innovative HSC-specific compounds.
Summary
An overview of different core molecules is presented together with an overview of targeting strategies toward different receptors on HSC, providing a versatile toolbox. Many therapeutics, ranging from small chemical entities and proteins to RNA- or DNA-modulating substances, have already been incorporated in these constructs in the recent years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Maybe a hundred years from now, people look back upon the present times as the era when drugs were administered without detailed control of their exact fate in the patient, and drugs were given to patients using statistics to estimate beforehand whether a person might benefit from that. Yet, times are changing, and the number of innovative formulations that allow cell-specific delivery and facilitate personalized medicine is rapidly expanding now. Today, many of these new cell-specific approaches are focused upon the treatment of cancers. Hundreds of monoclonal antibodies against many different cancer cell types are now being tested in different phases of (pre-)clinical trials (for review see [1, 2]). The treatment of many types of neoplasms obviously benefits from a cell-specific delivery of cell-killing or growth-arresting compounds, but similar, the treatment of many chronic diseases and in particular fibrotic diseases may also benefit from a cell-specific delivery. Fibrotic diseases, with the exception of idiopathic pulmonary fibrosis, are slowly progressive processes where several cell types and processes counterbalance each other, leading to a very complex situation in vivo [3••, 4••],. Many of the experimental antifibrotic drugs that perfectly work within the target cell in vitro are ineffective in vivo, either due to compensatory mechanisms or due to lack of uptake in target cells. The clinical trials examining Simtuzumab in patients with Primary Sclerosing Cholangitis (PSC) [5, 6], and non-alcoholic steatohepatitis (NASH) [7] are the most recent example of this. Clinical trials with this compound were unsuccessful; either uptake in target cells was too low [7] or the target of this antibody (lysyl oxidase-like protein 2) is not the (only) key player in disease progression [6]. Moreover, unlike the formation of tumors, tissue turnover and repair are normal physiological processes that occur in all normal tissues, leading to adverse effects of antifibrotic drugs. Such off-target effects are not very well accepted for chronic diseases in particular when the symptoms of the disease are not life-threatening yet, which is the case at the early stages of liver fibrosis. The intrahepatic delivery of therapeutics is therefore a key to treat diseases like liver fibrosis. Surprisingly, only a very few monoclonal antibodies have been tested to treat this disease, although there are many similarities with cancer [8]. Simtuzumab did reach clinical trials, but this is a rare case, in contrast to the hundreds of tumor-directed antibodies. Yet, to enhance delivery of drugs in the target area, many other strategies have been developed using a variety of drug carriers. The present review will focus on these developments, with special focus on the different drug carriers and targeting strategies that have been tested to enhance uptake in hepatic stellate cells (HSC), the key target cell for antifibrotic therapies.
Uptake of Drugs and Nanomedicines in the Liver
The liver contains several resident cell types that orchestrate together normal homeostasis, synthesize important plasma proteins and lipids, and take care of the clearance of metabolites, toxins, or foreign particles. To that end in particular, hepatocytes are endowed with many different transporter proteins that take up lipids, proteins, sugars, and chemical toxins [9, 10]. Uptake of plasma components by hepatocytes can occur by passive diffusion but also by active transport via many different transporters. Transporters on the sinusoidal site of hepatocytes, such as several types of organic anion transporters (OATs), organic cation transporters (OCTs), and the Na-Taurocholate Co-Transporting Polypeptide (NTCP), transport a wide variety of drugs from the blood into the hepatocyte through recognition of their physico-chemical properties, such as charge and lipophilicity, with often a broad overlap in substrate specificity (reviewed elsewhere [9, 10]. Next to these receptors, uptake of plasma glycoproteins by hepatocytes can occur via several lectin receptors such as the asialoglycoprotein receptor (ASGP-R) [11], and in particular, the ASGP-receptor that binds galactose residues has been applied in many studies to deliver proteins or other drug carriers to hepatocytes [11] by decorating core molecules with galactose or lactose moieties. However, even without drug targeting strategies, most drugs are taken up by the liver and uptake of these drugs mostly reflects uptake by hepatocytes.
Kupffer cells (KC) are liver-specific macrophages that also take up many different compounds. Compounds larger than 100 nm, foreign proteins, viruses, and bacterial products are taken up either non-specifically, or specifically via Fc receptors, complement receptors, the Coxsackie and adenovirus receptor (CAR), several types of scavenger receptors and lectin receptors like the mannose receptor [12, 13••], and other pattern recognizing receptors (PAMPs) or damage-associated membrane receptors (DAMPs) [14, 15]. In particular the mannose receptor [15] and scavenger receptors recognizing negatively charged proteins [16, 17] have been extensively used for cell-specific delivery of drugs to KC. KC represent approximately 80% of the total macrophage activity in the body [18], [19] and are therefore responsible for the clearance and immune presentation of most of the blood-born particles and foreign proteins. Even long-circulating compounds that are not immediately recognized by any uptake system eventually may end up in KC and macrophages in the spleen where the compounds are rapidly degraded. This has for instance been demonstrated for PEGylated liposomes [18]. PEGylation of compounds is done to induce enhance the half-life in plasma thus creating a long-circulating compound. However, repeated administrations of PEGylated compounds led to the accelerated blood clearance (ABC) phenomenon [18]. Unravelment of this phenomenon revealed evidence of macrophage activation by PEG liposomes and subsequent presentation of these constructs to the immune system, which led to rapid clearance from the blood [18]. Next to immune presentation, targeting to Kupffer cells or any other macrophage in the body also confers the risk of activating these macrophages thereby stimulating a local inflammatory process [14, 18, 19] or killing them with serious effects on the immune system. Many of the large constructs (> 100 nm) that are developed for drug-targeting purposes to tumors or other diseases are taken up by KC, eventually.
Sinusoidal endothelial cells (SEC) are aligning the sinusoids within the liver and via endothelial fenestrae (with diameters of around 200 nm), and the space of Disse and these SEC are exposed to plasma components on both sides. Blood cells can act as a plunger (as in a syringe) in the small sinusoids to push plasma via the fenestrae into the space of Disse to further facilitate contact of plasma components with hepatocytes, KC, and SEC. Together with the macrophages, liver sinusoidal endothelial cells represent the reticuloendothelial system (RES) [15, 20] that is responsible for the uptake of most large nanoparticles in the blood [21]. SEC takes up lipids and proteins non-specifically via transcytosis and pinocytosis mechanisms and by endocytosis via specific receptors such as scavenger receptors for negatively charged proteins [17, 22], and hyaluronic acid receptors [23]. Scavenger receptors and hyaluronic acid receptors have been frequently applied for the cell-specific delivery of drugs to SEC [22, 23].
Another cell type within the liver is the cholangiocyte that forms the bile ducts. These cells are important for the excretion of bile acids and other electrolytes secreted by hepatocytes, but they generally do not play a role in the uptake and intracellular processing of drugs or other plasma components. Cholangiocytes do play an important role in PSC by the secretion of proinflammatory and profibrogenic mediators that activate HSC, the key drivers of fibrogenesis, which makes them a target for antifibrotic therapies in particular for biliary fibrosis but also in advanced stages of other fibrotic liver diseases [4••, 24, 25]. The only known target receptor for drug carriers to cholangiocytes is the αvβ6-integrin receptor [4••, 24, 26]. Several compounds that bind to this receptor to allow delivery of drugs and imaging agents to cholangiocytes have been prepared [4••]. A synthetic peptide against this αvβ6-receptor (FBA-A20FMDV2) is now being tested in a clinical trial for imaging purposes (www.clinicaltrials.gov).
Last but not least, the liver contains hepatic stellate cells (HSC), portal fibroblasts, and myofibroblast-like cells. Fibroblasts are mostly located along bile ducts and blood vessels, and HSC are located in the space of Disse, in the vicinity of KC. In a quiescent state, these HSC store vitamin A [27]. In an activated state, induced by cell-damage or by soluble factors from adjacent cells, these cells strongly proliferate, acquire contractile elements, and become the major producers of extracellular matrix proteins, such as collagens, fibronectin, and laminin [27]. Fibroblasts and HSC play a key role in tissue repair, and the progression of liver fibrosis and these cells are therefore the main target cell for antifibrotic therapies. However, resting or activated-HSC have no particular role in the clearance or uptake of compounds from the circulation. Resting HSC are therefore not endowed with prominent uptake mechanisms, other than for vitamin A bound to retinol binding protein [28], and they consequently do not take up more drugs than any other random cell within the body. In fact, a drug has more chance ending up in neighboring hepatocytes, KC, or SLEC than in HSC. In the past years, several cell-specific carriers to HSC have therefore been developed directed at different receptors. The mannose-6-phosphate/insulin-like growth factor II (IGFII) receptor was the first receptor studied for this purpose [29], followed by the collagen type VI receptor [30], and the PDGF-β-receptor [17, 31]. A monoclonal antibody against the synaptophysin receptor also has been tested, but without follow-up [32], and in the recent years, many studies have been published using the vitamin A receptor upon HSC as target receptor for antifibrotic compounds [33,34,35,36,37]. A lipid nanoparticle functionalized with vitamin A and containing siRNA against heat shock protein 47 (HSP47) is now being tested in clinical phase 2 trials (www.clinicaltrials.gov), which makes it the first cell-specific nanoparticle against liver fibrosis reaching clinical trials.
Cell-Specific Delivery
As outlined above, all resident hepatic cells can be selectively reached by focusing on different target receptors and different drug-carrier systems [17, 38, 39]. The relevance of each cell type and hence the choice of the target and the carrier depends on the liver disease. Antiviral hepatitis therapies or hepatoprotective drugs need to be directed at hepatocytes, whereas autoimmune diseases and other inflammatory disease require uptake of drugs into Kupffer cells. Vasoactive compounds need to be delivered to SEC, whereas for biliary diseases, the cholangiocyte is the main target cell. The key target cell for antifibrotic drugs is obviously the hepatic stellate cell. Uptake by the wrong cell type makes drugs ineffective and may lead to adverse effects, or even worse, may have the contrary effect. For instance, whereas INFγ has significant antifibrotic effects in HSC [40, 41], its potent proinflammatory activities in macrophages create many pro-fibrotic stimuli in vivo [42], [43]. Also, therapeutical approaches that focus on apoptosis, survival, or induction of cell death of resident hepatic cells [44••] face the issue that while increased survival of hepatocytes may attenuate fibrogenesis, the very same effects in other cells may stimulate liver fibrosis or tumorrigenesis [44••, 45]. Also, the hepatotoxic effects of many tyrosine kinase inhibitors limit the clinical application of these drugs [46] despite their significant antifibrotic effects in HSC [46, 47].
Nanoparticles as Drug Carriers to Hepatic Cells: Benefits and Key Problems
A drug-carrier construct basically consists of at least three elements: a carrier (or core molecule), a homing device, and a drug. In the case of a monoclonal antibody, these three elements are represented in one molecule, with the Fab fragment as homing device and the therapeutic entity, the Fc region, and complement binding moiety as additional therapeutically effective components, and the big core molecule of 150 kD (in case of IgG) as structure that prevents renal clearance and ensures a long circulating half-life. In some constructs, a forth element is introduced: a linker or spacer between the drug carrier and the drug. This linker may be necessary for a stable binding of the drug to its carrier, or it is introduced to allow release of active drugs at the site of action. The linker technology to couple drugs to monoclonal antibodies is an important and rapidly evolving field [48, 49], but for other constructs such as liposomes this is less essential. Nevertheless, all drug-carrier constructs are sophisticated molecules involving a lot of engineering. This engineering creates many opportunities but can make the synthesis of constructs also very complex and costly and may introduce heterogeneity of the final product. Moreover, most carriers are large entities that confer the risk of uptake by macrophages and hence immunogenicity [38, 50]. A particular disadvantage of most drug-carrier constructs is that the oral route of administration is excluded. The development of delivery tools that create sustained release of proteins from a subcutaneous depot [51] may therefore be a valuable asset to address this problem. This is in particular relevant for chronic diseases that require prolonged treatment.
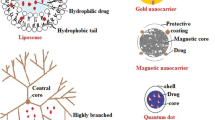
In the past decades, many other, non-antibody-based carriers have been explored. Studies have either examined viruses, liposomes, large proteins like albumin, antibodies, micelles, lipoplexes, and nanoparticles based on synthetic polymers, inorganic compounds such as gold and silica as drug carriers (Fig. 1). Each carrier offers its own possibilities, advantages, and disadvantages.

Schematic representation of the liver architecture and three mechanisms of passive targeting to liver cells (left hand panel), and an overview of the different core molecules, homing devices, and target receptors that are available for active delivery of therapeutic entities to HSC (right hand panel). The reports presented in this review make use of different passive targeting mechanisms and/or a combination of core molecules and homing devices to achieve accumulation of antifibrotic compounds in hepatic stellate cells
HSC, hepatic stellate cell; KC, Kupffer cell; SEC, sinusoidal endothelial cell; Hep, Hepatocyte; RBC, red blood cell
Nanoparticles generally have a size of 10–150 nm and can roughly be divided into two groups: organic and inorganic nanoparticles. Organic nanoparticles carriers comprise protein complexes, liposomes, solid lipid nanoparticles (SNL) and some polymers like polylactic acid (PLA) [52••]. Inorganic nanoparticles comprise dendrimers, gold nanoparticles, iron oxide quantum dots, polymers, carbon nanoparticles, and silica nanoparticles. Cell-specific delivery can be achieved by functionalization of the surface of these particles, and this approach has been applied in many studies in the recent years to achieve HSC delivery. For instance, inorganic nanoparticles like superparamagnetic iron oxide nanoparticles (SPIONs) have been functionalized with relaxin to deliver relaxin to HSC and simultaneously allow imaging of the target cell [53•]. Polyamidoamine (PAMAM) G5 dendrimers have been decorated with vitamin A to deliver an NO-donor to HSC [54], whereas the same dendrimer has been coated with RGD peptides to target the integrin αvβ3, which is also present on HSC [55]. These are examples of active targeting, where a compound is actively delivered to a designated target receptor. However, in particular, the larger drug carriers make use of passive targeting. In this case, accumulation in the target area is achieved by the enhanced permeability retention (EPR) effect [56]. The particle is not delivered to a designated target cell using a homing device but passively accumulates at the target area due to a local increase in vascular permeability (for a schematic representation see Fig. 1). This increased vascular permeability occurs during inflammatory processes or during angiogenesis in tumors and some chronic diseases. Following the extravasation of soluble plasma components, the relatively large size of nanoparticles combined with the low pressure in the interstitial space limits the return of such particles into the bloodstream that causes local retention. The “plunger effect” of blood cells (see above) may further add to the extravasation of particles. Similarly, the large size of pentameric IgM molecules (approximately 900 kD) leads to passive entrapment of IgM molecules in the inflammatory area, which is of course quite beneficial. For the treatment of liver fibrosis, several carriers have been designed that make use of this EPR effect. For instance, lipid nanoparticles (LNP) carrying siRNA that inhibit the production of procollagen α1(I) [57] or anti-col1α1 siRNA-loaded nanohydrogel particles [58]. Both carriers delivered siRNA to HSC in sufficient amounts to attenuate collagen production in fibrotic mouse models. However, passive accumulation in the target area does not mean uptake in the proper target cell, and not all diseases and tumors are characterized by a local enhanced vascular permeability. In fact, although the liver with its small sinusoids, its fenestrated endothelium and the open space of Disse underneath the endothelium cells, is perfectly equipped to create an EPR effect in normal physiological conditions, during liver fibrosis, the whole situation changes. Capillarization of sinusoids occurs during fibrosis which is associated with the closure of endothelial fenestrae and extracellular matrix deposition in the space of Disse [4, 27]. Due to this, the EPR effect will be hampered or even be blocked during fibrogenesis, causing reduced uptake of nanocarriers that make use of this EPR effect in the fibrotic liver [59].
The major advantage of larger nanoparticles is their payload; in contrast to small drug carriers like proteins or antibodies, they can incorporate high drug concentrations, and incorporation of drugs and additional compounds is rather easy. There is in general no need for chemical coupling of drugs to the carrier which ensures release of unmodified, pharmacologically active drugs after uptake by its target cell. Moreover, virtually, all drugs can be incorporated; lipophilic drugs can be non-covalently incorporated in the solid lipid core of lipid nanoparticles or in the lipid bilayer of liposomes or micelles and hydrophilic drugs in the aqueous phase of liposomes or micelles [60]. In contrast, small carriers like proteins and some polymers require chemical conjugation of drug and carrier. Not all drugs have a chemical entity that allows coupling, and conjugation may also affect the pharmacological effects of the drug or even the distribution of the carrier. Drugs incorporated in nanoparticles also do not alter the physico-chemical properties of the carrier itself, thus ensuring that the disposition of the carrier is not affected by its cargo, which may be a problem for instance when drug:IgG ratios are too high in antibody-drug conjugates [48, 49]. Also, mixture of drugs can be readily incorporated in large nanoparticles, allowing multi-drug therapies, which may turn out to be quite important for a complex disease like liver fibrosis. And last but not the least, incorporation of imaging agents, together with the drug, offers the option of a theranostic approach, that is, it opens opportunities for treatment and target imaging at the same time [61•].
However, a key problem for many types of nanoparticles is the formation of a corona [62]. Corona formation involves the opsonization of a nanoparticle with plasma components. All kind of plasma proteins and lipids can accumulate into the lipid bilayer of a liposome or may get non-covalently attached to silica, gold, or other inert inorganic particles [63]. Recent studies have shown that many different plasma proteins and lipids are more or less selectively opsonized by a nanoparticle, leading to an enrichment of certain proteins in the corona during their circulation time [63]. This opsonization greatly affects their body distribution: it can lead to enhanced uptake in non-target cells such as the opsonization of complement factor C3 into liposomes, leading to uptake in macrophages [64], whereas the binding of apo-lipoproteins shifts the uptake of nanoparticles to hepatocytes [15]. This corona formation can thus disrupt all cell-selective targeting strategies. Current research activities focus on the prevention of corona formation, e.g., by the coupling of PEG molecules to the liposomes or other nanoparticles [65], although this may lead to other problems such as the accelerated blood clearance phenomenon (see above). Fundamental knowledge on processes that affect corona formation is lacking, but it is becoming clear that physio-chemical properties of the particle itself are an important factor [62, 66]. The composition of plasma is also of great importance [66] and obviously this changes during disease, making it even more complicated. It is also becoming clear that the composition of the corona may affect the intracellular routing of nanoparticles within cells [66]. Nanoparticles can be internalized and degraded within the cell via clathrin- or caveolin-dependent pathways or other non-selective routes, dependent on the properties of the nanoparticle [67], and this will in turn affect the release profile and the pharmacological effects of the released drugs within the cell [67]. If more basic knowledge becomes available, strategies may be developed to modify the corona in such a way that it does not corrupt the delivery, yet even may be used to affect body distribution leading to enhanced delivery into the target area or cell. This strategy was applied in a recent study where retinol-binding protein 4 (RBP4) and albumin were recruited into the corona of a nanoparticle by using an amphiphilic polymer of positively charged polyethylenimine (PEI) and retinol [24]. Antisense oligonucleotides were also attached to PEI. The RBP4-retinol-albumin complex subsequently formed in plasma yielded a nanoparticle that effectively delivered antisense oligonucleotides in HSC in the CCL4-liver fibrosis model in mice [28], making in an elegant way use of corona formation. A schematic representation of the three mechanisms of passive targeting, with corona formation as one of them, is presented in Fig. 1.
Proteins as Drug Carriers to HSC
Paul Ehrlich introduced the “magic bullet” concept already in 1906 by proposing to use antibodies to enhance the effectiveness of antibiotics and to reduce their adverse effects, but it took until the year 2000 for the first antibody-drug conjugate (ADC) to reach the market, Gemtuzumab ozogamicin (Mylotarg) for the treatment of CD33-positive acute myeloid leukemia [49]. And even then, the construct was retracted in 2010 because of adverse effects. However, it was re-introduced in 2017 [49]. To date, five ADCs are approved by the FDA and EMA [49]; more are nowadays tested in clinical trials, and hundreds of ADCs are in the experimental or pre-clinical phase [49, 68] for antitumor therapies. A major step in the development of ADC’s was obviously the humanization of antibodies, preventing immunogenic responses against constructs, but the development of chemical linkers was also essential to prepare effective ADC’s. Due to lack of cell-specific targets for fibrotic diseases, no antibody-drug conjugates are available to treat fibrosis. Although many cells and cellular processes are derailed during fibrogenesis, the cells involved are all endogenous cells, and matrix turnover is a normal physiological process in all tissues. Cell-specific targets for monoclonal antibodies in fibrotic tissues remain to be found.
Yet, a relative enhancement of target receptors upon HSC appears to be equally applicable for drug-targeting purposes. In fact, an antibody with high affinity for its target may in these cases be less applicable than drug carriers with lower affinity, because interaction with the target receptor needs not to be maximal, but interaction with non-target cells needs to be minimal. This is for instance true for the PDGFβ receptor and the IGFII receptor, which are also expressed in other tissues but whose expression is very strongly upregulated on fibroblast-like cells during fibrogenesis, leading to a relatively high local accumulation in HSC of carriers directed at these receptors. Most likely, the high liver perfusion and an (residual) EPR effect in fibrotic livers may also help. Eventually, when cell-specific antibodies for fibrogenic cells will be found, much advantage can be gained by the experiences obtained in the cancer field [8]. Nanobodies, Fab fragments, or other modifications of Ig can be explored. Major problems specific for immunoglobulines include the limited options these molecules offer to attach drugs and their low payload to target cells. If a high drug-antibody ratio is obtained, it may significantly affect biodistribution and half-life of the antibody [49]. However, this area is rapidly expanding [48, 49, 68]. To allow stable coupling of drugs to immunoglobulines and at the same time release of pharmacological active drugs after internalization in the target cell, new linker strategies are now examined [48]. For instance, recently, a platinum-based linker XLS has been examined in several studies using protein-based carriers to HSC as well, with successful outcomes [47, 68].
Protein-based carriers, other than IgG, have been extensively applied to deliver antifibrotic drugs to hepatic stellate cells. Such carriers generally have a core protein, such as human serum albumin, with a size high enough to prevent renal clearance (> 65 kD) yet small enough to prevent uptake by the reticuloendothelial system (< 100 nm). Albumin allows a higher drug loading than IgG, Fab fragments, or nanobodies. Modified proteins may also be subject to immunogenic responses of the host. Indeed, immunogenicity toward a drug-carrier construct has been found in some studies in particular when heterologous albumin was used as carrier [51]. The change of human serum albumin into albumin derived from mice was found to reduce immunogenicity of the constructs in mice [51], but risks for prolonged treatment do exist in particular when sugar-moieties are used as homing device. Many different types of drugs have been targeted to HSC in the past decade (for an overview see [17, 39, 44••, 52••], but in recent years, the cell-specific delivery of tyrosine kinase inhibitors has been extensively explored, because of their significant antifibrotic affects [47], paralleled by adverse effects [46]. Because most kinase inhibitors do not have a chemical entity that allows covalent binding to its carrier, a linker between the carrier and the drug has been developed [68]. These platinum-based linkers have been applied in several studies to deliver tyrosine kinase inhibitors to HSC and other cells with good results in experimental animal models [47, 68].
Cell-Specific Delivery of Antifibrotic Drugs: Homing Devices
The alternative approach is active targeting which involves the coupling of homing moieties to the nanoparticle. Nanoparticles functionalized with folate or antibodies are well-studied constructs in the cancer field [69]. Focusing on the last 5 years, several innovative strategies have been examined in the field of HSC targeting (summarized in Fig. 1). A variety of liposomes have been engineered for the delivery of antifibrotic drugs to HSC, sometimes even delivering more than one drug at the same time. A number of receptor-recognizing peptides have been developed to direct proteins and nanoparticles to designated receptors on HSC. The first HSC-specific carrier that used a cyclic peptide was directed at the collagen type VI receptor [30], [70]. This RGD-based peptide was coupled to albumin to yield a HSC-selective drug carrier. Subsequently, a bicyclic peptide binding to the PDGF-β-receptor was developed (pPB) [31] that rapidly accumulated in HSC. This pPB was coupled to albumin to create a HSC-specific drug carrier [31]. Recently, this carrier was used to deliver the Rho-kinase inhibitor Y27632 to HSC leading to beneficial effects in vivo [47]. pPB has also been directly coupled to interferon γ to create a HSC-specific antifibrotic cytokine [71], whereas other groups coupled this pPB peptide to liposomes to deliver interferon γ to HSC [72]. The same peptide was used to deliver lipid nanoparticles containing siRNA to HSC [73]. In addition, a cyclic-RGD-based peptide was coupled to liposomes for the delivery of the hedgehog inhibitor vismodegib [74]. This latter construct was quite effective in reducing fibrosis in two experimental animal models of liver fibrosis in mice. In recent years, another receptor-recognizing peptide binding to the CXCR4 receptor has been developed [75•] which was coupled to polymeric biodegradable nanoparticles to create a carrier that allowed the simultaneous delivery of sorafenib and the MEK inhibitor selumetinib to HSC [75•]. This combination therapy displayed significant antifibrotic effects in a mouse model. The CXCR4 receptor was also used for the targeting of pirfenidone to HSC [76•]. In this latter case however, the nanoparticles (liposomes) were not functionalized with receptor-recognizing peptides but with the CXCR4 antagonist AMD3100 (plerixafor). The effect of pirfenidone, one of the few FDA-approved drugs against fibrosis, combined with blocking of the chemokine receptor CXCR4 may lead to synergistic effects and is another example of a successful combination therapy. Next to peptides and a receptor antagonist, sugar moieties have been used as homing devices as well. Mannose-6-phosphate coupled to albumin [29], binding to the IGF II-receptor has been used frequently for HSC-specific delivery of drugs (for review see [17, 39]. Vitamin A-loaded liposomes have gained much attention as HSC-specific carriers since the first publication by Sato et al. [33]. Using vitamin A-decorated nanoparticles, recent studies showed successful delivery to HSC of several compounds such as siRNA and anti-miR33 [77], BMP4 siRNA [78], the Rho-kinase inhibitor Y27632 [79], and Imatinib [37]. All these different types of drugs in vitamin A-coated liposomes nicely illustrate the versatility of liposomes as vehicles for drug delivery. Vitamin A has also been coupled to other nanoparticles to achieve HSC-specific delivery of antifibrotic compounds: next to the earlier mentioned dendrimers [54], chitosan-based nanoparticles have been modified with vitamin A and loaded with JQ1 and atorvastatin [80], both inhibiting HSC.
Other nanoparticle constructs that have been applied in recent years to reach the HSC in vivo for the delivery of antifibrotic drugs are carboxymethyl cellulose-based nanoparticles for the delivery of docetaxel to HSC. These PEGylated nanoparticles bind to serum albumin and the Secreted Protein Acidic and Rich in Cysteine (SPARC) which induces uptake in HSC and tumor cells [81], again a nice example of corona-directed uptake in target cells. Dibutyltin dichloride (DBTC)-based lipoplexes have been applied for the delivery of Foxf1 siRNA to HSC [82], and hyaluronic acid-based micelles have been used for the delivery of the angiotensin I receptor Losartan to the CD44 receptor on HSC [83]. Since these target receptors are not specific for HSC, the cellular specificity of these constructs need further examination.
Drugs to be Targeted
Next to small chemical entities with antifibrotic effects, that have been coupled and incorporated in many of the above-reviewed drug carriers, biological products like antisense oligonucleotides, siRNA, miRNA, and cytokines can be powerful tools to modify fibrosis. The therapeutic applications of siRNA have become clear in recent years, and many different miRNA [84] and siRNA molecules [22, 33, 35, 58, 73, 78] have been tested in experimental animal models of fibrosis. Delivery is the key issue for siRNA and miRNA molecules. Although cytokines play a key role in vivo in regulating fibrogenesis and are active in the picomolar range, their pleiotropic activities have strongly limited their therapeutical use. This may be solved using targeting strategies [71]. Also, the use of the CRISPR/Cas9 site-specific nuclease system has revolutionized the therapeutic options for several diseases, and may also provide new opportunities for the treatment of liver fibrosis. A recent study showing the antifibrotic effects of the knock down of the glial cell line-derived neurotrophic factor (GDNF) using the CRISPR/CAs9 in an adeno-viral delivery system [85] illustrates an exciting development. Because of this, the cell-specific delivery of biologicals has gained growing attention in recent years. The delivery of biologicals adds an additional set of requirements to their carriers [38]. They need to be protected from degradation mechanisms in plasma and in the target cell where the carrier constructs following endocytosis mostly end up in lysosomes. The acidic environment and proteolytic cleavage mechanisms in lysosomes are generally used to induce the release of chemical entities from their carriers, but biologicals are being destroyed there. For the delivery of genes, non-viral delivery systems have been developed (reviewed elsewhere; [86]), which escape from these degrading mechanisms, and viruses are also equipped with mechanisms that allow delivery of their payload to the nucleus, but toxicity, immunogenicity, transfection efficiency, and other factors limit their application. Nevertheless, tools are now available for the delivery of antisense oligonucleotides, miRNA, and siRNA molecules [33]. Challenges lie ahead in the area of cell-specific delivery of bioactive proteins. The CRISPR/Cas9 site-specific nuclease system offers new very innovative options for treatment, but requires an even more a sophisticated approach. In this respect, the recent fundamental studies in intracellular trafficking mechanisms of after the intracellular uptake of nanomedicines [66] are very important. In recent years, some studies have shown that cell-specific proteins can escape from the intracellular degradation mechanisms: the delivery of INFγ to the PDGF receptor is an example of this [71]. Maybe this offers opportunities for other cytokines as well.
Conclusions
It can be concluded that there are many different drug carriers to reach the different hepatic cells, in particular the hepatic stellate cell. This variety of carriers, each with their own advantages and disadvantages, offer a versatile toolbox for many different drugs and biologicals. Particular biologicals like siRNA and cytokines offer great opportunities for therapy but they do need steering to the proper cells and protection from endogenous clearing mechanisms. Liver fibrosis is a complex disease with many redundant systems; either a drug needs to be very potent or very pleiotropic or more than one drug needs to be applied, similar to the current antiviral and antitumor therapies. Developments in the field of nanomedicines may provide the tools to do just that; as outlined here, several studies have already shown that delivery of more than one compound to the target cell is feasible. Nanomedicines, including protein-based constructs, also offer a theranostic approach where treatment and monitoring of disease activity coincide. New advancements in this area have been made for the treatment of liver fibrosis in animal models [53•]. Tools for early diagnosis and monitoring of disease activity are essential for the development of an effective therapy [87]. The strategy of cell-specific drug carriers may thus remove major hurdles for antifibrotic therapies [88], i.e., low drug effectivity and adverse effects and also increase the success rate of clinical trials by facilitating patient stratification and providing new ways to monitor disease activity. In this way, cell-specific carriers may reduce the length of clinical trials (96 weeks for simtuzumab in PSC patients [5] and 240 weeks in a planned study in patients with NASH [7]), which is one of the largest issues in the development of new drugs against liver fibrosis [4, 87]. Upon entering the era of biologicals and personalized medicine, nanomedicines provide important opportunities to treat complex diseases of liver fibrosis.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Christofi T, Baritaki S, Falzone L, Libra M, Zaravinos A. Current perspectives in cancer immunotherapy. Cancers (Basel). 2019;11(10):E1472. https://doi.org/10.3390/cancers11101472.
Lucas AT, Robinson R, Schorzman AN, Piscitelli JA, Razo JF, Zamboni WC. Pharmacologic considerations in the disposition of antibodies and antibody-drug conjugates in preclinical models and in patients. Antibodies (Basel) 2019;8(1):https://doi.org/10.3390/antib8010003.
•• Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–22. Excellent review on current insights on the pathogenesis and options for treatment of fibrosis.
•• Schuppan D, Ashfaq-Khan M, Yang AT, Kim YO. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018;68–69:435–51. Excellent overview on current insights on the pathogenesis of liver fibrosis and targets for therapy.
Muir AJ, Levy C, Janssen HLA, Montano-Loza AJ, Shiffman ML, Caldwell S, et al. Simtuzumab for primary sclerosing cholangitis: phase 2 study results with insights on the natural history of the disease. Hepatology. 2019;69(2):684–98.
Fickert P. Is this the last requiem for Simtuzumab? Hepatology. 2019;69(2):476–9.
Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R, et al. Simtuzumab is ineffective for patients with bridging fibrosis or compensated cirrhosis caused by nonalcoholic Steatohepatitis. Gastroenterology. 2018;155(4):1140–53.
Prakash J, Pinzani M. Fibroblasts and extracellular matrix: targeting and therapeutic tools in fibrosis and cancer. Adv Drug Deliv Rev. 2017;121:1–2.
Schulze RJ, Schott MB, Casey CA, Tuma PL, McNiven MA. The cell biology of the hepatocyte: a membrane trafficking machine. J Cell Biol. 2019;218(7):2096–112.
Jetter A, Kullak-Ublick GA. Drugs and hepatic transporters: a review. Pharmacol Res. 2019;17:104234.
Ivanenkov YA, Majouga AG, Petrov RA, Petrov SA, Kovalev SV, Maklakova SY, et al. Synthesis and biological evaluation of novel doxorubicin-containing ASGP-R-targeted drug-conjugates. Bioorg Med Chem Lett. 2018;28(3):503–8.
Guillot A, Tacke F. Liver macrophages: old dogmas and new insights. Hepatol Commun. 2019;3(6):730–43.
•• Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66(6):1300–12. Excellent review highlighting the role of macrophages in liver fibrosis.
Peterson KR, Cottam MA, Kennedy AJ, Hasty AH. Macrophage-targeted therapeutics for metabolic disease. Trends Pharmacol Sci. 2018;39(6):536–46.
Bartneck M, Warzecha KT, Tacke F. Therapeutic targeting of liver inflammation and fibrosis by nanomedicine. Hepatobiliary Surg Nutr. 2014;3(6):364–76.
Ahmed M, Baumgartner R, Aldi S, Dusart P, Hedin U, Gustafsson B, et al. Human serum albumin-based probes for molecular targeting of macrophage scavenger receptors. Int J Nanomedicine. 2019;14:3723–41.
Poelstra K, Prakash J, Beljaars L. Drug targeting to the diseased liver. J Control Release. 2012;161(2):188–97.
Lai C, Li C, Luo X, Liu M, Liu X, Hu L, et al. Use of dual-ligand modification in Kupffer cell-targeted liposomes to examine the contribution of Kupffer cells to accelerated blood clearance phenomenon. Mol Pharm. 2018;15(7):2548–58.
Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–95.
Bartneck M, Keul HA, Wambach M, Bornemann J, Gbureck U, Chatain N, et al. Effects of nanoparticle surface-coupled peptides, functional endgroups, and charge on intracellular distribution and functionality of human primary reticuloendothelial cells. Nanomedicine. 2012;8(8):1282–92.
Koppert S, Buscher A, Babler A, Ghallab A, Buhl EM, Latz E, et al. Cellular clearance and biological activity of calciprotein particles depend on their maturation state and crystallinity. Front Immunol. 2018;9:1991.
Zhao Y, Gao H, He J, Jiang C, Lu J, Zhang W, et al. Co-delivery of LOX-1 siRNA and statin to endothelial cells and macrophages in the atherosclerotic lesions by a dual-targeting core-shell nanoplatform: a dual cell therapy to regress plaques. J Control Release. 2018;283:241–60.
Kim KS, Hur W, Park SJ, Hong SW, Choi JE, Goh EJ, et al. Bioimaging for targeted delivery of hyaluronic acid derivatives to the livers in cirrhotic mice using quantum dots. ACS Nano. 2010;4(6):3005–14.
Peng ZW, Ikenaga N, Liu SB, Sverdlov DY, Vaid KA, Dixit R, et al. Integrin alphavbeta6 critically regulates hepatic progenitor cell function and promotes ductular reaction, fibrosis, and tumorigenesis. Hepatology. 2016;63(1):217–32.
Locatelli L, Cadamuro M, Spirli C, Fiorotto R, Lecchi S, Morell CM, et al. Macrophage recruitment by fibrocystin-defective biliary epithelial cells promotes portal fibrosis in congenital hepatic fibrosis. Hepatology. 2016;63(3):965–82.
Popov Y, Patsenker E, Stickel F, Zaks J, Bhaskar KR, Niedobitek G, et al. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48(3):453–64.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411.
Zhang Z, Wang C, Zha Y, Hu W, Gao Z, Zang Y, et al. Corona-directed nucleic acid delivery into hepatic stellate cells for liver fibrosis therapy. ACS Nano. 2015;9(3):2405–19.
Beljaars L, Olinga P, Molema G, de Bleser P, Geerts A, Groothuis GM, et al. Characteristics of the hepatic stellate cell-selective carrier mannose 6-phosphate modified albumin (M6P(28)-HSA). Liver. 2001;21(5):320–8.
Beljaars L, Molema G, Schuppan D, Geerts A, De Bleser PJ, Weert B, et al. Successful targeting to rat hepatic stellate cells using albumin modified with cyclic peptides that recognize the collagen type VI receptor. J Biol Chem. 2000;275(17):12743–51.
Beljaars L, Weert B, Geerts A, Meijer DK, Poelstra K. The preferential homing of a platelet derived growth factor receptor-recognizing macromolecule to fibroblast-like cells in fibrotic tissue. Biochem Pharmacol. 2003;66(7):1307–17.
Douglass A, Wallace K, Parr R, Park J, Durward E, Broadbent I, et al. Antibody-targeted myofibroblast apoptosis reduces fibrosis during sustained liver injury. J Hepatol. 2008;49(1):88–98.
Sato Y, Murase K, Kato J, Kobune M, Sato T, Kawano Y, et al. Resolution of liver cirrhosis using vitamin A-coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat Biotechnol. 2008;26(4):431–42.
Li Y, Liu F, Ding F, Chen P, Tang M. Inhibition of liver fibrosis using vitamin A-coupled liposomes to deliver matrix metalloproteinase-2 siRNA in vitro. Mol Med Rep. 2015;12(3):3453–61.
Toriyabe N, Sakurai Y, Kato A, Yamamoto S, Tange K, Nakai Y, et al. The delivery of small interfering RNA to hepatic stellate cells using a lipid nanoparticle composed of a vitamin A-scaffold lipid-like material. J Pharm Sci. 2017;106(8):2046–52.
Qiao JB, Fan QQ, Xing L, Cui PF, He YJ, Zhu JC, et al. Vitamin A-decorated biocompatible micelles for chemogene therapy of liver fibrosis. J Control Release. 2018;283:113–25.
El-Mezayen NS, El-Hadidy WF, El-Refaie WM, Shalaby TI, Khattab MM, El-Khatib AS. Hepatic stellate cell-targeted imatinib nanomedicine versus conventional imatinib: a novel strategy with potent efficacy in experimental liver fibrosis. J Control Release. 2017;266:226–37.
Poelstra K, Beljaars L, Melgert BN. Cell-specific delivery of biologicals: problems, pitfalls and possibilities of antifibrotic compounds in the liver. Drug Discov Today. 2013;18(23–24):1237–42.
Schon HT, Bartneck M, Borkham-Kamphorst E, Nattermann J, Lammers T, Tacke F, et al. Pharmacological intervention in hepatic stellate cell activation and hepatic fibrosis. Front Pharmacol. 2016;7:33.
Wu X, Wu X, Ma Y, Shao F, Tan Y, Tan T, et al. CUG-binding protein 1 regulates HSC activation and liver fibrogenesis. Nat Commun. 2016;7:13498.
Vu TN, Chen X, Foda HD, Smaldone GC, Hasaneen NA. Interferon-gamma enhances the antifibrotic effects of pirfenidone by attenuating IPF lung fibroblast activation and differentiation. Respir Res. 2019;20(1):206 -019-1171-2.
Kawaratani H, Moriya K, Namisaki T, Uejima M, Kitade M, Takeda K, et al. Therapeutic strategies for alcoholic liver disease: focusing on inflammation and fibrosis (review). Int J Mol Med. 2017;40(2):263–70.
Hamidzadeh K, Christensen SM, Dalby E, Chandrasekaran P, Mosser DM. Macrophages and the recovery from acute and chronic inflammation. Annu Rev Physiol. 2017;79:567–92.
•• Tacke F, Weiskirchen R. An update on the recent advances in antifibrotic therapy. Expert Rev Gastroenterol Hepatol. 2018;12(11):1143–52. Excellent review showing the current state-of-the-art on potential targets and therapeutic interventions.
Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 2018;15(12):738–52.
Qu K, Liu T, Lin T, Zhang X, Cui R, Liu S, et al. Tyrosine kinase inhibitors: friends or foe in treatment of hepatic fibrosis? Oncotarget. 2016;7(41):67650–60.
Klein S, Frohn F, Magdaleno F, Reker-Smit C, Schierwagen R, Schierwagen I, et al. Rho-kinase inhibitor coupled to peptide-modified albumin carrier reduces portal pressure and increases renal perfusion in cirrhotic rats. Sci Rep. 2019;9(1):2256 -019-38678-5.
Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37.
Chau CH, Steeg PS, Figg WD. Antibody-drug conjugates for cancer. Lancet. 2019;394(10200):793–804.
Kavita U, Miller W, Ji QC, Pillutla RC. A fit-for-purpose method for the detection of human antibodies to surface-exposed components of BMS-986263, a lipid nanoparticle-based drug product containing a siRNA drug substance. AAPS J. 2019;21(5):92 -019-0360-8.
van Dijk F, Teekamp N, Beljaars L, Post E, Zuidema J, Steendam R, et al. Pharmacokinetics of a sustained release formulation of PDGFbeta-receptor directed carrier proteins to target the fibrotic liver. J Control Release. 2018;269:258–65.
•• Poilil Surendran S, George Thomas R, Moon MJ, Jeong YY. Nanoparticles for the treatment of liver fibrosis. Int J Nanomedicine. 2017;12:6997–7006. Excellent review highlighting the options for targeting compounds to hepatic cells.
• Nagorniewicz B, Mardhian DF, Booijink R, Storm G, Prakash J, Bansal R. Engineered Relaxin as theranostic nanomedicine to diagnose and ameliorate liver cirrhosis. Nanomedicine. 2019;17:106–18. First paper describing the use of a targeting ligand for therapy and imaging of liver fibrosis.
Duong HT, Dong Z, Su L, Boyer C, George J, Davis TP, et al. The use of nanoparticles to deliver nitric oxide to hepatic stellate cells for treating liver fibrosis and portal hypertension. Small. 2015;11(19):2291–304.
Li F, Yan H, Wang J, Li C, Wu J, Wu S, et al. Non-invasively differentiating extent of liver fibrosis by visualizing hepatic integrin alphavbeta3 expression with an MRI modality in mice. Biomaterials. 2016;102:162–74.
Ebrahim Attia AB, Oh P, Yang C, Tan JP, Rao N, Hedrick JL, et al. Insights into EPR effect versus lectin-mediated targeted delivery: biodegradable polycarbonate micellar nanoparticles with and without galactose surface decoration. Small. 2014;10(21):4281–6.
Jimenez Calvente C, Sehgal A, Popov Y, Kim YO, Zevallos V, Sahin U, et al. Specific hepatic delivery of procollagen alpha1(I) small interfering RNA in lipid-like nanoparticles resolves liver fibrosis. Hepatology. 2015;62(4):1285–97.
Leber N, Kaps L, Aslam M, Schupp J, Brose A, Schaffel D, et al. SiRNA-mediated in vivo gene knockdown by acid-degradable cationic nanohydrogel particles. J Control Release. 2017;248:10–23.
Ergen C, Niemietz PM, Heymann F, Baues M, Gremse F, Pola R, et al. Liver fibrosis affects the targeting properties of drug delivery systems to macrophage subsets in vivo. Biomaterials. 2019;206:49–60.
Dave V, Tak K, Sohgaura A, Gupta A, Sadhu V, Reddy KR. Lipid-polymer hybrid nanoparticles: synthesis strategies and biomedical applications. J Microbiol Methods. 2019;160:130–42.
• Baues M, Dasgupta A, Ehling J, Prakash J, Boor P, Tacke F, et al. Fibrosis imaging: Current concepts and future directions. Adv Drug Deliv Rev. 2017;121:9–26. Review describing the current state-of-the -art on imaging of liver fibrosis.
Sanchez-Moreno P, Buzon P, Boulaiz H, Peula-Garcia JM, Ortega-Vinuesa JL, Luque I, et al. Balancing the effect of corona on therapeutic efficacy and macrophage uptake of lipid nanocapsules. Biomaterials. 2015;61:266–78.
Escamilla-Rivera V, Solorio-Rodriguez A, Uribe-Ramirez M, Lozano O, Lucas S, Chagolla-Lopez A, et al. Plasma protein adsorption on Fe3O4-PEG nanoparticles activates the complement system and induces an inflammatory response. Int J Nanomedicine. 2019;14:2055–67.
Tavano R, Gabrielli L, Lubian E, Fedeli C, Visentin S, Polverino De Laureto P, et al. C1q-mediated complement activation and C3 Opsonization trigger recognition of stealth poly(2-methyl-2-oxazoline)-coated silica nanoparticles by human phagocytes. ACS Nano. 2018;12(6):5834–47.
Partikel K, Korte R, Stein NC, Mulac D, Herrmann FC, Humpf HU, et al. Effect of nanoparticle size and PEGylation on the protein corona of PLGA nanoparticles. Eur J Pharm Biopharm. 2019;141:70–80.
Francia V, Yang K, Deville S, Reker-Smit C, Nelissen I, Salvati A. Corona composition can affect the mechanisms cells use to internalize nanoparticles. ACS Nano. 2019;13(10):11107–21.
Voltan AR, Alarcon KM, Fusco-Almeida AM, Soares CP, Mendes-Giannini MJS, Chorilli M. Highlights in endocytosis of nanostructured systems. Curr Med Chem. 2017;24(18):1909–29.
Merkul E, Sijbrandi NJ, Muns JA, Aydin I, Adamzek K, Houthoff HJ, et al. First platinum(II)-based metal-organic linker technology (lx(R)) for a plug-and-play development of antibody-drug conjugates (ADCs). Expert Opin Drug Deliv. 2019;16(8):783–93.
Narmani A, Rezvani M, Farhood B, Darkhor P, Mohammadnejad J, Amini B, et al. Folic acid functionalized nanoparticles as pharmaceutical carriers in drug delivery systems. Drug Dev Res. 2019;80(4):404–24.
Meijer DK, Beljaars L, Molema G, Poelstra K. Disease-induced drug targeting using novel peptide-ligand albumins. J Control Release. 2001;72(1–3):157–64.
Bansal R, Prakash J, De Ruiter M, Poelstra K. Interferon gamma peptidomimetic targeted to hepatic stellate cells ameliorates acute and chronic liver fibrosis in vivo. J Control Release. 2014;179:18–24.
Li Q, Yan Z, Li F, Lu W, Wang J, Guo C. The improving effects on hepatic fibrosis of interferon-gamma liposomes targeted to hepatic stellate cells. Nanotechnology. 2012;23(26):265101–4484.
Jia Z, Gong Y, Pi Y, Liu X, Gao L, Kang L, et al. pPB peptide-mediated siRNA-loaded stable nucleic acid lipid nanoparticles on targeting therapy of hepatic fibrosis. Mol Pharm. 2018;15(1):53–62.
Li Y, Pu S, Liu Q, Li R, Zhang J, Wu T, et al. An integrin-based nanoparticle that targets activated hepatic stellate cells and alleviates liver fibrosis. J Control Release. 2019;303:77–90.
• Sung YC, Liu YC, Chao PH, Chang CC, Jin PR, Lin TT, et al. Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development. Theranostics. 2018;8(4):894–905. New carrier to HSC using a peptide and its application for dual targeting.
• Ullah A, Wang K, Wu P, Oupicky D, Sun M. CXCR4-targeted liposomal mediated co-delivery of pirfenidone and AMD3100 for the treatment of TGFbeta-induced HSC-T6 cells activation. Int J Nanomedicine. 2019;14:2927–44. New carrier to HSC using a receptor agonist and its application for dual targeting.
Furuhashi H, Tomita K, Teratani T, Shimizu M, Nishikawa M, Higashiyama M, et al. Vitamin A-coupled liposome system targeting free cholesterol accumulation in hepatic stellate cells offers a beneficial therapeutic strategy for liver fibrosis. Hepatol Res. 2018;48(5):397–407.
Omar R, Yang J, Alrushaid S, Burczynski FJ, Minuk GY, Gong Y. Inhibition of BMP4 and alpha smooth muscle actin expression in LX-2 hepatic stellate cells by BMP4-siRNA lipid based nanoparticle. J Pharm Pharm Sci. 2018;21(1):119–34.
Kuroda S, Tashiro H, Kimura Y, Hirata K, Tsutada M, Mikuriya Y, et al. Rho-kinase inhibitor targeting the liver prevents ischemia/reperfusion injury in the steatotic liver without major systemic adversity in rats. Liver Transpl. 2015;21(1):123–31.
Hassan R, Tammam SN, Safy SE, Abdel-Halim M, Asimakopoulou A, Weiskirchen R, et al. Prevention of hepatic stellate cell activation using JQ1- and atorvastatin-loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. Eur J Pharm Biopharm. 2019;134:96–106.
Chang CC, Yang Y, Gao DY, Cheng HT, Hoang B, Chao PH, et al. Docetaxel-carboxymethylcellulose nanoparticles ameliorate CCl4-induced hepatic fibrosis in mice. J Drug Target. 2018;26(5–6):516–24.
Abshagen K, Brensel M, Genz B, Roth K, Thomas M, Fehring V, et al. Foxf1 siRNA delivery to hepatic stellate cells by DBTC lipoplex formulations ameliorates fibrosis in livers of bile duct ligated mice. Curr Gene Ther. 2015;15(3):215–27.
Thomas RG, Moon MJ, Kim JH, Lee JH, Jeong YY. Effectiveness of losartan-loaded hyaluronic acid (HA) micelles for the reduction of advanced hepatic fibrosis in C3H/HeN mice model. PLoS One. 2015;10(12):e0145512.
Kumar V, Mahato RI. Delivery and targeting of miRNAs for treating liver fibrosis. Pharm Res. 2015;32(2):341–61.
Tao L, Ma W, Wu L, Xu M, Yang Y, Zhang W, et al. Glial cell line-derived neurotrophic factor (GDNF) mediates hepatic stellate cell activation via ALK5/Smad signalling. Gut. 2019;68(12):2214–27.
Sung YK, Kim SW. Recent advances in the development of gene delivery systems. Biomater Res. 2019;23:8 –0190156-z. eCollection 2019.
Torok NJ, Dranoff JA, Schuppan D, Friedman SL. Strategies and endpoints of antifibrotic drug trials: summary and recommendations from the AASLD emerging trends conference, Chicago, June 2014. Hepatology. 2015;62(2):627–34.
Poelstra K. Liver fibrosis in 2015: crucial steps towards an effective treatment. Nat Rev Gastroenterol Hepatol. 2016;13(2):67–8.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Chronic Liver Disease
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Poelstra, K. Innovative Nanotechnological Formulations to Reach the Hepatic Stellate Cell. Curr. Tissue Microenviron. Rep. 1, 13–22 (2020). https://doi.org/10.1007/s43152-020-00004-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43152-020-00004-x