
Abstract
Cathecolestrogens are estradiol metabolites produced during folliculogenesis in the mammalian ovary. 2-Hydroxyestradiol (2-OHE2) is one of the most abundant although its role remains unknown. The aim of this study is to investigate whether the presence of 2-OHE2 during the germinal vesicle-to-metaphase II transition affects oocyte meiotic and preimplantation developmental competence. Mouse cumulus-oocyte complexes (COCs), isolated from fully grown antral follicles, were in vitro–matured (IVM) in the presence of 2-OHE2 (0.1, 1, 10 or 100 nM) for 6 or 15 h; then, their meiotic and developmental competence was evaluated using a number of cytological quality markers. With the exception of the highest dose (100 nM), the addition of 2-OHE2 to the IVM medium, did not alter, compared with untreated control, the frequency of oocytes that reached the MII stage. Instead, IVM in the presence of 1 nM 2-OHE2 highly increased the rate of preimplantation development and blastocyst quality. To understand whether this positive effect could be attributed to the events occurring during meiosis resumption, we analysed a number of specific cytological quality markers of the asymmetric division, such as PB-I volume and position, presence and extension of the cortical F-actin cap, meiotic spindle shape and area, and microtubule organisation centre localisation. The results highlighted how the presence of 1 nM 2-OHE2 significantly improved the overall cytological organisation required for a correct asymmetric division. Our results contribute a first step to acknowledge a potential role of this estradiol metabolite during the GV-to-MII transition, contributing to the acquisition of oocytes developmental competence.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The acquisition of the oocyte developmental competence occurs inside the ovarian follicle thanks to a continuous cross-talk between the germ cell and the surrounding cumulus and granulosa cells. The follicle somatic cell components contribute crucial factors required, through several signalling pathways and regulatory loops, to complete oocyte growth and meiotic maturation [1, 2].
The importance to preserve the communication between oocyte and surrounding granulosa cells becomes evident when cumulus-oocyte complexes (COCs) are isolated from fully grown antral follicles and in vitro cultured [3, 4]. In the mouse, the efficiency of COCs in vitro maturation (IVM) varies depending on both strain (i.e., outbred or inbred) [5] and culture conditions [6, 7], although the meiotic and developmental competence of metaphase II (MII) oocytes obtained in vitro remains lower compared with that of ovulated MII oocytes [8, 9].
In antral follicles, granulosa cells are the major source of oestrogen production. These steroid hormones, present in all vertebrates [10] and some invertebrates [11], are the primary female sex hormones with a key role in the control of female reproduction [12]. The estradiol (E2)-oestrogen receptor system is functional in maintaining oocyte meiotic arrest, and, in association with an increase of the luteinising hormone, a decrease in E2 downregulates cGMP and cAMP inducing meiotic resumption and the extrusion of the first polar body (PB-I) [13, 14]. Several studies included E2 in the medium used for COCs IVM at different concentrations and time windows, showing inhibition of meiosis resumption [14, 15] and chromosome aberrations [16]. During this window of E2 downregulation, hydroxylases actively metabolise this hormone producing biologically active metabolites named catecholestrogens [17, 18], primarily at C-2 (2-hydroxyestradiol, 2-OHE2) or C-4 (4-hydroxyestradiol, 4-OHE2) positions, but also 6α-, 6β-, 7α-, 12β-, 15α-, 15β-, 16α-, and 16β-OHE2 [19,20,21].
Catecholestrogens have been shown to play a role in folliculogenesis both in Fish and Mammals. In teleosts (i.e., catfish and zebrafish), 2-OHE2 has a crucial role as component of the gonadotrophin cascade of regulation of oocyte maturation and ovulation [22, 23]. In vitro maturation of intact catfish follicles in the presence of 5 μM 2-OHE2 increased significantly the frequency of oocytes attaining germinal vesicle breakdown (GVBD); instead, it did not change it when oocytes were cultured in the absence of surrounding follicle cells [24].
In the mammalian ovary, oestrogen hydroxylases are well-represented showing a 100-fold increase from small antral to fully grown follicles and corpora lutea [25]. The produced 2-OHE2 and 4-OHE2 metabolites are detected in the follicular fluid of antral follicles; they increase concentration during follicle maturation [26] and have an influence on the differentiation and proliferation of granulosa [27] and luteal [28] cells. Also, they have been shown to stimulate progesterone production in small follicles [29], to enhance cAMP production and, as a consequence, improve FSH activity [27]. When injected, 4-OHE2 induced ovulation in immature rats [30] and improved the overall implantation success rate [31].
Overall, the studies described above highlight several roles for catecholestrogens during follicle maturation, and suggest a link with the female gamete meiotic and developmental competence. The objective of this study was to investigate how the presence of 2-OHE2 during the germinal vesicle-to-metaphase II transition affects oocytes meiotic and preimplantation developmental competence.
The main aim of the present study was to explore the effects of one of 2-OHE2 at concentrations ranging from 0.1 to 100 nM, during IVM of mouse fully grown antral follicles from the GV to the MII stage. Oocytes that reached MII were inseminated with capacitated sperm and their preimplantation developmental competence recorded. Our results indicate a dose-dependent effect of 2-OHE2, that, when present in the IVM culture medium at 1 nM concentration, induces a significant beneficial effect on both meiotic resumption and preimplantation embryonic development.
Materials and Methods
Animals and Reagents
Four 6-week-old female and 6-month-old male CD1 mice were purchased from Charles River (Como, Italy). Animals were maintained under controlled room conditions (22 °C, with 60% air moisture and 12/12 light/dark photoperiod), and investigations were conducted in accordance with the guiding principles of European (n. 2010/63/UE) and Italian (n. 26/2014) laws protecting animals used for scientific research. All chemicals used were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise stated. Ultrapure water used for preparing media and solutions was obtained with a MilliQ® IQ 7000 system (Merck) equipped with a Biopak® Ultrafiltration filter (Merck).
COC Isolation and In Vitro Maturation
Females were injected with 5 I.U. Folligon (Intervet Srl, Italy), and 48 h later, COCs were isolated from the ovary. Briefly, ovaries were placed in 1 ml HEPES-buffered isolation medium: α-MEM plus GlutaMAX (Life Technology; cat.no. 32561–029) supplemented with 6 mg/ml HEPES, 5% fetal bovine serum (FBS), 0.23 mM sodium pyruvate, 1 mg/ml Fetuin, 100 I.U./ml penicillin and 75 μg/ml streptomycin in a 30-mm Petri dish (Corning, Euroclone, Italy) under a stereomicroscope. Fully grown antral follicles were punctured from the ovarian surface with a thin and sharp-pulled sterile Pasteur pipette. Only intact COCs with layers of cumulus cells completely surrounding the oocyte were used in further experiments. After washing in fresh HEPES-buffered isolation medium, COCs were cultured in 100 μl drops of α-MEM supplemented with 5% FBS; 0.23 mM sodium pyruvate; 1 mg/ml fetuin; 100 I.U./ml penicillin and 75 μg/ml streptomycin; 50 mI.U./ml FSH; and 10 ng/ml epidermal growth factor (EGF) in the absence (IVM) or presence (IVM-2-OHE2) of 0.1 nM, 1 nM, 10 nM or 100 nM 2-OHE2 (prepared in dimethyl-sulfoxide, DMSO, at a maximum concentration of 0.002%. As reported earlier [32] and confirmed by our results (Tables 1S and 2S), the presence of DMSO in IVM medium, at concentrations up to 0.1%, did not have adverse effects on oocyte maturation and on further preimplantation development. Thus, DMSO was always present in our IVM medium, independently on the presence or absence of 2-OHE2.
IVM drops were layered with mineral oil and placed in an incubator at 37 °C, 100% humidity in 5% CO2, 5% O2 and 90% N2 for 6 h (MI stage) or 15 h (MII stage). At the end of the culture period, MI or MII (confirmed by the presence of PB-I) oocytes were used for further experiments.
In order to collect in vivo–matured oocytes, females were injected with 5 I.U. Folligon followed, 48 h later, by 5 I.U. Corulon (Intervet) and, after 6 h (MI) or 15 h (MII), gametes were isolated from ovarian surface or from the oviducts, respectively.
MII oocytes, with the exception of those used for in vitro fertilisation (IVF), were denuded of their cumulus cells by a brief treatment with 500 I.U. hyaluronidase.
In Vitro Fertilisation and Preimplantation Development
MII oocytes surrounded by their cumulus cells were inseminated with capacitated sperm (2 × 106 sperm/ml) as previously described [33]. Two hours after insemination, oocytes were transferred into 40 μl drops of Whittigham medium (2 μl/oocyte) for an additional hour. Then, presumptive zygotes (as determined by the presence of a second polar body, PB-II) were transferred into 40 μl drops of M16 medium (2 μl/oocyte) supplemented with 0.4% BSA, 2 mM glutamine (Gibco), 5 mM taurine and 0.23 mM pyruvate for preimplantation development. Embryonic developmental rate was evaluated at 24 (2-cell stage), 48 (4-cell) and 96 (blastocyst) hours post-insemination.
Immunofluorescence
For the analysis of the meiotic spindle, MI or MII oocytes were fixed in 4% paraformaldehyde (PFA) in a microtubule stabilisation buffer (137 mM NaCl, 5 mM KCl, 1.1 mM Na2HPO4, 0.4 mM KH2PO4, 2 mM MgCl2, 4 mM NaHCO3, 2 mM EGTA, 5 mM PIPES, 5.5 mM Glucose, 0.1 M Glycine) containing 1% Triton X-100 for 45 min at 37 °C in agitation. Fixed oocytes were washed 3 times, 5 min each, in a washing solution (WS: 1x PBS, 0.2% sodium azide; 0.2% powdered milk, 2% FBS, 1% BSA, 0.1% glycine) supplemented with 0.2% Tween 20 and stored at 4 °C in WS.
Double labelling of α- and γ-tubulin, raised in the same species, was done through sequential detection of both epitopes, i.e. primary and then secondary antibodies for the first epitope followed by primary and then secondary antibodies for the second epitope.
Oocytes were incubated with a mouse anti-α-tubulin antibody (cat. N. T9026; dilution 1:1000 in WS) for 1 h at 37 °C in agitation. Then, gametes were washed 3 times, 15 min each, in WS at room temperature in agitation and incubated with the secondary AlexaFluo488 goat anti-mouse IgG antibody (cat. N. A11001, Molecular Probes; dilution 1:1000); then the gametes were incubated with a mouse anti-γ-tubulin antibody (cat. N. T5326; dilution 1:1000 in WS) for 1 h at 37 °C in agitation, washed 3 times, 15 min each, in WS at room temperature in agitation and simultaneously incubated with the secondary AlexaFluo633 goat anti-mouse IgG antibody (cat. N. A21053, Molecular Probes; dilution 1:1000) and TRITC-conjugated phalloidin (cat. N. P1951; dilution 1:1000 in WS). After incubation, oocytes were washed 3 times, 15 min each, in WS, counterstained with 0.2 μg/ml 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; cat N. D8417) and mounted in Vectashield (cat. N. H-1000, Vector Laboratories).
Ninety-six hours post-insemination, blastocysts were fixed in freshly prepared 4% PFA for 30 min and then washed in 1x PBS containing 0.1% Tween 20 (PBT). Embryos were processed for immunolabelling using a rabbit polyclonal anti-human OCT4 (cat. N. ab19857, Abcam; diluted 1:400 in WS) or a rabbit anti-human CDX2 (cat. N. 3977, Cell Signaling Technology; diluted 1:100 in WS) antibodies. Blastocysts were incubated with primary antibodies for 1 h at 37 °C and then washed twice (30 min each) in WS under gentle agitation. Then, they were incubated with AlexaFluor555 goat anti-rabbit IgG (cat. N. A21428, Molecular Probes; diluted 1:500 in WS) secondary antibody for 1 h at 37 °C, followed by 2 washes (30 min each) under gentle agitation, counterstained with DAPI (0.2 μg/ml in PBS, 10 min) and mounted in Vectashield.
Samples were examined using a Leica TCS SP8 confocal microscope equipped with lasers for AlexaFluor488, AlexaFluor555, AlexaFluor633 and DAPI fluorochromes. Stacks were obtained with axial distances of 0.3 μm. Immunolocalisation of OCT4 and CDX2 on blastocysts was analysed with an Olympus BX60 fluorescence microscope (Olympus, Ibaraki, Japan) equipped with single-bandpass filters for DAPI, AlexaFluor488 and AlexaFluor555; images were captured with a DP72 camera (Olympus) controlled by the CellSens Dimension 1.4.1 software (Olympus) and processed with ImageJ (http://imagej.nih.gov/ij/).
First Polar Body Morphology and Size
CCs-free MII oocytes were photographed using an inverted Olympus IX71 microscope equipped with a JVC KY-F58 3-CCD camera and a micromanipulator. For each oocyte, images (under × 2x objective) were captured with a frontal and lateral PB-I orientation obtained moving the oocytes using the micromanipulator micropipette. Only intact PB-Is were considered for further analyses. Pictures were analysed using the CellSens Dimension 1.4.1 software and the PB-I volumes calculated drawing three lines corresponding to the three radii of each PB-I.
Amplitude Angle Between PB-I and Oocyte Chromosomes
CCs-free MII oocytes were stained with 0.05 μg/ml bisBenzimide H-33342 trihydrochloride (Hoechst 33342; cat. N. D2261) in M2 medium for 15 min. Then, by using an inverted Olympus IX71 fluorescence microscope equipped with a micromanipulation system, single oocytes were rotated until metaphase chromosomes of both PB-I and oocyte appeared on the same focus. Pictures were taken, under × 20 objective, with a JVC KY-F58 3-CCD camera and analysed using the CellSens Dimension 1.4.1 software. Two lines were drawn from the centroid of the oocyte towards either the oocyte’s or PB-I’s chromosomes and the resulting angle was measured.
F-Actin Cap Intensity Profile and Amplitude Angle
The F-actin cap intensity was calculated, on the phalloidin red channel image, using the ImageJ software. A line was drawn (see Fig. 3) that, from the cortical region (p), intersected the MI or MII chromosomes, passed through the centroid (c) and reached the opposite pole (d). The fluorescence intensity was measured as p/d ratio. MI or MII oocytes of the three experimental conditions were divided into two groups: group I, oocytes with intensity (I) comprised between 1.8 < I ≤ 21.8; and group II, 1.1 < I ≤ 1.8.
The amplitude angle of the F-actin cap was determined drawing two lines starting from the centroid of the oocyte and the ends of the F-actin cap defined by phalloidin staining (see Fig. 4a).
Spindle Area and Shape
Using the CellSens Dimension software, the spindle area, identified by the α-tubulin signal, was calculated considering the spindle as a barrel made of two trapezoids with a common side at the equator width, the shorter sides at the two spindle poles and the height as half the length between the two poles (Fig. 5).
Based on the ratio between the equator width and the mean of the widths at the two poles, a barrel and a rectangular configuration were observed.
MTOC Distribution
The microtubule organisation centre (MTOC) distribution was established based on the γ-tubulin signal. Two different patterns were identified: (i) a single focus, in which all the γ-tubulin signals were distributed as a single cluster, localised at each spindle pole; and (ii) multiple foci, in which the γ-tubulin signals were distributed as two or more clusters along the width of the two poles.
Statistical Analysis
Statistical analyses were carried out using the Sigma Stat 3.5 software. Data, obtained from at least five independent experiments, were analysed by the Student’s t test for comparing two conditions or by the one-way ANOVA for comparing more than two groups. In the presence of significant differences, one-way ANOVA test was followed by the Fisher LSD Method (post hoc test). When data were not normally distributed, statistical analysis was performed either by the Mann-Whitney test (when the comparison involved two samples) or by the Kruskal–Wallis test together with the Dunn’s multiple range test (for the comparison of more than two samples). Parametric and nonparametric data were expressed as the error standard ± mean (SEM). The analysis of the absolute frequencies was performed using a Fisher’s exact test. Differences were considered significant for p values ≤ 0.05.
Results
To test the effects of 2-OHE2 on oocyte developmental competence, COCs were in vitro–matured for 15 h in the presence of 0.1, 1, 10 or 100 nM 2-OHE2.
Oocytes In Vitro Maturation
Based on the presence of the PB-I, more than 90% of oocytes in all five experimental conditions reached the MII phase. Compared with the control in vitro–matured COCs (IVM) (Table 1), oocytes whose COCs were cultured in the presence of 2-OHE2 (IVM-2-OHE2), spontaneously resumed meiosis and reached MII without significant differences (p > 0.05), with the exception of those treated with the highest 2-OHE2 concentration (100 nM) in which a decreased MII rate was observed (90.4 ± 1.5%; p = 0.015). This latest experimental group also showed significantly higher frequency of fragmented oocytes (3.1 ± 1.4%; p = 0.002).
MII Oocyte Developmental Competence
Following in vitro insemination, MII oocytes of the five experimental groups reached the 2-cell stage with the same rate (p = 0.884) (Table 1). However, those treated with 2-OHE2 attained the 4-cell stage with significantly higher frequency (≥ 65.3%; p ≤ 0.047) compared with IVM (55.5%), with the exception of the highest 2-OHE2 dose (100 nM) in which the increment was not significant (62.5 ± 5.0%, p = 0.148). Compared with IVM (28.6 ± 1.6%), 32.4 ± 4.7% (p = 0.283), 49.6 ± 1.2% (p < 0.001), 47.8 ± 4.9% (p < 0.001) and 29.2 ± 1.6% (p = 0.875) of 0.1 nM, 1 nM, 10 nM or 100 nM IVM-2-OHE2-treated embryos reached the blastocyst stage, respectively.
Overall, these results indicate a highly significant improvement of the preimplantation developmental rate to blastocyst of those antral oocytes whose COCs were matured in the presence of 1 or 10 nM 2-OHE2. Comparison between ovulated MII oocytes (OVMII) and IVM or IVM-2-OHE2 showed that the former performed always significantly better (Table 1).
Blastocyst Cell Number
The quality of blastocysts obtained with the 1 or 10 nM 2-OHE2 experimental conditions was evaluated based on their total number of cells and on the number of trophectoderm (TE) or inner cell mass (ICM) blastomeres, compared with control IVM blastocysts or to those derived from OVMII. Embryos were processed for the immunofluorescence localisation of OCT4 and CDX2 proteins, markers of ICM and TE cells, respectively. As shown earlier [9], the total cell number of blastocysts derived from IVM-COCs (32.3 ± 1.6) was significantly lower than that of control OVMII oocytes (52.5 ± 1.0; p < 0.001) (Table 2). Compared with IVM blastocysts, the presence of 1 nM 2-OHE2 during COC maturation significantly (p = 0.011) increased the number of cells (43.2 ± 4.6), instead that of 10 nM IVM-2-OHE2 (34.4 ± 1.7) was not significantly different (p = 0.561). This higher number of cells could be attributed to an increased number of ICM blastomeres (9.9 ± 0.7 vs. 7.4 ± 0.6; p = 0.003).
In summary, the results described highlight a significant improvement of the preimplantation developmental rate when COCs were matured in the presence of 1 nM or 10 nM 2-OHE2, and better blastocyst quality with 1 nM concentration.
Next, we addressed the question of whether this observed improvement in the presence of 1 nM 2-OHE2 could be attributed to the latest events of meiotic resumption, when the first asymmetric division occurs culminating with PB-I extrusion. To this end, we employed a number of known cytological quality markers of the meiotic division to compare MI or MII oocytes matured in vitro, either in the absence or in the presence of 1 nM 2-OHE2, and those obtained by ovulation. Specifically, we analysed (i) the PB-I volume [6, 34,35,36,37,38,39,40,41,42,43] and its position in respect to the oocyte’s spindle [44]; (ii) the presence and extension of the cortical F-actin cap [45,46,47]; (iii) the meiotic spindle shape and area [48,49,50,51] and (iv) the microtubule organisation centre (MTOC) localisation [8, 48, 52].
PB-I Volume and Position
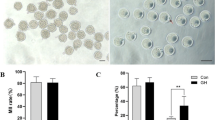
Based on the PB-I volume, we identified the presence of three main MII oocytes groups: group I, with a volume comprised between 1 × 103 and 15 × 103 μm3; group II, between 15 × 103 and 30 × 103 μm3 and group III, > 30 × 103 μm3 (Fig. 1).

PB-I size. MII oocytes from the in vitro–maturated (IVM), 1 nM IVM-2-OHE2 or ovulated (OV) groups were analysed to determine the PB-I volume (V) and divided (absolute percentage) into three groups (a): group I, with a volume comprised between 1 × 103 < V ≤ 15 × 103 μm3; group II, 15 × 103 < V ≤ 30 × 103 μm3 and group III, V > 30 × 103 μm3. (b) Following treatment with 2-OHE2, we observed a significant increase of MII oocytes with a correct group I PB-I size, although it remained lower compared with that of OVMII. In the same group, different superscript letters indicate the presence of a significant difference. Bar, 40 μm
Whilst all OVMII (p ≤ 0.001) oocytes belonged to group I, 60.6% and 75.2% of IVMMII and IVM-2-OHE2MII, respectively, the remaining 26.8% or 12.6% (IVMMII) and 23.0% or 1.8% (IVM-2-OHE2MII) were classified in group II (p = 0.601) or III (p = 0.003), respectively. Group III oocytes appeared similar to a 2-cell embryo, indicating the occurrence of an almost symmetrical division.
The PB-I position was calculated as the amplitude of the angle measured at the oocyte’s centroid between oocyte’s and PB-I’s chromosomes (Fig. 2a). IVM-2-OHE2MII oocytes showed significantly smaller angle compared with IVMMII (p = 0.044) oocytes, but similar (p = 0.399) to that measured in OVMII oocytes (Fig. 2b).

PB-I position relative to MII spindle. (a) Photograph of a fixed oocyte observed with bright-field and fluorescence, showing the relative position of PB-I to MII spindle. The chromosomes were stained with DAPI. The angle between the lines drawn from the centroid to the PB-I chromosomes or the oocyte spindle was measured and (b) the mean amplitude (± SEM) was calculated in the three experimental groups. Different superscript letters indicate the presence of a significant difference. The 2-OHE2 treatment improved significantly the positioning of the MII plate erasing the difference existing between IVMMII and OVMII. Bar, 30 μm
Presence and Extension of the Cortical F-Actin Cap
The eccentric position of the metaphase spindle and the consequent asymmetric meiotic division lead to the formation of two daughter cells with very different volumes. A crucial event during this cortical polarisation is the formation of a thick F-actin cap at the site of PB, just prior to its extrusion. Oocytes from OV, IVM or IVM-2-OHE2 groups were labelled with phalloidin after 6 h (MI) or 15 h (MII) of maturation. Based on the fluorescence intensity of the F-actin cap, we classified two main groups of oocytes (Fig. 3a): group I with an intensity comprised between 1.8 and 21.8; and a much fainter, group II, comprised between 1.1 and 1.8. All OVMI belonged to group I, compared with 53.8% (p < 0.001) of IVMMI oocytes, whilst the remaining 46.2% IVMMI oocytes showed a fainter, group II, F-actin cap (Fig. 3b). The presence of 2-OHE2 in the culture medium significantly increased (p = 0.016) to 86.7%; the number of MI oocytes with a clearly visible F-actin cap was not significantly (p = 0.112) different compared with that of OVMI oocytes (Fig. 3b). Similarly, the frequency of IVM-2-OHE2MII oocytes with marked F-actin cap (83.3%) resulted not significantly different (p = 0.059) compared with that of OVMII oocytes (97.5%) (Fig. 3c).

Relative fluorescence intensity of the F-actin cap. The F-actin cap intensity was calculated, on the phalloidin red channel image, drawing a line that, from the cortical region (p), intersects the MI or MII chromosomes, passes through the centroid (c) and reaches the opposite pole (d). The fluorescence intensity was measured as p/d ratio using the ImageJ software. MI or MII oocytes of the three experimental conditions were divided into two groups (a): group I, oocytes with an intensity (I) comprised between 1.8 < I ≤ 21.8; and group II, 1.1 < I ≤ 1.8. Following 2-OHE2 treatment, the matured MI (b) and MII (c) oocytes (absolute percentage) displayed an F-actin fluorescent intensity comparable with that observed in OVMI or OVMII oocytes. Different superscript letters indicate the presence of a significant difference. Bar, 20 μm
Then, we measured the amplitude of the angle formed between the oocyte centroid and the cortical F-actin cap ends in both in vitro– and in vivo–matured oocytes (Fig. 4a). Whilst all OVMI showed an angle comprised between 80° and 130°, 63.3% (p < 0.001) of IVMMI oocytes displayed an angle > 130° (Fig. 4b). When cultured in the presence of 2-OHE2, the majority (88.5%) of IVM-2-OHE2MI oocytes showed an angle similar to that of OVMI (p = 0.105; Fig. 4b), whereas only 11.5% oocytes had a wider (> 130°) angle. The same analysis performed on MII oocytes gave similar results (Fig. 4c).

F-actin cap extension. The extension of F-actin cap was measured as amplitude of the angle comprised between the two lines drawn from the centroid of the oocyte and the ends of the F-actin cap. MI or MII oocytes of IVM, IVM-2-OHE2 and OV were divided into two groups (a). Group I, oocytes with an angle amplitude (α) values 80° < α ≤ 130°; group II, oocytes with α values α > 130°. The MI or MII oocytes (absolute percentage) of IVM-2-OHE2 showing an F-actin extension comparable with that observed in OV oocytes (b–c). Different superscript letters indicate the presence of a significant difference. Bar, 20 μm
Meiotic Spindle Shape and Area
Oocytes from the three experimental groups were fixed after 6 h (MI) or 15 h (MII) of maturation, immunolabelled with anti-alpha tubulin to visualise their spindle and counterstained with DAPI.
The shape of the metaphase spindle was calculated as the ratio between the central width (Fig. 5a, y) and the mean between the width at the two poles (Fig. 5a, x and x′). Two main spindle shapes were observed: a barrel-like spindle (Fig. 5b) with a ratio value ≥ 1.8 and a rectangular-like spindle (Fig. 5c) with a ratio value < 1.8.

Meiotic spindle morphology. Representative fluorescent images of MI spindles with barrel- (a) or rectangular-shape (b), labelled with α-tubulin (green) and counterstained with DAPI (blue). Bar = 6 μm. (c) The spindle area was calculated as the sum of the areas of the two drawn trapezoids with a common side at the equator (y) and the shorter sides at the two spindle poles (x, x′)
All the OVMI oocytes displayed a barrel-like shape with an area of 173.6 ± 8.6 μm2 (Table 3). When compared with IVMMI, the latter showed a significant (p = 0.004) decreased frequency (75%) of oocytes with a barrel-like spindle. The maturation of COCs in the presence of 2-OHE2 decreased the frequency of oocytes with a rectangular-like spindle to values not significantly different (13.3%; p = 0.077) compared with that of OVMI.
Similarly, all OVMII oocytes showed a barrel-like spindle, whereas the two IVM conditions produced both barrel- and rectangular-like spindles. Again, IVM in the presence of 2-OHE2 gave a significantly (p = 0.011) higher frequency (81%) of oocytes with barrel-like spindle compared with those matured in the absence of the hormone (53.8%).
MTOC Localisation
Gamma-tubulin, a component of the microtubule organisation centres (MTOCs), plays a key role in spindle formation during meiosis [53]. Here, we analysed the MTOCs localisation in MI and MII oocytes obtained from the three experimental conditions. Double immunostaining with α- and γ-tubulin showed two main patterns of MTOCs localisation: one with all MTOCs clustered at each of the two spindle poles (single signal) (Fig. 6a); the other, with the MTOCs dispersed at the two spindle poles (multiple signals) (Fig. 6b).

MTOC localisation. Representative fluorescent images of MTOCs localisation in MI spindles with barrel- (a) or rectangular-shape (b). Double immunostaining with α- (green) and γ- (red) tubulin, the latter displaying two main patterns of MTOCs localisation: one with all MTOCs clustered (a), the other, with MTOCs dispersed (b) at the spindle poles. Chromosomes are counterstained with DAPI (blue). Bar, 6 μm
The great majority of control OVMI (97.2%) or OVMII (93.3%) oocytes displayed only a single signal pattern of MTOC localisation associated to a barrel-like spindle (Table 4). Instead, only 33.3% of IVMMI or 14.3% IVMMII oocytes showed the combination of a barrel-like spindle and a single MTOC signal. These frequencies increased significantly when COCs were matured in the presence of 2-OHE2 with 57.9% (p = 0.039) of IVM-2-OHE2MI and 57.1% (p < 0.001) of IVM-2-OHE2MII displaying a barrel-like spindle and a single MTOC signal.
Discussion
This study demonstrates a dose-dependent response to the presence of 2-OHE2 during the GV-to-MII transition on the oocyte meiotic and developmental competence (Fig. 7). Although the overall quality of oocytes cultured in the presence of 2-OHE2 remained lower compared with that of control OV oocytes, the presence of this catecholestrogen clearly produced a highly significant improvement when compared with COCs that were matured in the absence of 2-OHE2. Our results show that whilst the rate of meiotic resumption remained unchanged at doses comprised between 0.1and 10 nM, it decreased significantly at the highest dose employed. It was the combined observation of the developmental rate and blastocyst quality that highlighted the highly significant improvement obtained following GV-to-MII maturation in the presence of 1 nM dose, with a sharp 21% increase of embryos that completed preimplantation development. Furthermore, these blastocysts displayed higher number of blastomeres, a sign of an intensified cell cycle rate [9, 54, 55].

Graphical summary of the results highlighting the improvement of the meiotic and developmental competence of oocytes matured in the presence of 1 nM 2-OHE2 when compared with oocytes matured in the absence of the catecholestrogen (IVM) and towards the acquisition of properties pertaining to ovulated oocytes (OV)
This improved developmental competence contradicts earlier studies with bovine COCs that, when matured in the presence of 3 μM of this catecholestrogen, display a 5-fold decrease of development to blastocyst [56]. Also, COC maturation in the presence of a 2-OHE2 metabolite (2-methoxyestradiol, 2-ME2) caused spindle aberrations, chromosome congression failure and non-disjunction at the time of mouse oocytes meiosis resumption [57] and had negative effects on bovine oocyte developmental competence [56]. The improved results we obtained might find an explanation in the doses used in our experiments (0.1–100 nM), which are closer to the 15 nM physiological condition described in pig preovulatory follicles [26].
To understand the reason of the recorded oocyte developmental competence improvement, we used a number of known cytological quality markers of meiotic resumption. All these markers showed that the presence of 2-OHE2 smoothed the GV-to-MII transition, favouring a more physiological asymmetric division (Fig. 7). Further studies will be needed to understand the mechanism of action of this catecholestrogen, although three of its described features may give us clues to explain our results. One of these refers to the fact that 2-OHE2 contains a phenolic functional group that has been shown, in smooth muscles, to work as a potent antioxidant eliminating free radicals [58, 59] and protecting membrane phospholipids against peroxidase [60]. Indeed, COCs cultured in the presence of antioxidants (i.e. sodium citrate, α-lipoic acid and acetyl-l-carnitine) complete the GV-to-MII transition with significant reduced number of oocytes with abnormal spindles [61]. In alternative, 2-OHE2 could work as an E2 antagonist. Earlier studies showed that culturing catfish post-vitellogenic follicles in the presence of 1–20 μM 2-OHE2 induced a 6–17-fold increase in the number of oocytes that resumed meiosis [22,23,24, 62]. This improvement correlated with increased steroid production [63, 64], inhibition of aromatase activity [65] and decreased follicular cAMP, which would result in the removal of the inhibitory effects of E2 on oocyte maturation and meiotic resumption. Another possible explanation on how 2-OHE2 improves the female gamete quality is related with its role in the production of nitric oxide, a key factor that regulates germinal vesicle breakdown and PB-I extrusion in mouse oocytes [66]. Interestingly, a recent study showed that 10 nM 2-OHE2 induced an increase of nitric oxide production by ovine uterine artery endothelial cells, whereas 100 nM was unable to produce this effect [67].
In conclusion, we demonstrated that, when present at 1 nM concentration during mouse COC in vitro maturation, 2-OHE2 has a beneficial effect on the meiotic and developmental competence of the enclosed oocytes. Our results contribute a first step to acknowledge a potential role of this estradiol metabolite during the latest stages of mammalian folliculogenesis.
References
Zuccotti M, Merico V, Cecconi S, Redi CA, Garagna S. What does it take to make a developmentally competent mammalian egg? Hum Reprod Update. 2011;17:525–40. https://doi.org/10.1093/HUMUPD/DMR009.
Russell DL, Gilchrist RB, Brown HM, Thompson JG. Bidirectional communication between cumulus cells and the oocyte: old hands and new players? Theriogenology. 2016;86:62–8. https://doi.org/10.1016/J.THERIOGENOLOGY.2016.04.019.
Gilchrist RB. Recent insights into oocyte–follicle cell interactions provide opportunities for the development of new approaches to in vitro maturation. Reprod Fertil Dev. 2011;23:23–31. https://doi.org/10.1071/RD10225.
Da Broi MG, Giorgi VSI, Wang F, Keefe DL, Albertini D, Navarro PA. Influence of follicular fluid and cumulus cells on oocyte quality: clinical implications. J Assist Reprod Genet. 2018;35:735–51. https://doi.org/10.1007/S10815-018-1143-3.
Suzuki O, Asano T, Yamamoto Y, Takano K, Koura M. Development in vitro of preimplantation embryos from 55 mouse strains. Reprod Fertil Dev. 1996;8:975–80. https://doi.org/10.1071/RD9960975.
Cavalera F, Simovic M, Zanoni M, Merico V, Garagna S, Zuccotti M. IVM of mouse fully grown germinal vesicle oocytes upon a feeder layer of selected cumulus cells enhances their developmental competence. Reprod Fertil Dev. 2019;29:1068. https://doi.org/10.1071/RD18444.
Zuccotti M, Merico V, Rebuzzini P, Belli M, Vigone G, Mulas F, et al. 3D culture of ovarian follicles: a system towards their engineering? Int J Dev Biol. 2015;59(4–6):211–6. https://doi.org/10.1387/ijdb.150172mz.
Barrett SL, Albertini DF. Cumulus cell contact during oocyte maturation in mice regulates meiotic spindle positioning and enhances developmental competence. J Assist Reprod Genet. 2010;27(1):29–39. https://doi.org/10.1007/s10815-009-9376-9.
Sanfins A, Plancha CE, Albertini DF. Pre-implantation developmental potential from in vivo and in vitro matured mouse oocytes: a cytoskeletal perspective on oocyte quality. J Assist Reprod Genet. 2015;32(1):127–36. https://doi.org/10.1007/s10815-014-0363-4.
Eick GN, Thornton JW. Evolution of steroid receptors from an estrogen-sensitive ancestral receptor. Mol Cell Endocrinol. 2011;334(1–2):31–8. https://doi.org/10.1016/j.mce.2010.09.003.
Lafont R, Mathieu M. Steroids in aquatic invertebrates. Ecotoxicology. 2007;16(1):109–30. https://doi.org/10.1007/s10646-006-0113-1.
Findlay JK, Liew SH, Simpson ER, Korach KS. Estrogen signaling in the regulation of female reproductive functions. Handb Exp Pharmacol. 2010;198:29–35. https://doi.org/10.1007/978-3-642-02062-9_2.
Eppig JJ, Koide SL. Effects of progesterone and oestradiol-17beta on the spontaneous meiotic maturation of mouse oocytes. J Reprod Fertil. 1978;53(1):99–101. https://doi.org/10.1530/jrf.0.0530099.
Smith DM, Tenney DY. Effects of steroids on mouse oocyte maturation in vitro. J Reprod Fertil. 1980;60(2):331–8. https://doi.org/10.1530/jrf.0.0600331.
Richter JD, McGaughey RW. Specificity of inhibition by steroids of porcine oocytes maturation in vitro. J Exp Zool. 1979;209(1):81–90.
Beker AR, Colenbrander B, Bevers MM. Effect of 17-beta-estradiol on the in vitro maturation of bovine oocytes. Theriogenology. 2002;58(9):1663–73. https://doi.org/10.1016/S0093-691X(02)01082-8.
Ball P, Knuppen R. Catecholoestrogens (2-and 4-hydroxyoestrogens): chemistry, biogenesis, metabolism, occurrence and physiological significance. Acta Endocrinol Suppl (Copenh). 1980;232:1–127.
Martucci CP, Fishman J. P450 enzymes of estrogen metabolism. Pharmacol Ther. 1993;57(2–3):237–57. https://doi.org/10.1016/0163-7258(93)90057-K.
Zhu BT, Conney AH. Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis. 1998;19(1):1–27. https://doi.org/10.1093/carcin/19.1.1.
Lee AJ, Kosh JW, Conney AH, Zhu BT. Characterization of the NADPH-dependent metabolism of 17beta-estradiol to multiple metabolites by human liver microsomes and selectively expressed human cytochrome P450 3A4 and 3A5. J Pharmacol Exp Ther. 2001;298:420–32.
Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227:115–24. https://doi.org/10.1016/j.canlet.2004.10.007.
Mishra A, Joy KP. Effects of gonadotrophin in vivo and 2-hydroxyoestradiol-17 in vitro on follicular steroid hormone profile associated with oocyte maturation in the catfish Heteropneustes fossilis. J Endocrinol. 2006;189:341–53. https://doi.org/10.1677/joe.1.06686.
Mishra A, Joy KP. Involvement of mitogen-activated protein kinase in 2-hydroxyestradiol-17beta-induced oocyte maturation in the catfish Heteropneustes fossilis and a note on possible interaction with protein phosphatases. Gen Comp Endocrinol. 2006;147:329–35. https://doi.org/10.1016/j.ygcen.2006.02.002.
Mishra A, Joy KP. 2-Hydroxyestradiol-17beta-induced oocyte maturation: involvement of cAMP-protein kinase A and okadaic acid-sensitive protein phosphatases, and their interplay in oocyte maturation in the catfish Heteropneustes fossilis. J Exp Biol. 2006;209:2567–75. https://doi.org/10.1242/jeb.02270.
Hammond JM, Hersey RM, Walega MA, Weisz J. Catecholestrogen production by porcine ovarian cells. Endocrinology. 1986;118(6):2292–9. https://doi.org/10.1210/endo-118-6-2292.
Basini G, Bussolati S, Santini SE, Bianchi F, Careri M, Mangia A, et al. Hydroxyestrogens inhibit angiogenesis in swine ovarian follicles. J Endocrinol. 2008;199(1):127–35. https://doi.org/10.1677/JOE-08-0258.
Spicer LJ, Hammond JM. Catecholestrogens inhibit proliferation and DNA synthesis of porcine granulosa cells in vitro: comparison with estradiol, 5 alpha-dihydrotestosterone, gonadotropins and catecholamines. Mol Cell Endocrinol. 1989;64(1):119–26.
Tekpetey FR, Armstrong DT. Catecholestrogen modulation of steroid production by rat luteal cells: mechanism of action. Mol Cell Endocrinol. 1994;101(1–2):49–57. https://doi.org/10.1016/0303-7207(94)90218-6.
Spicer LJ, Kao LC, Strauss JF, Hammond JM. 2-Hydroxyestradiol enhanced progesterone production by porcine granulosa cells: dependence on de novo cholesterol synthesis and stimulation of cholesterol side-chain cleavage activity and cytochrome P450scc messenger ribonucleic acid levels. Endocrinology. 1990;127:2763–70. https://doi.org/10.1210/endo-127-6-2763.
Emons G, Ball P. Induction of ovulation in immature female rats by a single injection of 4-hydroxyoestradiol-dibenzoate. Acta Endocrinol. 1982;100(1):105–8. https://doi.org/10.1530/acta.0.1000105.
Paria BC, Ma W, Andrenyak DM, Schmid PC, Schmid HHO, Moody DE, et al. Effects of cannabinoids on preimplantation mouse embryo development and implantation are mediated by brain-type cannabinoid receptors. Biol Reprod. 1998;58(6):1490–5. https://doi.org/10.1095/biolreprod58.6.1490.
Nogueira D, Cortvrindt R, Everaerdt B, Smitz J. Effects of long-term in vitro exposure to phosphodiesterase type-3 inhibitors on follicle and oocyte development. Reproduction. 2005;130(2):177–86. https://doi.org/10.1530/rep.1.00652.
Zuccotti M, Giorgi Rossi P, Martinez A, Garagna S, Forabosco A, Redi CA. Meiotic and developmental competence of mouse antral oocytes. Biol Reprod. 1998;58(3):700–4. https://doi.org/10.1095/biolreprod58.3.700.
Xia P. Intracytoplasmic sperm injection: correlation of oocyte grade based on polar body perivitelline space and cytoplasmic inclusions with fertilization rate and embryo quality. Hum Reprod. 1997;12:1750–5. https://doi.org/10.1093/HUMREP/12.8.1750.
Ebner T, Yaman C, Moser M, Sommergruber M, Feichtinger O, Tews G. Prognostic value of first polar body morphology on fertilization rate and embryo quality in intracytoplasmic sperm injection. Hum Reprod. 2000;15:427–30. https://doi.org/10.1093/HUMREP/15.2.427.
Mikkelsen AL, Lindenberg S. Morphology of in-vitro matured oocytes: impact on fertility potential and embryo quality. Hum Reprod. 2001;16:1714–8. https://doi.org/10.1093/HUMREP/16.8.1714.
Wang WH, Meng L, Hackett RJ, Odenbourg R, Keefe DL. The spindle observation and its relationship with fertilization after intracytoplasmic sperm injection in living human oocytes. Fertil Steril. 2001;75(2):348–53. https://doi.org/10.1016/s0015-0282(00)01692-7.
Rienzi L, Ubaldi FM, Iacobelli M, Minasi MG, Romano S, Ferrero S, et al. Significance of metaphase II human oocyte morphology on ICSI outcome. Fertil Steril. 2008;90:1692–700. https://doi.org/10.1016/J.FERTNSTERT.2007.09.024.
Rienzi L, Vajta G, Ubaldi F. Predictive value of oocyte morphology in human IVF: a systematic review of the literature. Hum Reprod Update. 2011;17:34–45. https://doi.org/10.1093/HUMUPD/DMQ029.
Navarro PA, de Arau’jo MM, de Arau’jo CM, Rocha M, dos Reis R, Martins W. Relationship between first polar body morphology before intracytoplasmic sperm injection and fertilization rate cleavage rate and embryo quality. Int J Gynaecol Obstet. 2009;104:226–9. https://doi.org/10.1016/J.IJGO.2008.11.008.
Yi K, Rubinstein B, Li R. Symmetry breaking and polarity establishment during mouse oocyte maturation. Philos Trans R Soc Lond Ser B Biol Sci. 2013;368:20130002. https://doi.org/10.1098/RSTB.2013.0002.
Almonacid M, Terret ME, Verlhac MH. Control of nucleus positioning in mouse oocytes. Semin Cell Dev Biol. 2018;82:34–40. https://doi.org/10.1016/J.SEMCDB.2017.08.010.
Cavalera F, Zanoni M, Merico V, et al. Chromatin organization and timing of polar body I extrusion identify developmentally competent mouse oocytes. Int J Dev Biol. 2019;63(3–4-5):245–51. https://doi.org/10.1387/ijdb.180362sg.
Cooke S, Tyler JP, Driscoll GL. Meiotic spindle location and identification and its effect on embryonic cleavage plane and early development. Hum Reprod. 2003;18(11):2397–405. https://doi.org/10.1093/humrep/deg447.
Webb M, Howlett SK, Maro B. Parthenogenesis and cytoskeletal organization in ageing mouse eggs. J Embryol Exp Morpholog. 1986;95:131–45.
Kim NH, Moon SJ, Prather RS, Day BN. Cytoskeletal alteration in aged porcine oocytes and parthenogenesis. Mol Reprod Dev. 1996;43(4):513–8.
Coticchio G, Guglielmo MC, Albertini DF, Dal Canto M, Mignini Renzini M, de Ponti E, et al. Contributions of the actin cytoskeleton to the emergence of polarity during maturation in human oocytes. Mol Hum Reprod. 2014;20(3):200–7. https://doi.org/10.1093/molehr/gat085.
Sanfins A, Lee GY, Plancha CE, Overstrom EW, Albertini DF. Distinctions in meiotic spindle structure and assembly during in vitro and in vivo maturation of mouse oocytes. Biol Reprod. 2003;69(6):2059–67. https://doi.org/10.1095/biolreprod.103.020537.
Battaglia DE, Goodwin P, Klein NA. Influence of maternal age on meiotic spindle assembly in oocytes from naturally cycling women. Hum Reprod. 1996;11:2217–22. https://doi.org/10.1093/oxfordjournals.humrep.a019080.
Wang Q, Sun QY. Evaluation of quality: morphological, cellular and molecular predictors. Reprod Fertil Dev. 2007;19(1):1–12. https://doi.org/10.1071/RD06103.
Coticchio G, Sereni E, Serrao L, Mazzone S, Iadarola I, Borini A. What criteria for the definition of oocyte quality? Ann N Y Acad Sci. 2004;1034:132–44. https://doi.org/10.1196/annals.1335.016.
Ibáñez E, Sanfins A, Combelles CM, Overström EW, Albertini DF. Genetic strain variations in the metaphase-II phenotyte of mouse oocytes matured in vivo or in vitro. Reproduction. 2005;130(6):845–55. https://doi.org/10.1530/rep.1.00558.
Zimmerman WC, Sillibourne J, Rosa J, Doxsey SJ. Mitosis-specific anchoring of gamma tubulin complexes by pericentrin controls spindle organization and mitotic entry. Mol Biol Cell. 2004 Aug;15(8):3642–57.
Matsuura K, Hayashi N, Takiue C, Hirata R, Habara T, Naruse K. Blastocyst quality scoring based on morphologic grading correlates with cell number. Fertil Steril. 2010;94(3):1135–7. https://doi.org/10.1016/j.fertnstert.2009.11.003.
Kong X, Yang S, Gong F, et al. The relationship between cell number, division behavior and developmental potential of cleavage stage human embryos: a time-lapse study. PLoS One. 2016;11(4):e0153697. https://doi.org/10.1371/journal.pone.0153697.
Lattanzi ML, Santos CB, Mudry MD, Baranao JL. Exposure of bovine oocytes to the endogenous metabolite 2-methoxyestradiol during in vitro maturation inhibits early embryonic development. Biol Reprod. 2003;69(6):1793–800. https://doi.org/10.1095/biolreprod.103.020743.
Eichenlaub-Ritter U, Winterscheidt U, Vogt E, Shen Y, Tinneberg HR, Sorensen R. 2-methoxyestradiol induces spindle aberrations, chromosome congression failure, and nondisjunction in mouse oocytes. Biol Reprod. 2007;76(5):784–93. https://doi.org/10.1095/biolreprod.106.055111.
Gelbke HP, Ball P, Knuppen R. 2-Hydroxyoestrogens: chemistry, biogenesis, metabolism and physiological significance. In: Briggs MH, Christie GA, editors. Advances in steroid biochemistry and pharmacology, vol. 6. New York: Academic Press; 1977. p. 81–154.
Niki E, Nakano M. Estrogens as antioxidants. Methods Enzymol. 1990;186:330–3. https://doi.org/10.1016/0076-6879(90)86126-g.
Dubey RK, Tyurina YY, Tyurin VA, et al. Estrogen and tamoxifen metabolites protect smoth muscle cell membrane phospholipids against peroxidation and inhibit cell growth. Circ Res. 1999;84(2):229–39. https://doi.org/10.1161/01.RES.84.2.229.
Liang LF, Qi ST, Xian YX, Huang L, Sun XF, Wang WH. Protective effect of antioxidants on the pre maturation aging of mouse oocytes. Sci Rep. 2017;7(1):1434. https://doi.org/10.1038/s41598-017-01609-3.
Senthilkumaran B, Joy KP. Periovulatory changes in catfish ovarian oestradiol-17beta, oestrogen-2-hydroxylase and catechol-O-methyltransferase during GnRH analogue-induced ovulation and in vitro induction of oocyte maturation by catecholoestrogens. J Endocrinol. 2001;168:239–47. https://doi.org/10.1677/joe.0.1680239.
Chourasia TK, Joy KP. Seasonal variation in tissue estrogen-2/4-hydroxylases (EH) and in vitro effects of steroids on ovarian EH activity in the catfish Heteropneustes fossilis. Steroids. 2010;75:1097–105. https://doi.org/10.1016/j.steroids.2010.07.004.
Chourasia TK, Joy KP. In vitro effects of 2-hydroxyestradiol-17β on ovarian follicular steroid secretion in the catfish Heteropneustes fossilis and identification of the receptor and signaling mechanisms. Gen Comp Endocrinol. 2012;175:500–13. https://doi.org/10.1016/j.ygcen.2011.12.007.
Chourasia TK, Joy KP. Estrogen-2/4-hydroxylase activity is stimulated during germinal vesicle breakdown induced by hCG, IGF-1, GH and insulin in the catfish Heteropneustes fossilis. Gen Comp Endocrinol. 2008;155:413–21. https://doi.org/10.1016/j.ygcen.2007.07.005.
Huo LJ, Liang CG, Yu LZ, Zhong ZS, Yang ZM, Fan HY, et al. Inducible nitric oxide synthase-derived nitric oxide regulates germinal vesicle breakdown and first polar body emission in the mouse oocyte. Reproduction. 2005;129(4):403–9. https://doi.org/10.1530/rep.1.0542.
Landeros RV, Pastore MB, Magness RR. Effects of the catechol and methoxy metabolites of 17β-estradiol on nitric oxide production by ovine uterine artery endothelial cells. Reprod Sci. 2019;26(4):459–68. https://doi.org/10.1177/1933719118783265.
Acknowledgments
Open access funding provided by Università degli Studi di Pavia within the CRUI-CARE Agreement. The authors thank Merck-MilliQ Laboratory Water Solutions for support and Kinesis for supplying the plastic ware necessary to perform this study.
Funding
This work was made possible through the support from the Italian Ministry of Education, University and Research (MIUR) Dipartimenti di Eccellenza Program (2018–2022) to the Department of Biology and Biotechnology ‘L. Spallanzani’, University of Pavia, and a grant from the University of Pavia (FRG 2018).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 87 kb).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Merico, V., Zanoni, M., Parada-Bustamante, A. et al. In Vitro Maturation of Fully Grown Mouse Antral Follicles in the Presence of 1 nM 2-Hydroxyestradiol Improves Oocytes’ Developmental Competence. Reprod. Sci. 28, 121–133 (2021). https://doi.org/10.1007/s43032-020-00276-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-020-00276-6