
Abstract
The common opossum (Didelphis marsupialis) is a marsupial widely distributed in the Neotropics, where it lives in urbanized environments. The apparent scarcity and fragmentation of available habitat, and high rates of vehicle collisions, may represent barriers to dispersal. To assess the functional connectivity of this species and its potential use as a model in urban ecology, we evaluated the structure and genetic diversity of opossums in the metropolitan area of the Aburrá Valley (AMVA), Colombia, using sequences of the mitochondrial Cytochrome B (Cytb) gene and nine autosomal microsatellite loci. Although Cytb presented low levels of variation, microsatellite markers revealed high genetic diversity (He = 0.852 and Ho = 0.698). The geographic distribution of mitochondrial lineages and a spatial principal component analysis (sPCA) of microsatellites showed the absence of genetic structure and effective barriers to dispersal of opossums in the study area. This scenario suggests that hostile landscapes such as those in urban environments may be suitable enough to allow the effective connectivity of some species such as the common opossum and others with high dispersal ability in urbanized areas. Nonetheless, it is necessary to evaluate the impact of releases of rescued fauna made by the environmental authority on the population structure of urban opossums.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Habitat loss and landscape fragmentation are major threats to biological diversity (Huxel and Hastings 1999), leading to loss of natural corridors and reduction of suitable habitat patches (Herrera 2011). Habitat conversion for human use in urban areas can lead to disturbances in (1) species movement and dispersal, (2) available resources within the matrix, and (3) the abiotic environment of patches (Driscoll et al. 2013). These disturbances directly influence connectivity and can affect species abundance, community composition, population sizes, and ecological processes within native vegetation patches.
From an evolutionary standpoint, habitat fragmentation and degradation can alter patterns of natural selection, modify gene flow, and reduce the effective size of populations, leading to a decrease in genetic diversity and increased inbreeding, which may result in local extinctions (Templeton et al. 1990; White et al. 1999; Ruell et al. 2012; Munshi-South et al. 2013). However, some wild species can survive in urbanized environments, where they find abundant resources throughout the year (Ives et al. 2015), and even experience an increase in dispersal ability, thus reducing the probability of isolation, bottlenecks, and genetic drift (Miles et al. 2019). Likewise, transformed urban landscapes with gardens, green and wooded areas can even promote dispersal, eliminate possible barriers, and facilitate success in urban species (Holderegger and Di Giulio 2010).
The common opossum, Didelphis marsupialis (Linnaeus, 1758) is a neotropical marsupial species (family Didelphidae) distributed from southern Mexico to northern Argentina (IUCN 2021) in tropical and subtropical forests, secondary forests and near human settlements such as houses and agricultural lands. It is a nocturnal species with high dispersal ability, as it does not occupy its den for more than five days. It is an opportunistic omnivore, and its diet can easily change depending on the availability of different food types including anthropogenic sources (Ringier 1961). Even though this is species is frequently observed in urbanized areas, its abundance may show seasonal and regional variation (Adler et al. 1997; Cruz-Salazar et al. 2016), and its distribution may be influenced by fragmentation and levels of habitat disturbance. The common opossum is not considered a threatened species according to the IUCN (IUCN 2021), but it is a frequent victim of vehicle collisions and acts of aggression in urban areas since it is frequently stigmatized and viewed as a pest (Delgado 2007; Figueroa et al. 2009). For these reasons, several protection and conservation programs for the common opossum have been proposed in Colombia, including rescues and releases that do not take into consideration the possible origin of these animals, the connectivity between populations, or the existing differences between them (Figueroa et al. 2009).
Previous population studies of this species have focused on abundance, reproductive habits, and their parasite-host relationship (Tyndale-Biscoe and Mackenzie 1976; Adler et al. 1997; Ramírez and Osorio 2014; Saldaña et al. 2019). Yet, genetic data in populations in southern Mexico have revealed intermediate levels of genetic variation coupled with low levels of inbreeding and an overall negative effect of habitat fragmentation on gene flow (Cruz-Salazar et al. 2014; Cruz-Salazar and Ruiz-Montoya 2022). Given the high prevalence of the common opossum in urban environments, which contrasts with frequent cases of aggression and accidents, it is important to study the composition and connectivity of the populations of this species to understand its use of urban environments. Here we studied the diversity and genetic structure of the common opossum in the Aburrá Valley Metropolitan Area (AMVA), a highly populated city in northwestern Colombia. Given the heterogeneity and complexity of this urban matrix, we explored potential discontinuities in the distribution of genetic variation to recognize the existence of possible barriers to dispersal.
Materials and methods
Area description
This study was carried out in the urban area of AMVA, a valley located in the Central Cordillera of Colombia, with approximately 1300 m above the sea level at the lowest points. This region is approximately 60 km long and 6–8 km wide and it is crossed by the Medellín River from south to north. It is shared by ten municipalities and a population of 4.1 million people (CCM 2021). Despite its high population density, AMVA has several protected forest areas, riparian forest, and urban parks that are home to various species of wildlife including common opossums. The city of Medellín (the largest municipality) has almost 450 species of birds, more than 90 species of wild mammals, and more than 2,600 species of vascular plants, demonstrating a shared use of the territory by an extensive human population and a high diversity of fauna and flora (Medellín-Alcaldía 2017).
Sampling
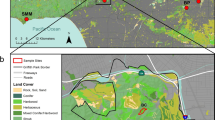
Since the common opossum is a synanthropic species present in both forested urban areas, and residential areas, this species is frequently impacted by injuries and deaths derived from accidents and aggressions associated with the human-animal conflict. Samples were then taken from 66 individuals rescued by the environmental authority in urban areas of six of the ten municipalities that make up the AMVA (Copacabana, Bello, Medellín, Envigado, Itagüí and La Estrella; Fig. 1) between the months of September and December 2021 (Supplementary File 1). The study sample comprised blood samples taken from six live animals and 60 skin biopsies (ear or tail) from animals euthanized by environmental authorities due to poor health conditions (Table 1). The samples were stored in 96% ethanol in 1.5 μl Eppendorf® tubes. DNA was extracted using the commercial DNeasy Blood and Tissue kit (Qiagen, Valencia, California, USA) and a standard salting out method, respectively. In cases of juvenile/infantile couples sampled on the same day and location (potential siblings), only one member of each couple was included in the study sample to prevent any effect of inbreeding caused by the family structure.

Location of opossum sampling sites in the Aburrá Valley Metropolitan Area (AMVA) are shown in orange color. The green dots depict the release sites of animals rescued by the environmental authority
Records of 642 common opossums rescued and then released in the AMVA between 2020 and 2022 were provided by the local environmental authority (Corantioquia) and used to identify any overall geographical bias in the location of released animals (Supplementary File 2). This is important since, depending on the release areas, translocations may alter the original genetic structure of the studied population.
Sequencing and analysis of the mitochondrial Cytochrome B gene
A 770 bp fragment of the Cytochrome B (Cytb) gene was amplified and sequenced using the primers Didm1413F (5′-AACCTATGGCATGAAAAACCACCATYG-3′) and Didm15007R (5′-AAATAGAAAATACCATTCTGGCTTGAT-3′). These primers were designed for this study with the Primer3 on-line software (http://primer3.ut.ee), using reference sequences of five species of Didelphis (Accession codes Z29573 and MT892661-MT892665) and other sequences of Cytb of D. marsupialis available in GenBank. DNA amplification reactions were carried out in a 15 μl final volume, containing 2 μl of DNA, 1X Taq Buffer, 7.5 μg/μl BSA, 0.2 mM dNTPs, 0.2 μM of each primer (F and R), 1U of Taq polymerase and 2 mM MgCl2 (Thermo Fisher Scientific Ltd., Waltham, Massachusetts, USA). Amplification was performed at 94 °C for 5 min, followed by 34 cycles of 94 °C for 35 s, 58.3 °C for 35 s and 72 °C for 1 min, with a final extension at 72 °C for 10 min. The amplicons were purified with Exonuclease I and FastAP Alkaline Phosphatase (Thermo Fisher Scientific Ltd.) by incubating the samples at 37 °C for 30 min and then at 95 °C for five min. Both strands (H and L) were then sequenced in a commercial laboratory following a standard Sanger methodology.
The sequences were edited with Geneious Prime v2020.0.5 (https://www.geneious.com) and aligned with ClustalW in MEGA X11 (Tamura et al. 2021). The alignment was then exported to DNAsp v5.1.1.1.1.8 (Librado and Rozas 2009) to estimate the number of haplotypes (h), number of polymorphic sites (S), haplotype diversity (Hd), nucleotide diversity (π) and, apply the Tajima’s D (Tajima 1989) and Fu’s F neutrality tests (Fu 1996). Hd indicates the probability that two randomly chosen haplotypes are different, whereas π estimates the probability that two randomly chosen homologous sites are different (Excoffier and Lischer 2010). D and F provide evidence of historical demographic changes, i.e., either expansion (negative values) or contraction (positive).
To analyze geographic distribution patterns and relationships among the haplotypes obtained in this study, nineteen reference sequences of D. marsupialis and three of D. virginiana were retrieved from GenBank (Clark et al. 2016) and aligned with our sequences (Supplementary File 3). A Bayesian phylogenetic tree of 50 million steps was then constructed in the BEAST v2.5 sampling every 1,000 steps (Bouckaert et al. 2019). Monophyly of D. marsupialis was forced and D. virginiana was used as the outgroup. A Yule process and a GTR mutational model selected using jModelTest2 in CIPRES Science Gateway v3.1 (http://www.phylo.org/) were implemented. Five thousand trees (10%) were discarded as burn-in.
Genotyping and microsatellite analysis
Individual amplification protocols for nine microsatellite markers previously designed for opossums (Lavergne et al. 1999; Dias et al. 2019; Fike et al. 2009) were tested, standardized (Supplementary File 4) and genotyped in 49 individuals. Each 15 μl reaction contained 2 μl of DNA, 1X Taq Buffer, 7.5 μg/μl BSA, 0.2 mM dNTPs, 0.2 μM forward primer labeled with a fluorochrome (Tamra, Fam, or Hex), 0.2 μM reverse primer, 1U Taq polymerase and a variable volume of MgCl2 (Thermo Fisher Scientific Ltd.). PCR conditions were 94 °C for 5 min, followed by 30 cycles of 94 °C for 35 s, alignment temperature for 30 s and 72 °C for 45 s, and a final extension step of 72 °C for 7 min. Products from different loci were combined into three groups of three loci. The products in each group were distinguishable by color or allelic range. Allele sizes were identified by capillary electrophoresis on an ABI 3730 DNA kit with a LIZ-500 size standard.
Allele bins were delimited utilizing Geneious Prime v2020.0.5. The subsequent analyses were performed in R v4.2.1 (R Core Team 2023). The Probability of Identity between unrelated (PI) and sibling pairs (PIsibs) were calculated with 100 permutations in the PopGenUtils package. The Polymorphic information content (PIC) as an estimator of informativeness was computed for each microsatellite using POLYSAT (Clark and Jasieniuk 2011). Pairwise linkage disequilibrium was assessed using the POPPR (Kamvar et al. 2014). The influence of null alleles was evaluated in PopGenReport package (Adamack and Gruber 2014). The total number of alleles per locus (Na), effective number of alleles per locus (Ne), expected heterozygosity (He), observed heterozygosity (Ho) and inbreeding coefficient (Fis) were calculated along with deviation from Hardy–Weinberg equilibrium (HWE) using the PEGAS package (Paradis 2010). Na and Ne are estimators of polymorphism, whereas He, Ho and Fis indicate the levels of genetic diversity and inbreeding.
Spatial genetic structure analysis
A spatial principal component analysis (sPCA) was performed to test for either the presence of spatial groups or clines of genetic differentiation (positive autocorrelations) as it is typically observed within patches and islands, or local structure where differences between close neighbors are stronger than expected by chance (negative autocorrelations) (Jombart et al. 2008). This analysis does not require neither a previous definition of genetic groups nor assumptions of Hardy–Weinberg equilibrium and Linkage Equilibrium. Unlike conventional PCA, sPCA maximizes the genetic variance and the spatial autocorrelation as measured by the Moran's I. A Monte Carlo test was also applied to evaluate local and global structures contrasting the eigen values of the selected eigen vectors against a null distribution constructed from 999 permutations, where the null hypothesis is that the allele frequencies of the individuals are randomly distributed in space. Furthermore, a Discriminant Analysis of Principal Components (DAPC) was aimed to identify the number of genetically differentiated clusters, without incorporating spatial considerations. All three analyses were carried out with adegentet v.2.1.1 in R v4.2.1 (Jombart 2015). The selected eigen vectors were mapped using Qgis 3.4 to visualize these patterns spatially (Neteler and Mitasova 2002). Finally, we conducted a Mantel test in ade4 package (Dray and Dufour 2007) to assess the hypothesis of Isolation by Distance, contrasting pairwise geographic distance vs Euclidian genetics distances.
Results
Mitochondrial data analysis
We obtained a total of 57 mitochondrial sequences with four distinct haplotypes in the study sample (Supplementary File 3; GenBank accession numbers PP889393–PP889449). These sequences present five polymorphic sites, with low values of haplotype diversity (Hd = 0.280 ± 0.072 SD) and nucleotide diversity (π = 0.0029). Neutrality tests (D = − 1.176, F = − 0.547) yielded negative values consistent with a population expansion process but were not significantly different from zero (P > 0.10).
The phylogenetic tree recovered two well-supported haplogroups in the sampled opossums (PP > 0.99), named haplogroups 1 and 2 (Fig. 2). The haplogroup Hap_1 comprised 50 individuals (87.7%) from all six municipalities within AMVA, as well as two identical sequences obtained from the same individual in Panama (KT437726 and MG491975). The haplogroup Hap_2 was exclusively represented by seven specimens sampled in Medellín and Envigado. We also identified differentiated groups geographically restricted to Guyana (KT153570, KT153571 and KT153572), Mexico (HM589701, HM589702 and KM659273-KM659279), Peru (KJ129895 and AJ606420) and Brazil (group 1: JF280998-JF281002; group 2: JF281004-JF281006, KT447521, DQ236278, NC057518, OL800243, OL800249, OP381007 and U34665; and group 3: OL800237, OL800239, OL800240-OL800242, OL800244-OL800248, OL800250, OL800252, OL800253, OL800256 and OL800258) with high Posterior Probabilities (PP > 0. 96) (Fig. 2). However, the statistical support of basal branches was poor and therefore the relationships among the identified haplogroups remains undefined.

Bayesian phylogenetic reconstruction of the mitochondrial Cytochrome B gene of Didelphis marsupialis. Sequences retrieved from GenBank are marked with a dot and their major groups were collapsed for visual simplification. Due to poor probability support in the dichotomic reticulation within the two haplogroups in the study sample (Hap_1 and Hap_2), they were also collapsed
Microsatellite marker analysis
We identified a total of 142 alleles at nine loci with a mean of 16.4 alleles per locus. Locus DM3 presented the highest number of alleles (Na = 25), while the lowest number was OP48 (Na = 11) (Table 2). The mean effective number of alleles per locus (Ne) was 9.3, where loci MNUD41 and DM3 presented the lowest (2.6) and highest (20.0) values, respectively. The combined PI and PIsibs were 1-49E−14 and 4.90E−05, respectively. Polymorphic information content (PIC) was high at all loci, with locus DM3 having the highest PIC value (0.937) and locus MNUD41 the lowest value (0.604). The probability of null alleles ranged from 0.001 (MNUD41) to 0.354 (DM1). In addition, the loci pairs DM1-DM3, DM3-DM5, and DM5-DM9 were found to be in linkage disequilibrium. The mean observed heterozygosity (Ho) was 0.696, ranging from 0.408 (DM1) to 0.796 (OP48). The mean expected heterozygosity (He) was 0.858, ranging from 0.620 (MNUD41) to 0.950 (DM3). Hardy–Weinberg disequilibrium was significant in five loci, all with heterozygosity deficit. The mean inbreeding coefficient (Fis) value was 0.184 and ranged between 0.012 (MNUD41) to 0.530 (DM1). Due to the high chance error introduced by the elevated levels of null alleles in DM1, this locus was excluded from subsequent analyses.
Analysis of spatial genetic structure
The spatial distribution of the observed heterozygosity levels shows a slight trend toward higher values (Ho ≥ 0.8) toward the north of AMVA (Fig. 3). However, the sPCA failed to reveal any type of structure at the spatial level (Fig. 4). This analysis showed that the axes 1 and 48 explain most of the genetic variation at the global and local levels, respectively (Supplementary File 5); whereas the eigen value of axis 1 had the highest positive eigen value (λ1 = 0.121) and positive spatial autocorrelation (Moran's I = 0.528), axis 48 presented the most negative eigen value (λ48 = − 0.076) and had negative spatial autocorrelation (Moran's I = − 0.343)(Supplementary File 5). Similarly, the Monte Carlo tests did not support the existence of any global (P = 0.09) or local (P = 0.33) spatial structure as shown in Supplementary File 5 (a–d). These results were consistent with the DAPC analysis, which revealed that the optimal number of clusters is one, as determined through the Bayesian Information Criterion (BIC = 0.68) (Supplementary File 5). Overall, our data show that allele frequencies are randomly distributed throughout the urban landscape. Finally, de Mantel test of Isolation by Distance was not significant (P = 0.93) (Supplementary File 5).

Distribution of observed heterozygosity values (Ho) in AMVA. Release sites of individuals in AMVA are shown in dark green

Spatial genetic structure of Didelphis marsupialis individuals based on spatial principal component analysis (sPCA). The size of each box represents the probability that an individual belongs to a defined cluster. A Axis sPCA1 for global structure; B axis sPCA48 for local structure. Green dots represent release sites of individuals in AMVA
Discussion
To the best of our knowledge, ours is the first study to evaluate diversity and spatial genetic structure of D. marsupialis in urban environments. We evaluated the genetic characteristics of this species using mitochondrial and nuclear markers in AMVA, a highly heterogeneous and densely populated environment.
Despite the valuable information from mitochondrial sequences, the low haplotype and nucleotide diversity of Cytb contrasts with a high diversity in microsatellite markers. Despite the reduced diversity of the Cytb sequences in D. marsupialis, the observed variation was sufficient to recognize two well-defined haplogroups in AMVA (Hap_1 and Hap_2), as well as other geographically restricted lineages in Mexico, Guyana, Peru and Brazil. The phylogenetic analysis revealed the presence of the Hap_1 haplogroup in both AMVA and Panama, suggesting the wide distribution of this lineage in northern South America and the transition area to North America.
The negative values of Fu's F and Tajima's D (although non-significant) were consistent with constant or increasing population sizes. This is interesting because despite the high number of records of deaths and accidents of D. marsupialis in AMVA, the species seems to thrive with a stable population. Future analyses might be supplemented with other mitochondrial markers presumably more polymorphic than Cytb such as D-loop, for deeper phylogeographic and demographic analyses of D. marsupialis across its distribution range.
Microsatellite loci in the urban common opossum population of AMVA showed high levels of diversity. Four of these markers (DM1, DM3, DM4 and DM5) were previously evaluated in a wild population of D. marsupialis in French Guiana (Lavergne et al. 1999). Despite geographic and landscape differences, this set of markers presented high levels of expected heterozygosity (0.933 and 0.903 in French Guiana and AMVA, respectively) and deficit of observed heterozygosity (0.720 and 0.670) in both studies. High levels of diversity and heterozygosity deficit were also observed in southern Mexico (Cruz-Salazar and Ruiz-Montoya 2022). Such pattern suggests a natural trend toward high diversity and natural levels of inbreeding in D. marsupialis, but it must be seen with caution due to the impact of null alleles that may lead to underestimate the observed levels of heterozygosity in microsatellite loci as detected here in the locus DM1, but also in Mexico and Guyana using the same locus (Lavergne et al. 1999; Cruz-Salazar and Ruiz-Montoya 2022). The future use of this locus in population analyses might require sequencing of this region to inspect any mispairing between the primers and their target region and, redesigning such primers as needed.
Both mitochondrial and nuclear markers failed to detect any signature of genetic structure in AMVA. The wide distribution of the two mitochondrial haplotypes in AMVA, and all the multiple tests using microsatellite loci failed to detect the effect of any potential barriers to dispersal and were consistent with high genetic exchange across AMVA. Although the home range of common opossums is relatively small in relation to the extent of the AMVA [0.16 km2 for females and 1.23 km2 for males (Sunquist and Austad 1987)], the area that they occupy is quite dynamic and often changes as these animals do not remain in their dens for more than five days, thus facilitating their dispersal and connectivity over large distances. In addition, the opportunistic, generalist feeding behavior of common opossums allow them to adapt to different environments, thus being an ecologically flexible species with high dispersal ability (Kasparian et al. 2004; Chemisquy et al. 2021). Therefore, future studies in contrasting environments (e. g. urban vs rural) will be necessary to understand the impact of landscape disturbance, since dispersal patterns and inbreeding may differ in contrasting habitats as shown in rural populations of D. marsupialis in southern Mexico (Cruz-Salazar and Ruiz-Montoya 2022).
The lack of any population structure in both mitochondrial and nuclear loci is also consistent with no sex-biased dispersal, as it has also been shown in D. virginiana in agricultural landscapes (Beatty et al. 2012). This contrasts with male-biased dispersal documented in field studies of both D. marsupialis and D. virginiana (Ryser 1995; Flórez-Oliveros and Vivas-Serna 2020), and future genetic analyses aimed to assess the social structure of D. marsupialis will be necessary to better assess this hypothesis.
Finally, the translocation of rescued animals in AMVA may potentially promote artificial dispersal and therefore impact on our results. Historical records reveal multiple releases made by the environmental authority in peri-urban and rural areas, mainly in the South and Southeast of AMVA, which evidently can facilitate gene flow, homogenize gene pools, and maximize genetic diversity in the area (Crispo et al. 2011). Since released individuals can have multiple origins, one would expect that the areas where releases occur would exhibit the highest levels of heterozygosity. However, the highest heterozygosity values (Ho ≥ 0.8) do not coincide with the release sites and are located further North. It will then be necessary to contrast our findings with parallel studies in other urban or rural areas where animals have not been released, to distinguish the different factors that may be facilitating the dispersal of this species.
Conclusions
The genetic data from this study are the first to report spatial genetic diversity and structure for D. marsupialis in Colombia using molecular markers. Our results showed high levels of genetic diversity and absence of genetic structuring of D. marsupialis in a densely populated urban matrix such as the one in the Aburrá Valley, which is consistent with high dispersal and lack of effective barriers to dispersal. These results may have been influenced to some extent by null alleles and animal translocations. Therefore, it is necessary to design future studies to specifically identify the effects of ecological factors on the composition and genetic structure of D. marsupialis populations. Other species that are not subject to translocations or releases, or species with different dispersal abilities and levels of adaptation to habitat disturbance could be quite useful to evaluate functional connectivity processes in urban environments.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Data availability statement
Data is contained within the article or supplementary material. The data presented in this study are available in Supplementary files 1 – 5.
References
Adamack AT, Gruber B (2014) PopGenReport: simplifying basic population genetic analyses in R. Methods Ecol Evol 5(4):384–387. https://doi.org/10.1111/2041-210X.12158
Adler GH, Arboledo JJ, Travi BL (1997) Population dynamics of Didelphis marsupialis in northern Colombia. Stud Neotrop Fauna Environ 32:7–11. https://doi.org/10.1076/snfe.32.1.7.13462
Beatty WS, Beasley JC, Dharmarajan G, Rhodes OE Jr (2012) Genetic structure of a Virginia opossum (Didelphis virginiana) population inhabiting a fragmented agricultural ecosystem. Can J Zool 90(1):101–109. https://doi.org/10.1139/z11-119
Bouckaert R, Vaughan TG, Barido-Sottani J, Duchêne S, Fourment M, Gavryushkina A, Drummond AJ (2019) BEAST 2.5: an advanced software platform for Bayesian evolutionary analysis. PLoS Comput Biol 15(4):e1006650. https://doi.org/10.1371/journal.pcbi.1006650
CCM - Cámara de Comercio de Medellín (2021) Perfil socioeconómico del Valle de Aburrá. Cámara de Comercio de Medellín, Medellín. https://www.studocu.com/co/document/servicio-nacional-de-aprendizaje/finanzas/perfil-socioeconomico-del-valle-de-aburra-2021/72113551
Chemisquy MA, Morínigo FM, Fameli A, González-Ittig RE (2021) Genetic diversity of the white-eared opossum Didelphis albiventris (Didelphimorphia: Didelphidae) in Argentina. Mastozool Neotrop 28(1):534
Clark LV, Jasieniuk M (2011) POLYSAT: an R package for polyploid microsatellite analysis. Mol Ecol Resour 11(3):562–566. https://doi.org/10.1111/j.1755-0998.2011.02985.x
Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2016) GenBank. Nucl Acids Res 44(D1):D67–D72. https://doi.org/10.1093/nar/gkv1276
Crispo E, Moore JS, Lee-Yaw JA, Gray SM, Haller BC (2011) Broken barriers: human-induced changes to gene flow and introgression in animals: an examination of the ways in which humans increase genetic exchange among populations and species and the consequences for biodiversity. BioEssays 33(7):508–518. https://doi.org/10.1002/bies.201000154
Cruz-Salazar B, Ruiz-Montoya L (2022) Population genetics of the common opossum, Didelphis marsupialis (Marsupialia: Didelphimorphia: Didelphidae), in southeastern Mexico. Stud Neotrop Fauna Environ 57(3):218–226. https://doi.org/10.1080/01650521.2020.1844971
Cruz-Salazar B, Ruiz-Montoya L, Navarrete-Gutiérrez D, Espinoza-Medinilla EE, Vázquez-Domínguez E, Vázquez LB (2014) Genetic diversity and relative abundance of Didelphis marsupialis and Didelphis virginiana in Chiapas, Mexico. Rev Mex Biodiv 85(1):251–261. https://doi.org/10.7550/rmb.36116
Cruz-Salazar B, Ruiz-Montoya L, Navarrete-Gutiérrez D, Vázquez LB (2016) Influence of the composition and structure of modified landscapes on abundance of two marsupials during the dry season. Therya 7(3):393–406
Delgado VC (2007) Muerte de mamíferos por vehículos en la vía del Escobero, Envigado (Antioquia), Colombia. Actual Biol 29(87):229–233
Dias IM, Amato G, Cunha HM, DeSalle R, Paglia AP, Peterson JK, Fonseca CG (2019) Isolation, characterization, and cross-species amplification of new microsatellite markers for three opossum species of the Didelphidae family. Conserv Genet Resour 1(1):405–410. https://doi.org/10.1007/s12686-009-9094-8
Dray S, Dufour AB (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw 22:1–20
Driscoll DA, Banks SC, Barton PS, Lindenmayer DB, Smith AL (2013) Conceptual domain of the matrix in fragmented landscapes. Trends Ecol Evol 28(10):605–613. https://doi.org/10.1016/j.tree.2013.06.010
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Figueroa C, Brieva C, Trujillo M, Moreno O (2009) Experiencias sobre manejo y crianza de zarigüeyas (Didelphis albiventris). Rev Mem Conf Intern Med Aprovech Fauna Exot Silv No Convenc 5(1):69–73
Fike JA, Beasley JC, Rhodes OE (2009) Isolation of 21 polymorphic microsatellite markers for the Virginia opossum (Didelphis virginiana). Mol Ecol 9(4):1200–1202. https://doi.org/10.1111/j.1755-0998.2009.02606.x
Flórez-Oliveros FJ, Vivas-Serna C (2020) Zarigüeyas (chuchas comunes) marmosas y colicortos en Colombia. FUNDZAR, Medellín, Colombia. https://www.metropol.gov.co/Documentos_SalaPrensa/LIBRO%20ZARIGUEYA%20VERSION_ESPA%C3%91OL.pdf
Fu YX (1996) New statistical tests of neutrality for DNA samples from a population. Genetics 143(1):557–570. https://doi.org/10.1093/genetics/143.1.557
Herrera JM (2011) El papel de la matriz en el mantenimiento de la biodiversidad en hábitats fragmentados. De la teoría ecológica al desarrollo de estrategias de conservación. Ecosistemas 20:2–3
Holderegger R, Di Giulio M (2010) The genetic effects of roads: a review of empirical evidence. Basic Appl Ecol 11(6):522–531. https://doi.org/10.1016/j.baae.2010.06.006
Huxel GR, Hastings A (1999) Habitat loss, fragmentation, and restoration. Restor Ecol 7(3):309–315. https://doi.org/10.1046/j.1526-100X.1999.72024.x
IUCN— The IUCN Red List of Threatened Species (2021):e.T40501A197310576. https://doi.org/10.2305/IUCN.UK.2021-1.RLTS.T40501A197310576.en. Accessed 17 Feb 2022
Ives CD, Lentini PE, Threlfall CG, Ikin K, Shanahan DF, Garrard GE, Bekessy SA, Fuller RA, Mumaw L, Rayner L et al (2015) Cities are hotspots for threatened species. Glob Ecol Biogeogr 25(1):117–126. https://doi.org/10.1111/geb.12404
Jombart T, Devillard S, Dufour AB, Pontier D (2008) Revealing cryptic spatial patterns in genetic variability by a new multi-variate method. Heredity 101(1):92–103. https://doi.org/10.1038/hdy.2008.34
Jombart T (2015) A tutorial for the spatial analysis of principal components (sPCA) using adegenet 2.0.0. MRC Centre for Outbreak Analysis and Modelling, pp 1–50. https://adegenet.r-forge.r-project.org/files/tutorial-spca.pdf
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kasparian MA, Hellgreen EC, Ginger SM, Lavesque LP, Clark JE, Winkerman L (2004) Population characteristics of Virginia opossum in the cross timbers during raccoon reduction. Am Midl Nat 151:154–163. https://doi.org/10.1674/0003-0031(2004)151[0154:PCOVOI]2.0.CO;2
Lavergne A, Douady C, Catzeflis FM (1999) Isolation and characterization of microsatellite loci in Didelphis marsupialis (Marsupialia: Didelphidae). Mol Ecol 8(3):517–518
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Medellín-Alcaldía (2017) Biodiversidad y servicios ecosistémicos en Medellín: Cartilla para maestros. Alcaldía de Medellín, Medellín, Colombia. http://repository.humboldt.org.co/bitstream/handle/20.500.11761/36091/cartilla%2014-07-14.pdf?sequence=2&isAllowed=y
Miles LS, Rivkin LR, Johnson MTJ, Munshi-South J, Verrelli BC (2019) Gene flow and genetic drift in urban environments. Mol Ecol 28(18):4138–4151. https://doi.org/10.1111/mec.15221
Munshi-South J, Zak Y, Pehek E (2013) Conservation genetics of extremely isolated urban populations of the northern dusky salamander (Desmognathus fuscus) in New York City. PeerJ 1:e64. https://doi.org/10.7717/peerj.64
Neteler MY, Mitasova H (2002) Open source GIS: a GRASS GIS approach, vol 689. Springer, New York
Paradis E (2010) pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26(3):419–420. https://doi.org/10.1093/bioinformatics/btp696
R Core Team (2023) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/
Ramírez GF, Osorio JH (2014) Identificación de helmintos en zarigüeyas (Didelphis marsupialis) en el Suroccidente Colombiano. Biosalud 13(1):37–44
Ringier HJ (1961) Review of Oligocene Didelphis marsupialis. J Paleontol 35:218–228
Ruell EW, Riley SPD, Douglas MR, Antolin MF, Pollinger JR, Tracey JA, Lyren LM, Boydston EE, Fisher RN, Crooks KR (2012) Urban habitat fragmentation and genetic population structure of bobcats in coastal southern California. Am Midl Nat 168(2):265–280. https://doi.org/10.1674/0003-0031-168.2.265
Ryser J (1995) Activity, movement and home range of Virginia opossum (Didelphis virginiana) in Florida. Bull Fla Mus Nat Hist 38(6):177–194
Saldaña I, Cadavid A, Gómez D (2019) Abundancia relativa y patrones de actividad de Didelphis marsupialis en un área periurbana de Medellín, Colombia. Rev MVZ Cordoba 24(3):7366–7371. https://doi.org/10.21897/rmvz.1352
Sunquist MS, Austad F (1987) Movement patterns and home range in the common opossum (Didelphis marsupialis). J Mammal 68(1):173–176. https://doi.org/10.2307/1381069
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595. https://doi.org/10.1093/genetics/123.3.585
Tamura K, Stecher G, Kumar S (2021) MEGA11: molecular evolutionary genetics analysis version 11. Mol Biol Evol 38(7):3022–3027. https://doi.org/10.1093/molbev/msab120
Templeton AR, Shaw K, Routman E, Davis SK (1990) The genetic consequences of habitat fragmentation. Anal Mo Bot Gard 77(1):13. https://doi.org/10.2307/2399621
Tyndale-Biscoe CH, Mackenzie RB (1976) Reproduction in Didelphis marsupialis and D. albiventris in Colombia. J Mammal 57(2):249–265. https://doi.org/10.2307/1379686
White GM, Boshier DH, Powell W (1999) Genetic variation within a fragmented population of Swietenia humilis Zucc. Mol Ecol 8(11):1899–1909. https://doi.org/10.1046/j.1365-294x.1999.00790.x
Acknowledgements
We thank the medical staff of AMVA for their support with the sampling procedures and access to individual records of sampled opossums. Special thanks to Omer Campo for his technical support during the implementation of molecular methods, and Andrea Hinek for reviewing a preliminary version of this manuscript. We are very grateful to Fundación Zarigüeya, Claudia Ceballos, and students from the laboratory for aiding in the optimization of field techniques. We also acknowledge the environmental authority of the Área Metropolitana del Valle de Aburrá (AMVA) for funding this work through the interinstitutional agreement grant No. 926, 2021.
Funding
Open Access funding provided by Colombia Consortium.
Author information
Authors and Affiliations
Contributions
J. Herrera-Pérez, and I.D. Soto-Calderón designed the research; I.D. Soto-Calderón acquired the funding; C. Henao-Sáenz conducted the experiments; C. Henao-Sáenz, J. Herrera-Pérez, and I.D. Soto-Calderón. analyzed the results and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no financial or proprietary interests in any material discussed in this article.
Institutional review board statement
This research was endorsed by the Ministry of Environment of Colombia through the Non-Commercial Scientific Research Permit granted to Universidad de Antioquia (Resolution 0524 of May 27, 2014). The animal study protocols were approved by the Ethics Committee for Animal Experimentation of the University of Antioquia on October the 5th, 2021.
Informed consent
Not applicable.
Additional information
Handling editor: Nicola S. Heckeberg.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
42991_2024_434_MOESM1_ESM.xlsx
Supplementary file1 The Supplementary data files are available on the online version of this paper: Supplementary File 1 details the coordinates of the sites where the 66 studied animals were rescued in AMVA. Supplementary File 2 is an Excel file containing the records of common opossums released in AMVA between 2020 and 2022. Supplementary File 3 contains the Cytochrome B sequences. Supplementary File 4 is also an Excel file with the microsatellite primers and amplification conditions. Supplementary File 5 provides further details of the sPCA, DAPC and Mantel test (XLSX 26 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Henao-Sáenz, C., Herrera-Pérez, J. & Soto-Calderón, I.D. Diversity and spatial genetic structure of the common opossum (Didelphis marsupialis) in an urban matrix of Northwestern Colombia. Mamm Biol (2024). https://doi.org/10.1007/s42991-024-00434-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42991-024-00434-2