
Abstract
Ascidian-derived microorganisms are a significant source of pharmacologically active metabolites with interesting structural properties. When discovering bioactive molecules from ascidian-derived fungi, two new phenols, roussoelins A (1) and B (2), and ten known polyketides (3–12) were isolated from the ascidian-derived fungus Roussoella siamensis SYSU-MS4723. The planar structure of compounds 1 and 2 was established by analysis of HR-ESIMS and NMR data. The conformational analysis of the new compounds was assigned according to coupling constants and selective gradient NOESY experiments, and absolute configurations were completed by the modified Mosher’s method. Among the isolated compounds, 1, 2, and 9 showed moderate antioxidant capacity.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Marine organisms have been a significant natural source for the discovery of multiple pharmacologically active molecules with various structures (Blunt et al. 2017, 2018; Carroll et al. 2019; Jiang et al. 2020; Liu et al. 2019). Among them, about 150 molecules with a wide range of bioactivities have been discovered from ascidian-derived microorganisms (Bugni and Ireland 2004; Chen et al. 2018; Donia et al. 2006). For instance, the lomaiviticins A and B with an intricate dimeric diazobenzofluorene glycoside structure and antitumor activity were discovered from ascidian-derived Actinomycetes Micromonospora lomaivitiensis (He et al. 2001). The ascidian-associated fungus Eurotiomycetes strain 110,162 produced an anti-mycobacterial oxazinin A that contained a unique dimeric structure (Lin et al. 2014b). Another ascidian-derived fungus Trichobotrys effuse 4729 yielded an anti-glioma trichobamide A that was a pyrrocidine alkaloid containing a novel tetrahydro-5H-furo[2,3-b]pyrrol-5-one moiety (Chen et al. 2019b).
Since the first report in 1997 from Crews’ research group describing the chemical investigation of a fungus Pithomyces sp. (isolated from the Indo-Pacific tunicate Oxycorynia fascicularis) to afford polekeides (pitholides A–D) (Wang et al. 1997), a total of 52 new metabolites have been reported from 22 research papers involved in ascidian-derived fungi (Belofsky et al. 2000; Bugni et al. 2000; Chen et al. 2019a, b; Dewapriya et al. 2017, 2018; Garo et al. 2003; Ivanets et al. 2018; Li et al. 2020; Lin et al. 2014a; MalmstrøM et al. 2000; Montenegro et al. 2012; Motohashi et al. 2009; Murshid et al. 2016; Niaz et al. 2019; Shaala and Youssef 2015; Smetanina et al. 2004; Song et al. 2019; Sumilat et al. 2017; Xin et al. 2007; Yamazaki et al. 2015; Yurchenko et al. 2017). There were 21 strains (including one strain of unidentified fungus) belonging to eight genera (Acremonium, Aspergillus, Humicola, Penicillium, Pithomyces, Talaromyces, Trichobotrys, and Trichoderma). Penicillium (34.6%, 18) and Aspergillus (28.8%, 15) each represents more than 25% of the total and are the dominant producers of new metabolites, whose contributions together comprise more than half of the total. These new metabolites with various structures (including polyketide, alkaloid, sesquiterpene, merosesquiterpene, peptide, cerebroside) displayed numerous biological activities, including cytotoxicity (Chen et al. 2019b), antibacterial activity (Dewapriya et al. 2018), antifungal activity (Murshid et al. 2016), anti-inflammatory activity (Belofsky et al. 2000; Chen et al. 2019a), enzyme inhibitor activity (Yamazaki et al. 2015), and other activities (Lin et al. 2014a).
Though 25 genera fungi of 19 families in two phyla have been derived from the ascidian, eight genera have been chemically investigated and the number of reports describing natural products from ascidian-derived fungi is still low. Recently, we focused on bioactive secondary metabolites from ascidian-derived fungi isolated from the South China Sea (Chen et al. 2019a, b; Niaz et al. 2019). As we continue to discover bioactive molecules from ascidian-derived fungi, two new 5-(3-hydroxybutan-2-yl)benzene-1,3-diol, roussoelins A (1) and B (2), together with ten known polyketides (3–12) were obtained from the ascidian-derived fungus Roussoella siamensis SYSU-MS4723 (Fig. 1), whose secondary metabolites were studied for the first time from a genus of an ascidian-derived fungi. The conformational analysis was assigned according to coupling constants and selective gradient NOESY experiments, and absolute configurations were finally identified by a modified version of Mosher’s method (Ohtani et al. 1991). The cytotoxicity, anti-inflammatory, and antioxidant activity of these molecules are reported herein.

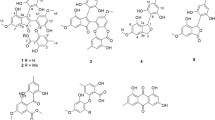
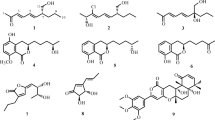
Chemical structures of 1–12
Results and discussion
The EtOAc extract of R. siamensis SYSU-MS4723 was subjected to repeated silica gel and Sephadex LH-20 column chromatography, followed by semipreparative HPLC, to afford two new phenols, roussoelins A (1) and B (2), and ten known polyketides (3–12).
Roussoelin A (1) was isolated as a colorless oil. The molecular formula C10H14O3 was assigned by the negative HR-ESIMS ions at m/z 181.08712 [M−H]− (calcd. for C10H13O3, 181.08702) (Supplementary Fig. S1), indicating four degrees of unsaturation. The IR spectrum (Supplementary Fig. S2) of 1 revealed the presence of a hydroxy (3346 cm−1) group. The 1H NMR data (Supplementary Fig. S3) (Table 1) revealed three aromatic protons [δH 6.13 (2H, d, J = 2.2 Hz); 6.10 (1H, t, J = 2.2 Hz)], indicating a 1,3,5-trisubstituted aromatic ring; two methyls [δH 3.69 (1H, dq, J = 8.3, 6.3 Hz); 2.39 (1H, m)]; and two methyl groups [δH 1.00 (3H, d, J = 6.3 Hz); 1.26(1H, t, J = 6.9 Hz)]. The 13C NMR (Supplementary Fig. S4) and HSQC data (Table 1) of 1 showed the presence of 10 carbons. Among them, six sp2 hybridized carbons (δC 101.5, 107.2, 107.2, 148.8, 159.4, 159.4) belonged to a benzene ring, while there were four remaining sp3 hybridized carbons, one of them (δC 73.4) directly connected with a heteroatom. The planar structure of 1 was mainly identified by 1H-1H COSY (Supplementary Fig. S5), HSQC (Supplementary Fig. S6), and HMBC (Supplementary Fig. S7) spectra (Fig. 2). A 3-hydroxybutan-2-yl group was deduced by the 1H-1H COSY cross peak between H-1 and H-2, H-2 and H-3, H-3 and H-10, together with HMBC correlations from H-1 to C-2 and C-3, H-10 to C-2 and C-3. Key HMBC correlations from H-10 and H-3 to C-4 suggested that the 3-hydroxybutan-2-yl group was linked to C-4 of an aromatic ring. Two hydroxyl groups were located on C-6 (δC 159.4) and C-8 (δC 159.4) of an aromatic ring according to the chemical shift and the HMBC correlations from H-7 to C-6 and C-8. The planar structure of 1 was elucidated as 5-(3-hydroxybutan-2-yl)benzene-1,3-diol.

Key 1H-1H COSY (red line) and HMBC (blue arrow) correlations of compounds 1 and 2 (color figure online)
The relative configuration of C-2 and C-3 in roussoelin A was established through selective NOESY correlations and coupling constants. A large coupling constant (3JH-2,H-3 = 8.3 Hz) between protons H-2 and H-3 was observed, indicating they should be in an anti conformation (Chlipala et al. 2010; Matsumori et al. 1999). In the analysis of anti conformation of roussoelin A, only two of the six possible relative conformations (blue and red color) for C-2 and C-3 were satisfied with the coupling constant (Fig. 3). A 1D selective gradient NOESY experiment revealed that H3-1 and H3-10 do not have an NOE correlation (Supplementary Figs. S8, S9), indicating a relative configuration of 2S*,3S*. The absolute configuration of the secondary alcohol was resolved by a modified version of Mosher’s method. The (R) and (S)-MTPA chloride reacted with 1, respectively, and esterification occurred at the C-2 hydroxy group to produce the corresponding (S)-MTPA ester (1a) and (R)-MTPA ester (1b). The chemical shifts for H-1, H-3, and H-10 of 1a and 1b were measured as δH 1.18, 3.09, and 1.27 for 1a and δH 1.20, 3.04, and 1.24 for 1b, respectively. The observed differences of chemical shifts (∆δ = δS − δR) (Fig. 4) indicated that the C-2 absolute configuration is S. Hence, compound 1 was identified as shown in Fig. 1 and named as roussoelin A.

Newman projection for C-2 and C-3 of compounds 1 and 2. Six possible relative conformations are shown: (top) 2S*,3S* and (bottom) 2R*,3S* (LG large coupling constant, SM small coupling constant)

∆δ = δS − δR values in ppm obtained from the MTPA esters of 1 and 2
Roussoelin B (2) was also obtained as a colorless oil and had the same molecular formula (C10H14O3) as roussoelin A (1) established by the HR-ESIMS ions at m/z 181.08712 [M−H]− (calcd. for C10H13O3, 181.08702). Compound 2 shared the same planar structure as 1, and was further identified by 2D NMR spectra (1H-1H COSY, HSQC, and HMBC) (Fig. 2). The chemical shift variation of C-1 (δC 22.0, δH 1.00 for 1; δC 20.1, δH 1.10 for 2), C-2 (δC 73.4, δH 3.69 for 1; δC 72.8, δH 3.82 for 2), C-3 (δC 49.6, δH 2.39 for 1; δC 48.4, δH 2.58 for 2), and C-10 (δC 18.7, δH 1.26 for 1; δC16.9, δH 1.18 for 2), together with the different specific rotations ([α]\(\begin{array}{c}{20}\\ {\text{D}}\end{array}\) −6.6 (c 0.20, MeOH) of 1; [α]\(\begin{array}{c}{20}\\ {\text{D}}\end{array}\) +18.5 (c 0.20, MeOH) of 2) suggested that 2 was a stereoisomer of 1. Similarly, the protons H-2 and H-3 were in an anti conformation on the base of a relative large coupling constant (3JH-2, H-3 = 6.3 Hz). Only two of the six possible relative conformations for C-2 and C-3 were satisfied (Fig. 3). A selective NOE experiment revealed that H3-1 and H3-10 have a strong NOE correlation (Supplementary Figs. S17, S18), indicating a relative configuration of 2R*,3S*. The stereostructure of C-2, bearing a secondary hydroxy group, was identified as R on the base of the modified Mosher’s method compared to the chemical shifts for H-1, H-3, and H-10 (1a δH 1.19, 3.02, and 1.22; 1b δH 1.08, 3.03, and 1.28) (Fig. 4). Thus, roussoelin B (2) was 2-epimer of roussoelin A.
The known compounds, 4-hydroxyscytalone (3) (Cimmino et al. 2016), 4,6,8-trihydroxy-3,4-dihydronaphthalen-1(2H)-one (6-hydroxyisosclerone) (4) (Yan et al. 2008), acremonone F (5) (Angelie et al. 2002), xestodecalactone A (6) (Angelie et al. 2002), corynechromone K (7) (Dong-Lin et al. 2015), corynechromone A (8) (Dong-Lin et al. 2015), (3Z,5S,6E,8S,9S,10R)-8-chloro-5,8,9,10-tetrahydro-5,9-dihydroxy10-methyl-2H-oxecin-2-one (9) (Greve et al. 2008; Zheng et al. 2015), modiolide A (10) (Greve et al. 2008), curvulide B1 (11) (Greve et al. 2008), and curvulide B2 (12) (Greve et al. 2008) were verified by 1H and 13C NMR, ESI–MS, and optical rotation data analysis, as well as comparison of spectroscopic data with literature.
All isolated compounds were tested for their anti-inflammatory activity in vitro by inhibition of LPS-activated NO production in RAW264.7 cells with the Griess assay and their cytotoxicity using MCF-7 (breast cancer), HepG2 (liver cancer), and A549 (lung cancer) human cell lines. None of them showed inhibition activity or cytotoxicity at 50 μmol/L. Compounds 1–12 were also evaluated using the total antioxidant capacity assay kit with a rapid ABTS method. Only compounds 1, 2, and 9 showed moderate total antioxidant capacity (0.65 of 1; 0.61 of 2; 0.32 of 9) with Trolox as a positive control (Fig. 5). Phenolic compounds (including cinnamic acids, benzoic acids, flavonoids, proanthocyanidins, coumarins, stilbenes, lignans, and lignins) are the most widespread class of metabolites in nature (Pereira et al. 2009). The antioxidant capacity of phenolic compounds 1 and 2 should be attributed to their ability to chelate metal ions involved in the production of free radicals and suggests that chemical protection of symbiotic microbes are benefitial to ascidians screening UV or inhibiting enzymes involved in radical generation (Cos et al. 1998).

Antioxidant capacity of compounds 1, 2, and 9 as determined by ABTS
Materials and methods
General experimental procedures
Optical rotations were measured on an MCP 200 polarimeter (Anton Paar, China). Infrared spectroscopy was performed on a Fourier transformation infrared spectrometer coupled with infrared microscope EQUINOX 55 (Bruker, Germany). 1D and 2D NMR data were measured on Bruker Avance 400 or 600 MHz spectrometers (Bruker, Germany) using tetramethylsilane (TMS) as the internal standard. Electrospray mass spectrometry (ESIMS) was obtained on an ACQUITY QDA (Waters Corporation, USA). High resolution electrospray mass spectrometry (HR-ESIMS) was tested on an LTQ-Orbitrap LC–MS spectrometer (Thermo Corporation, USA). Column chromatography was carried out on silica gel with 200–300 mesh (Qingdao Marine Chemical Factory, China) and Sephadex LH-20 (GE Healthcare, UK). High performance liquid chromatography (HPLC) was performed on on Essentia LC-16 with an SPD-16 Detector (Shimadzu, China).
Fungal material
In this study, the fungus SYSU-MS4723 was isolated from an ascidian Styela plicata, which was collected in the Mirs Bay (22°33′22.1′′N, 114°27′09.3′′E), Shenzhen, Guangdong Province, China, in April 2016. Purified fungus was isolated from ascidian on the base of the standard protocol (Kjer et al. 2010). The strain was identified to be R. siamensis SYSU-MS4723 on the base of morphological characteristics and the ITS region (Raja et al. 2017). The sequence data of the fungal strain have been submitted and deposited at GenBank with accession no. MH465397. The voucher specimen was preserved on potato dextrose agar slants at 4 °C at the School of Marine Sciences, Sun Yat-Sen University.
Extraction and isolation
The strain SYSU-MS4723 was cultured in autoclaved solid-substrate rice medium on sixty Erlenmeyer flasks (each flask containing 60 ml rice and 60 ml 3% artificial sea water) for 30 days under static conditions and daylight. Following incubation, the fungal solid-substrate rice medium was extracted three times with MeOH solvent to afford the crude extract. The crude extract was then extracted three times with EtOAc solvent and evaporated under reduced pressure to give a dark brown residue (18.5 g). The EtOAc extract residue was then subjected to flash column chromatography on silica gel eluted by a gradient of petroleum ether/EtOAc from 100:0 to 0:100 to separate into seven fractions (Fr. A–Fr. G). Fraction B was divided into five subfractions Fr.B.1–Fr.B.5 by Sephadex LH-20 (CC, 3 × 50 cm) eluting with MeOH-CH2Cl2 (v/v, 1:1). Fr.B.3 was subsequently performed on silica gel CC eluted by PE-EtOAc (v/v, 70:30) to give Fr.B.3.1–Fr.B.3.6. Then compound 6 (3 mg) was purified from Fr.B.3.3 subjected to Sephadex LH-20 (CC, 3 × 50 cm) and eluted with MeOH-CH2Cl2 (v/v, 1:1). Fr.B.3.4 was purified by the semi-preparative PR-HPLC (MeOH-H2O, v/v, 75:25, 1.5 ml/min, ultimate C18 column 10 × 250 nm, 5 μm) to yield compound 7 (3 mg, tR = 15.5 min). Compound 8 (3 mg) was directly purified from Fr.B.4 performed on silica gel CC by elution with PE-EtOAc (v/v, 70:30), while compounds 3 (4 mg) and 4 (5 mg) were isolated from Fr.B.3.5 using the silica gel CC eluted by MeOH-CH2Cl2 (v/v, 3:97). Then Fr. C was subjected to Sephadex LH-20 (MeOH-CH2Cl2, v/v, 1:1) to produce Fr.C.1–Fr.C.6, and Fr.C.4 was chromatographed on a silica gel with MeOH-CH2Cl2 (4:96) to afford five subfractions (Fr.C.4.1–Fr.C.4.5). The new compounds 1 (4 mg, tR = 17 min) and 2 (4 mg, tR = 18 min) were purified by semi-preparative PR-HPLC (MeOH-H2O, v/v, 75:25, 1.5 ml/min, ACE 5 C18-PFP column 250 × 10 mm, 5 μm) from Fr.C.4.4. The fourth fraction D was applied to a Sephadex LH-20 (MeOH-CH2Cl2, v/v, 1:1) to yield Fr.D.1–Fr.D.5. Subsequently, compounds 11 and 12 (3 mg, tR = 23.5 min; 2 mg, tR = 24.3 min) were purified from Fr.D.5 by semi-preparative PR-HPLC (MeOH-H2O, v/v, 70:30, 1.5 ml/min, ACE 5C18-AR column 250 × 10 mm, 5 μm). Fr. E was also applied to Sephadex LH-20 (MeOH-CH2Cl2, v/v, 1:1) to yield Fr.E.1–Fr.E.5. Fr.E.4 was chromatographed on a silica gel column with PE-EtOAc (v/v, 50:50) to give four subfractions (Fr.E.4.1–Fr.E.4.5). Fr.E.4.3 was performed on silica gel CC eluted by MeOH-CH2Cl2 (v/v, 5:95) to afford 5 (3 mg) and 9 (6 mg). And Fr.E.4.5 was subject to silica gel CC eluted by MeOH-CH2Cl2 (v/v, 5:95) to obtained 10 (4 mg).
Roussoelin A (1): colorless oil; [α]\(\begin{array}{c}{20}\\ {\text{D}}\end{array}\) −6.6 (c 0.20, MeOH); IR (neat) vmax 3346, 2978, 2918, 2850, 1601, 1462, 1329, 1151, 1084, 989, 931, 839, 700 cm−1; 1H NMR (400 MHz, CD3OD) and 13C NMR (100 MHz, CD3OD) data see Table 1; HR-ESIMS m/z 181.08712 [M−H]− (calcd. for C10H13O3, 181.08702).
Roussoelin B (2): colorless oil; [α]\(\begin{array}{c}{20}\\ {\text{D}}\end{array}\) 18.5 (c 0.20, MeOH); IR (neat) vmax cm−1 3329, 2972, 2924, 1603, 1454, 1342, 1149, 997, 841, 700; 1H NMR (400 MHz, CD3OD) and 13C NMR (100 MHz, CD3OD) data see Table 1; HR-ESIMS m/z 181.08712 [M−H]− (calcd. for C10H13O3, 181.08702).
Preparation of (S)-MTPA ester and (R)-MTPA ester
(S)-MTPA ester (1a) and (R)-MTPA ester (1b)
Compound 1 (1.0 mg) dissolved in pyridine-d5 (0.5 ml) in an NMR tube, and then (R)-MPTACl (5.0 μl) was added to react at room temperature for 24 h. Then the 1H NMR spectrum of the (S)-MTPA ester derivative (1a) was measured directly on the reaction mixture (Hoye et al. 2007; Zhang et al. 2017). 1H NMR (selected signals, pyridine-d5, 400 MHz) δH: 1.18 (3H, d, H-1), 3.09 (1H, m, H-3), 1.27 (3H, d, H-10).
Similarly, another reaction of 1 (1.0 mg), (S)-MPTACl (5.0 μl), and pyridine-d5 (0.5 ml) was performed as described above for 1a to afford 1b. 1H NMR (selected signals, pyridine-d5, 400 MHz) δH: 1.20 (3H, d, H-1), 3.04 (1H, m, H-3), 1.24 (3H, d, H-10).
(S)-MTPA ester (2a) and (R)-MTPA ester (2b)
(S)-MTPA ester (2a) and (R)-MTPA ester (2b) were obtained by refering to the above method. 1H NMR (selected signals, pyridine-d5, 400 MHz) 2a δH: 1.19 (3H, d, H-1), 3.02 (1H, m, H-3), 1.22 (3H, d, H-10). 2b δH: 1.08 (3H, d, H-1), 3.03 (1H, m, H-3), 1.28 (3H, d, H-10).
Cytotoxic assay
All compounds were tested for cytotoxicity against MCF-7 (breast cancer), HepG2 (liver cancer), and A549 (lung cancer) human cancer cell lines. Human cancer cell lines were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China). The cytotoxicity assay was based on the MTT method according to previously reported procedures (Chen et al. 2016).
Anti-inflammatory assay
All compounds were tested for their anti-inflammatory activity on the basis of previously reported procedures (Zhang et al. 2019).
Total antioxidant capacity assay
Total antioxidant capacity assay kit with a rapid ABTS method (Beyotime Institute of Biotechnology, China) was used to evaluate the total antioxidant capacity based on the manufacturer’s instructions. Samples were incubated at 25 °C for 6 min and then were recorded at 414 nm using a multimode reader (Thermo Fisher Scientific, USA).
References
Angelie ER, Markus H, Gernot B, Victor W, Albrecht B, Udo G, Michael W, Jorg M, Karsten S, Sudarsono S (2002) Online analysis of xestodecalactones A–C, novel bioactive metabolites from the fungus Penicillium cf. montanense and their subsequent isolation from the sponge Xestospongia exigua. J Nat Prod 65:1598–1604
Belofsky GN, Anguera M, Jensen PR, Fenical W, Köck M (2000) Oxepinamides A–C and fumiquinazolines H-I: bioactive metabolites from a marine isolate of a fungus of the genus Acremonium. Chem Eur J 6:1355–1360
Blunt JW, Copp BR, Keyzers RA, Munro MHG, Prinsep MR (2017) Marine natural products. Nat Prod Rep 34:235–294
Blunt JW, Carroll AR, Copp BR, Davis RA, Keyzers RA, Prinsep MR (2018) Marine natural products. Nat Prod Rep 35:8–53
Bugni TS, Ireland CM (2004) Marine-derived fungi: a chemically and biologically diverse group of microorganisms. Nat Prod Rep 21:143–163
Bugni TS, Abbanat D, Bernan VS, Maiese WM, Greenstein M, Van Wagoner RM, Ireland CM (2000) Yanuthones: novel metabolites from a marine isolate of Aspergillus niger. J Org Chem 65:7195–7200
Carroll AR, Copp BR, Davis RA, Keyzers RA, Prinsep MR (2019) Marine natural products. Nat Prod Rep 36:122–173
Chen S, Chen D, Cai R, Cui H, Long Y, Lu Y, Li C, She Z (2016) Cytotoxic and antibacterial preussomerins from the mangrove endophytic fungus Lasiodiplodia theobromae ZJ-HQ1. J Nat Prod 79:2397–2402
Chen L, Hu J, Xu J, Shao C, Wang G (2018) Biological and chemical diversity of ascidian-associated microorganisms. Mar Drugs 16:362
Chen S, Shen H, Zhang P, Cheng H, Dai X, Liu L (2019a) Anti-glioma trichobamide A with an unprecedented tetrahydro-5H-furo[2,3-b]pyrrol-5-one functionality from ascidian-derived fungus Trichobotrys effuse 4729. Chem Commun 55:1438–1441
Chen S, Jiang M, Chen B, Salaenoi J, Niaz S-I, He J, Liu L (2019b) Penicamide A, a unique N,N′-ketal quinazolinone alkaloid from ascidian-derived fungus Penicillium sp. 4829. Mar Drugs 17:522
Chlipala GE, Tri PH, Van Hung N, Krunic A, Shim SH, Soejarto DD, Orjala J (2010) Nhatrangins A and B, aplysiatoxin-related metabolites from the marine cyanobacterium lyngbya majuscula from Vietnam. J Nat Prod 73:784–787
Cimmino A, Maddau L, Masi M, Evidente M, Linaldeddu BT, Evidente A (2016) Further secondary metabolites produced by Diplodia corticola, a fungal pathogen involved in cork oak decline. Tetrahedron 72:6788–6793
Cos P, Ying L, Calomme M, Hu JP, Cimanga K, Van Poel B, Pieters L, Vlietinck AJ, Berghe DV (1998) Structure−activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J Nat Prod 61:71–76
Dewapriya P, Prasad P, Damodar R, Salim AA, Capon RJ (2017) Talarolide A, a cyclic heptapeptide hydroxamate from an Australian marine tunicate-associated Fungus, Talaromyces sp. (CMB-TU011). Org Lett 19:2046–2049
Dewapriya P, Khalil ZG, Prasad P, Salim AA, Cruz-Morales P, Marcellin E, Capon RJ (2018) Talaropeptides A–D: structure and biosynthesis of extensively N-methylated linear peptides from an Australian marine tunicate-derived Talaromyces sp. Front Chem 6:394
Dong-Lin Z, Chang-Lun S, Li-She G, Mei W, Chang-Yun W (2015) Chromone derivatives from a sponge-derived strain of the fungus Corynespora cassiicola. J Nat Prod 78:286–293
Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, Schmidt EW (2006) Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat Chem Biol 2:729–735
Garo E, Starks CM, Jensen PR, Fenical W, Lobkovsky E, Clardy J (2003) Trichodermamides A and B, cytotoxic modified dipeptides from the marine-derived fungus Trichoderma virens. J Nat Prod 66:423–426
Greve H, Schupp PE, Kehraus S, Konig GM (2008) Ten-membered lactones from the marine-derived fungus Curvularia sp. J Nat Prod 71:1651–1653
He H, Ding W, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT (2001) Lomaiviticins A and B, potent antitumor antibiotics from micromonospora lomaivitiensis. J Am Chem Soc 123:5362–5363
Hoye TR, Jeffrey CS, Shao F (2007) Mosher ester analysis for the determination of absolute configuration of stereogenic (chiral) carbinol carbons. Nat Protoc 2:2451–2458
Ivanets EV, Yurchenko AN, Smetanina OF, Rasin AB, Zhuravleva OI, Pivkin MV, Popov RS, von Amsberg G, Afiyatullov SS, Dyshlovoy SA (2018) Asperindoles A–D and a p-terphenyl derivative from the ascidian-derived fungus Aspergillus sp. KMM 4676. Mar Drugs 16:232
Jiang M, Wu Z, Guo H, Liu L, Chen S (2020) A Review of terpenes from marine-derived fungi: 2015–2019. Mar Drugs 18:321
Kjer J, Debbab A, Aly AH, Proksch P (2010) Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat Protoc 5:479–490
Li X, Li L, Li X-M, Li H-L, Wang B-G (2020) Ustusaustin A: a new neuraminidase inhibitory meroterpene from the ascidian-derived endophytic fungus Aspergillus ustus TK-5. Nat Prod Res. https://doi.org/10.1080/14786419.2020.1752211
Lin Z, Koch M, Abdel Aziz MH, Galindo-Murillo R, Tianero MD, Cheatham TE, Barrows LR, Reilly CA, Schmidt EW (2014a) Oxazinin A, a pseudodimeric natural product of mixed biosynthetic origin from a filamentous fungus. Org Lett 16:4774–4777
Lin Z, Koch M, Aziz M, Galindomurillo R, Tianero MD, Cheatham TE, Barrows LR, Reilly CA, Schmidt EW (2014b) Oxazinin A, a pseudodimeric natural product of mixed biosynthetic origin from a filamentous fungus. Org Lett 16:4774–4777
Liu L, Zheng Y-Y, Shao C-L, Wang C-Y (2019) Metabolites from marine invertebrates and their symbiotic microorganisms: molecular diversity discovery, mining, and application. Mar Life Sci Tech 1:60–94
MalmstrøM J, Christophersen C, Frisvad JC (2000) Secondary metabolites characteristic of Penicillium citrinum, Penicillium steckii and related species. Phytochemistry 54:301–309
Matsumori N, Kaneno D, Murata M, Nakamura H, Tachibana K (1999) Stereochemical determination of acyclic structures based on carbon–proton spin-coupling constants. a method of configuration analysis for natural products. J Org Chem 64:866–876
Montenegro TG, Rodrigues FA, Jimenez PC, Angelim AL, Melo VM, Rodrigues EF, de Oliveira MdCF, Costa-Lotufo LV (2012) Cytotoxic activity of fungal strains isolated from the ascidian Eudistoma vannamei. Chem Biodivers 9:2203–2209
Motohashi K, Hashimoto J, Inaba S, Khan ST, Komaki H, Nagai A, Takagi M, Shin-ya K (2009) New sesquiterpenes, JBIR-27 and -28, isolated from a tunicate-derived fungus, Penicillium sp. SS080624SCf1. J Antibiot 62:247–250
Murshid SSA, Badr JM, Youssef DTA (2016) Penicillosides A and B: new cerebrosides from the marine-derived fungus Penicillium species. Rev Bras Farmacogn 26:29–33
Niaz SI, Zhang P, Shen H, Li J, Chen B, Chen S, Liu L, He J (2019) Two new isochromane derivatives penisochromanes A and B from ascidian-derived fungus Penicillium sp. 4829. Nat Prod Res 33:1262–1268
Ohtani I, Kusumi T, Kashman Y, Kakisawa H (1991) High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J Am Chem Soc 113:4092–4096
Pereira DM, Valentão P, Pereira JA, Andrade PB (2009) Phenolics: from chemistry to biology. Molecules 14:2202–2211
Raja HA, Miller AN, Pearce CJ, Oberlies NH (2017) Fungal identification using molecular tools: a primer for the natural products research community. J Nat Prod 80:756–770
Shaala LA, Youssef DT (2015) Identification and bioactivity of compounds from the fungus Penicillium sp. CYE-87 isolated from a marine tunicate. Mar Drugs 13:1698–1709
Smetanina O, Kuznetsova T, Gerasimenko A, Kalinovsky A, Pivkin M, Dmitrenok P, Elyakov G (2004) Metabolites of the marine fungus Humicola fuscoatra KMM 4629. Russ Chem B+ 53:2643–2646
Song Q, Li X-M, Hu X-Y, Li X, Chi L-P, Li H-L, Wang B-G (2019) Antibacterial metabolites from Ascidian-derived fungus Aspergillus clavatus AS-107. Phytochem Lett 34:30–34
Sumilat DA, Yamazaki H, Endo K, Rotinsulu H, Wewengkang DS, Ukai K, Namikoshi M (2017) A new biphenyl ether derivative produced by Indonesian ascidian-derived Penicillium albobiverticillium. J Nat Med 71:776–779
Wang GYS, Borgeson BM, Crews P (1997) Pitholides A–D, polyketides from a marine tunicate-derived culture of Pithomyces sp. Tetrahedron Lett 38:8449–8452
Xin Z-H, Li T, Zhu T-j, Wang W-L, Du L, Fang Y-c, Gu Q-Q, Zhu W-M (2007) Isocoumarin derivatives from the sea squirt-derived fungus penicillium stoloniferum QY2-10 and the halotolerant fungus Penicillium notatum B-52. Arch Pharm Res 30:816–819
Yamazaki H, Nakayama W, Takahashi O, Kirikoshi R, Izumikawa Y, Iwasaki K, Toraiwa K, Ukai K, Rotinsulu H, Wewengkang DS, Sumilat DA, Mangindaan RE, Namikoshi M (2015) Verruculides A and B, two new protein tyrosine phosphatase 1B inhibitors from an Indonesian ascidian-derived Penicillium verruculosum. Bioorg Med Chem Lett 25:3087–3090
Yan DJ, Chuan SH, Hua LJ, Shu TY, Rong S, Le W, Ping ZY, Mei WL, Ze SK, Chun Ren W (1029A) Ymf 1029A-E, preussomerin analogues from the fresh-water-derived fungus YMF 1.01029. J Nat Prod 71:952–956
Yurchenko A, Ivanets E, Smetanina O, Pivkin M, Dyshlovoi S, Von Amsberg G, Afiyatullov SS (2017) Metabolites of the marine fungus Aspergillus candidus KMM 4676 associated with a kuril colonial ascidian. Chem Nat Compd+ 53:747–749
Zhang P, Li Y, Jia C, Lang J, Niaz S-I, Li J, Yuan J, Yu J, Chen S, Liu L (2017) Antiviral and anti-inflammatory meroterpenoids: stachybonoids A–F from the crinoid-derived fungus Stachybotrys chartarum 952. RSC Adv 7:49910–49916
Zhang P, Deng Y, Lin X, Chen B, Li J, Liu H, Chen S, Liu L (2019) Anti-inflammatory mono- and dimeric sorbicillinoids from the marine-derived fungus Trichoderma reesei 4670. J Nat Prod 82:947–957
Zheng CJ, Shao CL, Chen M, Niu ZG, Zhao DL, Wang CY (2015) Merosesquiterpenoids and ten-membered macrolides from a soft coral-derived Lophiostoma sp. fungus. Chem Biodivers 12:1407–1414
Acknowledgements
We thank the National Natural Science Foundation of China [Grant no. 41806155]; Guangdong MEPP Fund [no.GDOE (2019) A21]; the Natural Science Foundation of Guangdong Province, China (2018A030310304) for generous support.
Author information
Authors and Affiliations
Contributions
SC and LL conceived and designed the experiments; SC and HS performed the experiments; YD, HG, MJ, ZW, HY participated in the experimental process and result discussion. SC analyzed the data and wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Animal and human rights statement
This article does not contain any studies with human participants or animals performed by the authors.
Additional information
Edited by Chengchao Chen.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, S., Shen, H., Deng, Y. et al. Roussoelins A and B: two phenols with antioxidant capacity from ascidian-derived fungus Roussoella siamensis SYSU-MS4723. Mar Life Sci Technol 3, 69–76 (2021). https://doi.org/10.1007/s42995-020-00066-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42995-020-00066-8