
Abstract
Fanconi anemia (FA) is a multisystem disease, characterized by the triad of physical abnormalities, bone marrow failure, and increased risk for malignancy. In the past few years, data has accumulated regarding fertility issues in FA patients, mostly due to gonadal dysfunction, which is prevalent in FA patients reaching puberty. It seems that attenuated FA phenotype lacking the classical manifestations often is presented with POI or azoospermia. Searching the literature, we summarized data regarding FA patients presenting as suffering from sub/infertility due to gonadal dysfunction, with or without other FA symptoms. We present a summary of the patients having biallelic pathogenic variants in FA genes FANCA, FANCM, BRCA2, and XRCC2 that presented with gonadal dysfunction with or without other phenotypic features of FA. Some were in mosaic, while some are considered hypomorphic, enabling residual protein function. There are also a few descriptions of POI associated with monoallelic pathogenic variants in FANCA, BRCA2, and FANCL. We conclude that the diagnosis of FA in gonadal dysfunction patients is of utmost importance due to its actionability. Follow-up strategies in FA patients are designed to discover early stages of leukemias and solid tumors and thus save lives. The feasibility of next-generation sequencing (NGS) can now ease this diagnostic procedure. An open question is the justification of performing NGS for all isolated azoospermia/POI patients.
Similar content being viewed by others
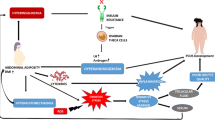

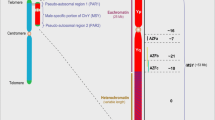
Availability of Data and Material
All resources are specified in the article and in Table 1.
Code Availability
Not applicable.
References
Fiesco-Roa MO, Giri N, McReynolds LJ, Best AF, Alter BP. Genotype-phenotype associations in Fanconi anemia: a literature review. Blood Rev. 2019;37:100589.
Mehta PA, Tolar J, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, Amemiya A. In: GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2021. 2002 Feb 14 [updated 2018 Mar 8].
Alter BP. Bone marrow failure: a child is not just a small adult (but an adult can have a childhood disease). Hematology Am Soc Hematol Educ Program. 2005;2005:96–103.
Tan IB, Cutcutache I, Zang ZJ, Iqbal J, Yap SF, Hwang W, et al. Fanconi's anemia in adulthood: chemoradiation-induced bone marrow failure and a novel FANCA mutation identified by targeted deep sequencing. J Clin Oncol. 2011;29:e591–4.
Krausz C, Riera-Escamilla A, Chianese C, Moreno-Mendoza D, Ars E, Rajmil O, et al. From exome analysis in idiopathic azoospermia to the identification of a high-risk subgroup for occult Fanconi anemia. Genet Med. 2019;21:189–94.
Tsui V, Crismani W. The Fanconi anemia pathway and fertility. Trends Genet. 2019;35:199–214.
Petryk A, Kanakatti Shankar R, Giri N, Hollenberg AN, Rutter MM, Nathan B, et al. Endocrine disorders in Fanconi anemia: recommendations for screening and treatment. J Clin Endocrinol Metab. 2015;100:803–11.
Bonfim C, Ribeiro L, Nichele S, Bitencourt M, Loth G, Koliski A, et al. Long-term survival, organ function, and malignancy after hematopoietic stem cell transplantation for fanconi anemia. Biol Blood Marrow Transplant. 2016;22:1257–63.
Yang X, Zhang X, Jiao J, Zhang F, Pan Y, Wang Q, et al. Rare variants in FANCA induce premature ovarian insufficiency. Hum Genet. 2019;138:1227–36.
Bogliolo M, Bluteau D, Lespinasse J, Pujol R, Vasquez N, d'Enghien CD, et al. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet Med. 2018;20:458–63.
Catucci I, Osorio A, Arver B, Neidhardt G, Bogliolo M, Zanardi F, et al. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet Med. 2018;20:452–7.
Fouquet B, Pawlikowska P, Caburet S, Guigon C, Mäkinen M, Tanner L, et al. A homozygous FANCM mutation underlies a familial case of non-syndromic primary ovarian insufficiency. Elife. 2017;6:e30490.
Kasak L, Punab M, Nagirnaja L, Grigorova M, Minajeva A, Lopes AM, et al. Bi-allelic recessive loss-of-function variants in FANCM cause non-obstructive azoospermia. Am J Hum Genet. 2018;103:200–12.
Yin H, Ma H, Hussain S, Zhang H, Xie X, Jiang L, et al. A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet Med. 2019;21:62–70.
Weinberg-Shukron A, Rachmiel M, Renbaum P, Gulsuner S, Walsh T, Lobel O, et al. Essential role of BRCA2 in ovarian development and function. N Engl J Med. 2018;379:1042–9.
Miao Y, Wang P, Xie B, Yang M, Li S, Cui Z, et al. BRCA2 deficiency is a potential driver for human primary ovarian insufficiency. Cell Death Dis. 2019;10:474.
Turchetti D, Zuntini R, Tricarico R, Bellacosa A. BRCA2 in ovarian development and function. N Engl J Med. 2019;380:1086–7.
Caburet S, Heddar A, Dardillac E, Creux H, Lambert M, Messiaen S, et al. Homozygous hypomorphic BRCA2 variant in primary ovarian insufficiency without cancer or Fanconi anaemia trait. J Med Genet. 2020. https://doi.org/10.1136/jmedgenet-2019-106672.
Qin Y, Zhang F, Chen ZJ. BRCA2 in ovarian development and function. N Engl J Med. 2019;380:1086.
Winship AL, Willson C, Hansen KR, Hutt KJ, Hickey M. Do BRCA1 and BRCA2 gene mutation carriers have a reduced ovarian reserve? Protocol for a prospective observational study. BMJ Open. 2019;9:e033810.
Park JY, Virts EL, Jankowska A, Wiek C, Othman M, Chakraborty SC, et al. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J Med Genet. 2016;53:672–80.
Shamseldin HE, Elfaki M, Alkuraya FS. Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J Med Genet. 2012;49:184–6.
Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly consanguineous population. Am J Hum Genet. 2019;105:879.
Alhathal N, Maddirevula S, Coskun S, Alali H, Assoum M, Morris T, et al. A genomics approach to male infertility. Genet Med. 2020;22:1967–75.
Yang Y, Guo J, Dai L, Zhu Y, Hu H, Tan L, et al. XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J Med Genet. 2018;55:628–36.
Zhang YX, Li HY, He WB, Tu C, Du J, Li W, et al. XRCC2 mutation causes premature ovarian insufficiency as well as non-obstructive azoospermia in humans. Clin Genet. 2019;95:442–3.
Zlotogora J, Dagan J, Ganen A, Abu-Libdeh M, Ben-Neriah Z, Cohen T. A syndrome including thumb malformations, microcephaly, short stature, and hypogonadism. J Med Genet. 1997;34:813–6.
Israeli ministry of health: Recommendations for carrier screening. In: https://www.health.gov.il/Subjects/Genetics/prof/Pages/default.aspx.
Yang Y, Guo T, Liu R, Ke H, Xu W, Zhao S, et al. FANCL gene mutations in premature ovarian insufficiency. Hum Mutat. 2020;41:1033–41.
Bieth E, Hamdi SM, Mieusset R. Genetics of the congenital absence of the vas deferens. Hum Genet. 2021;140(1):59–76.
Alvarez-Mora MI, Todeschini AL, Caburet S, Perets LP, Mila M, Younis JS, et al. An exome-wide exploration of cases of primary ovarian insufficiency uncovers novel sequence variants and candidate genes. Clin Genet. 2020;98(3):293–8.
Cocuzza M, Alvarenga C, Pagani R. The epidemiology and etiology of azoospermia. Clinics (Sao Paulo). 2013;68(Suppl 1):15–26.
Acknowledgements
We thank Mrs. Sarah Cohen for the language editing.
Author information
Authors and Affiliations
Contributions
Both authors took part in review design, execution, analysis, manuscript drafting, and critical discussion.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Daum, H., Zlotogora, J. Fanconi Anemia Gene Variants in Patients with Gonadal Dysfunction. Reprod. Sci. 29, 1408–1413 (2022). https://doi.org/10.1007/s43032-021-00582-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-021-00582-7