
Abstract
The grey wolf (Canis lupus) is one of the most challenging species to conserve in our modern and crowded world. Due to various factors, most European wolf populations are currently growing. In Hungary, numbers have increased since the 2000s. Although spontaneous recolonisation from Slovakia is considered to be the most likely mechanism by the majority of experts, some stakeholders claim that hand-reared individuals have been released. To determine the origin of wolves in northern Hungary, we analysed samples of free-ranging wolves collected in Slovakia and Hungary as well as samples from wolves in private enclosures in the region. We also included reference samples from domestic dogs. All samples were genotyped at 14 canine autosomal tetranucleotide microsatellite loci (STR) and analysed using multivariate, Bayesian methods. Hungarian wolf samples were also analysed using kinship methods. In the free-ranging wolf samples, all loci were polymorphic with 3–12 alleles. The overall observed (Ho) and unbiased expected (uHE) heterozygosities were 0.60–0.66 and 0.69–0.71, respectively. Parental and sibling relationships were also found among Hungarian individuals: three generations of a pack in the Bükk Mountains were identified. Samples from free-ranging wolves clustered separately from those of captive wolves and dogs. However, genetic similarities were found between Slovakian and Hungarian wolf samples. Our analyses indicate a Slovakian origin of the sampled Hungarian wolves, and we found no evidence that individuals originating in captivity have played any role in the recolonisation process. Kinship relationships and moderate genetic diversity suggest that there is ongoing gene flow across the Slovakian–Hungarian border.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Large carnivores are among the most challenging groups of animals concerning their protection in our modern and crowded world (Chapron et al. 2014). Their conservation in most European countries is dependent upon not only suitable ecological conditions but also the attitudes and actions of local communities and stakeholders, particularly livestock farmers and hunters (Linnell et al. 1999; Berger 2006; Boitani et al. 2015). Large carnivores play an important role in regulating ecosystems (Ripple et al. 2014) but, due to hunting and persecution as well as major habitat changes, large carnivore populations have declined worldwide during the last two centuries and the ranges of many species have contracted and been fragmented (Ceballos and Ehrlich 2002; Laliberte and Ripple 2004). In Europe, however, populations of several large carnivore species are now increasing (Deinet et al. 2013), mainly due to conservation programmes, legal protection, favourable public opinion, habitat restoration and recovery of prey populations (Chapron et al. 2014; Cimatti et al. 2021).
The grey wolf (Canis lupus) was persecuted in Europe for centuries and by the twentieth century was exterminated in much of its former range. Its numbers probably reached a minimum in the 1940s–1960s (Chapron et al. 2014). During the last 20 years, the species has shown natural recovery and reappeared in areas from which it had previously been eradicated (e.g. Western European countries, Scandinavia), although densities vary widely (Salvatori and Linnell 2005). Wolves have considerable dispersal capabilities (Wabakken et al. 2007; Ciucci et al. 2009) and are ecologically flexible (Mech and Boitani 2003). They can therefore survive in a broad range of habitats, assuming that sufficient food is available and any hunting or persecution is within sustainable limits. The major limiting factors are thought to be anthropogenic pressures and the presence of appropriate breeding sites (Salvatori and Linnell 2005).
The Carpathian Mountains represent the largest high-mountain system of Central and Eastern Europe, with a key role in the phylogeography of many species (Schmitt 2009; Frank et al. 2017). This region harbours one of the largest wolf populations in Europe, estimated to number ca. 3500–3800 individuals, mostly in Romania, Slovakia and Poland (Linnell and Cretois 2018). The Carpathian Basin in neighbouring Hungary may serve as an area of expansion for this population, where dispersing wolves can settle.
Wolves have faced similar changes in Hungary as in other parts of Europe. Their numbers and range decreased significantly by the end of the nineteenth century (Demeter 1984). Sporadic occurrences were recorded during the twentieth century in several parts of the country (Demeter 1984; Faragó 1989). These were concentrated in the northern part of the country, close to the Slovakian border, where suitable habitats form a continuous corridor and facilitate expansion in the last decades (Köck et al. 2014). Wolf occurrences are more sporadic in other areas because of various barriers that make movement more difficult.
In 2005, there were estimated to be a total of 3–6 single individuals in northern Hungary, which were thought to belong to the Carpathian population (Salvatori and Linnell 2005). More recently, around 12–18 wolves were reported to be living in Bükk National Park and surrounding areas of the North Hungarian Mountains based on individual sightings and signs (Wallendums 2018). Due to the sudden and dispersed appearances of single individuals and small groups relatively far from the putative source population in Slovakia, some local stakeholders and experts—game managers, hunters, foresters and even some national park wardens—suspected the release of hand-reared wolves or wolf-dog hybrids from private enclosures and game parks as the origin of Hungarian occurrences (Kovács 2018; Fluck 2020). Although there are no empirical data to support this opinion, it has become more and more accepted by local managers. This has important consequences for conservation actions because the spontaneous recolonisation of a strictly protected species must be tolerated and even supported by current nature conservation regulations, whereas the release of hand-reared individuals is not permitted. While the origin and genetic background of hand-reared individuals may be diverse, these are expected to be genetically divergent from the autochthonous wolf population, and if such individuals are detected in nature, their removal would be permitted. Therefore, clarification of the origin of wolves currently occurring in northern Hungary is crucial for the conservation of the species.
Today, genetic methods are in widespread use to study wolf dispersal and genetic identity throughout Europe (e.g. De Groot et al. 2016; Hindrikson et al. 2017). Non-invasive and tissue samples can be used to assess genetic diversity and population structure as well as to measure gene flow between subpopulations and identify potential risks associated with demographic change and inbreeding (Duchamp et al. 2012; Shafer et al. 2015). Genetic studies of the Carpathian wolves have largely focussed on the northern populations in Poland and Slovakia (Pilot et al. 2006, 2010; Gula et al. 2009; Czarnomska et al. 2013; Bakan et al. 2014; Rigg et al. 2014; Hulva et al. 2018) and the eastern populations in Romania and Ukraine (Ericson et al. 2020). The first genetic survey of grey wolves in Hungary was done in 2004–2006 in the region of Aggtelek, which verified the presence of resident wolves in the Slovakian–Hungarian borderland (Hausknecht et al. 2010).
The main goal of our research was to explore the origin of wolves appearing in the North Hungarian Mountains. In particular, we sought to answer the following questions: (1) are wolves in northern Hungary dispersing individuals from the source population in Slovakia, which could be the most probable place of origin due to its proximity and the advantageous ecological conditions, or were animals released from local private enclosures? Gene flow can occur from other populations than the Slovakian, so if different genotypes are detected, reference samples from other neighbouring populations can be included in future analyses. (2) Besides is there evidence of non-Carpathian wolf genotypes or intraspecific hybrids? (3) Finally, if there are breeding individuals in the area, can their offspring be detected?
Materials and methods
Sampling
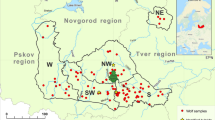
Tissue and non-invasive samples of free-ranging wolves in the Western Carpathians of northern Slovakia (Low Tatras, VeľkáFatra, Western Tatras; n = 15), northern Hungary (Börzsöny, Karancs Mountains, Heves-Borsod Hills, Bükk Mountains; n = 34) and north-eastern Hungary (Great Plain; n = 1) were collected between 2016 and 2020 (Fig. 1). We also used samples from captive wolves of the Foundation for the Preservation of European Wildlife (n = 9), collected between 2018 and 2019, and samples of village and pedigree dogs from the region (n = 14). Non-invasive samples (scat and urine) were mostly obtained during snow-tracking, and tissue samples were obtained from road-killed wolves and dogs. Blood and saliva samples were obtained from captive wolves and dogs. The pedigree of the captive wolves was not known. Blood was collected in EDTA-coated collection tubes (Greiner Bio-One GmbH, Austria) and saliva using swabs (Copan Diagnostics Inc., USA). Scat, urine, blood and saliva were stored at − 20 °C; tissue samples were preserved in 96% ethanol and stored at − 20 °C until processing.

Locations of sampling sites and the number of samples in Hungary and Slovakia. 1—Low Tatras (n = 6), 2—Vel’káFatra (n = 6), 3—Western Tatras (n = 3), 4—Börzsöny Mountains (n = 5), 5—Karancs Mountains (n = 1), 6—Heves-Borsod Hills (n = 4), 7—Bükk Mountains (n = 24), 8—Great Plain (n = 1)
DNA preparation and genotyping
Total genomic DNA was extracted from tissue samples using the QIAamp DNA Investigator Kit (QIAGEN GmbH, Germany) and the MagCore Genomic DNA Tissue Kit (RBC Bioscience Corp. Taiwan); from the scat samples using the QIAamp DNA Stool Mini Kit (QIAGEN GmbH, Germany); and from blood using the MagCore Genomic DNA Whole Blood Kit (RBC Bioscience Corp. Taiwan) according to the manufacturers’ protocols. DNA extraction from urine was performed according to Valiere and Taberlet (2000). Samples were genotyped at 14 tetranucleotide microsatellite loci: c2001, c2054, FH2538, PEZ3, PEZ8, PEZ19 (Vilà et al. 2003), FH2004, FH2010, FH2088, FH2107, FH2309, FH3313, FH3377, PEZ02 (Dayton et al. 2009). Sex was determined by the amelogenin gene (Yan et al. 2013). Primers were divided into multiplex reactions. Multiplex PCRs were set up in a total volume of 25 μl, containing 1 × QIAGEN Multiplex Master Mix (QIAGEN GmbH, Germany), 60–240 ng of template DNA, each primer in optimum concentration (0.04–0.30 mM), and filled up with water. Amplification reactions were performed with the following cycling conditions: an initial activation at 95 °C for 15 min followed by 40 cycles of 95 °C for 30 s, 58 °C for 60 s, and 72 °C for 60 s, and a final extension at 60 °C for 30 min. A LifeECO Thermal Cycler (Hangzhou Bioer Technology Co. Ltd., China) was used for amplification reactions. Amplified PCR products were separated on an ABI Prism 3100 Genetic Analyser using LIZ500 Size Standard (Applied Biosystems, USA). The PeakScanner ver. 1.0 (Applied Biosystems, USA) was used to analyse electropherograms and score allele sizes.
Programme MICROCHECKER ver. 2.3.4 (Van Oosterhout et al. 2004) was used to assess the presence of null alleles, scoring errors and large allele dropout. We used GenAlEx ver. 6.5 (Peakall and Smouse 2012) to estimate allele frequency by locus and population, the number of alleles (Na), the number of effective alleles (Ne), observed and unbiased expected heterozygosity values (Ho and uHE, respectively). Allelic richness (AR) values were computed with FSTAT ver. 2.9.3.2 (Goudet 1995).
Assessing genetic structure
To identify parental and kinship relations among Hungarian wolves and to define family groups (packs), we used the parentage assignment package Colony2 (Jones and Wang 2010). As the sex of individuals was known, this was considered during kinship reconstructions.
The Bayesian clustering method implemented in STRUCTURE ver. 2.3.4 (Pritchard et al. 2000) was used to infer the most probable number of genetic clusters without a priori definition of populations to assess potential admixture between Hungarian wolves and other wolves and dogs. The Bayesian clustering method and Markov Chain Monte Carlo (MCMC) simulation were run using an admixture model and correlated allele frequencies. STRUCTURE was run with simulations of 10 runs for each value of K, with a burn-in period of 250,000 iterations and 750,000 replications for a number of genetic clusters (K) from one to seven. We estimated the number of clusters (K) by calculating the second-order rate of change in log-likelihood values (ΔK) in STRUCTURE Harvester ver. 0.6.94 (Earl and vonHoldt 2012), as well as the four supervised estimators of K from threshold values ranging from 0.5 to 0.8 (Puechmaille 2016) in StructureSelector (Li and Liu 2018).
We used also discriminant analysis of principal components (DAPC) implemented in adegenet ver. 2.1.1 (Jombart 2008), which identifies clusters of individuals without using any population genetic model (Jombart et al. 2010). We used the find.clusters() function for the identification of the optimal number of clusters based on the Bayesian information criterion (BIC). The adegenet package was run with R ver. 4.0.1 (R Core Team 2018).
Additionally, a principal coordinate analysis (PCoA) was employed on individual multilocus genotypes to visualise their clustering. PCoA was created from pairwise genetic distance values with the help of GenAlEx ver. 6.5 (Peakall and Smouse 2012).
We detected the direction of differentiation with the diveRsity package (Keenan et al. 2013) and used this information to infer relative migration between pairs of populations (Sundqvist et al. 2016). To plot the relative migration level between populations, the Nm method (Alcala et al. 2014) implemented in diveRsity ver. 1.9.90 was used in R ver. 4.0.1 (R Core Team 2018). To determine significant relative migrations, bootstrapping was employed with 1000 replications (Keenan et al. 2013).
Results
Genetic diversity
Null alleles were detected at three loci (FH2309, FH3313, FH3377) in the group of dogs, at three loci (FH2107, FH2004, FH2010) in Hungarian wolf samples, at one locus (PEZ02) in the Slovakian wolf samples and a single locus in the captive wolf samples (FH3377). No other PCR errors (scoring error, large allele dropout, false alleles) were detected in the data set. We obtained a complete 14-locus genetic profile for all samples.
All analysed loci were polymorphic in the free-ranging wolves, with the number of alleles per locus ranging from three (PEZ19) in both Hungarian and Slovakian wolves to 11 (FH2107, FH3313) in Hungarian wolves and 12 (FH2107) in Slovakian wolves, with an average of 6.29 in Hungarian and 5.93 in Slovakian samples. The mean observed heterozygosity (Ho) was 0.60–0.66 and the mean unbiased expected heterozygosity (uHE) was 0.69–0.71 in the two free-ranging groups. The average number of alleles was 5.93–6.29 and the number of effective alleles was 3.51–3.72 (Table 1).
We identified 15 individual free-ranging wolves among 15 samples collected in Slovakia and 25 individuals among 35 samples from Hungary. Several individuals were identified multiple times: four animals were detected from two samples each, one from three samples and one from five. Sex determination revealed 15 males and 10 females among the Hungarian wolves.
Genetic structure
The programme Colony2 detected several siblings and offspring-parent relationships among the sampled Hungarian wolves. Based on the “Best Cluster” result, three generations were identified for a wolf pack in the Bükk Mountains. The sex determination of these individuals revealed four males and one female. Genetic incompatibilities suggested that males denoted with genotypes 1 and 2 are full siblings, whereas the male with genotype 2 is the father of the female with genotype 3. This female is the mother of two males denoted with genotypes 4 and 5 (Fig. 2). Furthermore, two three-individual sibling relationships were found among the Hungarian samples, but they could not be connected to other samples. To reduce bias caused by including closely related genotypes, first-order relatives were excluded from subsequent analyses.

Reconstructed pedigree of free-ranging wolves in the Bükk Mountains, Hungary. Estimated kinship probabilities are shown above detected haplotypes (numbered 1–5)
The programme STRUCTURE detected the highest average log-likelihood values for six genetic clusters, and the second-order rate of change in log-likelihood values was the highest for two genetic clusters, K = 2 (Fig. 3). In this case, free-ranging Slovakian and Hungarian wolves clustered together forming one group, whereas dogs and captive wolves formed the other cluster. The four supervised estimators of Puechmaille indicated the presence of four clusters throughout all threshold values, K = 4 (Fig. 3). In this case, dogs and captive wolves formed two distinct groups and separated well from the other samples, Slovakian wolves formed another group clustered with some Hungarian samples and the rest of the Hungarian samples formed the fourth group (Fig. 3).

The results of the Bayesian clustering of free-ranging wolves in Slovakia and Hungary, captive wolves and dogs. A The mean log-likelihood values for each value of the number of clusters (LnP(K)). B The probability of the models according to cluster size based on the second-order rate of change in log-likelihood values (Delta K). C The optimal number of clusters based on supervised estimators (MedMed K, MedMean K, MaxMed K, MaxMean K). D Bar plot of membership probabilities from K = 2 to K = 4
DAPC also showed the lowest BIC scores for K = 4. Similarly to the results from STRUCTURE, dogs and captive wolves formed two distinct groups and separated well from the other samples. Free-ranging wolves formed two clusters that corresponded to Slovakian and Hungarian samples, but again, some Hungarian samples clustered with Slovakian wolves (Fig. 4). The Principal Coordinates Analysis (PCoA) confirmed this picture (Fig. 5).

Discriminant analysis of principal components (DAPC) to identify clusters of individuals without using a population genetic model. A Bayesian information criterion (BIC) according to the number of clusters in the DAPC. The most likely number of clusters is where BIC is lowest. B DAPC scatter plot showing genetic separation of free-ranging wolves (HU, SLO), dogs and captive wolves; DA eigenvalues are shown in the upper left corner. C Analysis of direction of differentiation using the diveRsity package indicates that there is significant migration only between Slovakian (group 2, SLO) and Hungarian (group 3, HU) free-ranging wolves

Principal coordinate analysis showing genetic differentiation of dogs (blue diamonds), free-ranging wolves (grey triangles and brown squares) and captive wolves (yellow dots)
Using diveRsity, a significant relative movement was found only between Slovakian and Hungarian free-ranging wolves. All other possible migration rates were small and non-significant. Migration between free-ranging wolves seemed to be unidirectional: from Slovakia to Hungary (Fig. 4).
Discussion
This study is the first to investigate the origin of grey wolves in Hungary using microsatellite loci. The canine microsatellite markers we used were suitable for the grey wolf and showed sufficient polymorphisms to assess genetic diversity and genetic structure.
We found moderate levels of genetic diversity in Hungarian wolves (Ho = 0.60; uHE = 0.69). Similar levels of heterozygosity were found in Slovakia by Rigg et al. (2014) (Ho = 0.65, HE = 0.64), Szewczyk et al. (2019) (Ho = 0.65, uHE = 0.678) and Hulva et al. (2018) (Ho = 0.694, HE = 0.733) and in Serbia, including the southern-most portion of the Carpathians, by Đan et al. (2016) (Ho = 0.69; HE = 0.75). Bakan et al. (2014) also reported heterozygosity of Slovakian (Ho = 0.539; HE = 0.707) and Serbian (Ho = 0.526; HE = 0.637) samples. These findings are consistent with studies of the species elsewhere in Europe (Hindrikson et al. 2017) except in Italy, where heterozygosity was found to be lower (Ho = 0.57; uHE = 0.58) as a result of the population passing through a severe genetic bottleneck (Fabbri et al. 2014).
The results of our analyses of the genetic structure suggest that the Slovakian population probably contributed to the gene pool of Hungarian wolves, likely via natural dispersal, but it may not be the only population involved in the recolonisation of Hungary. Our results do not support the hypothesis that the presence of free-ranging wolves in northern Hungary is the result of releases from zoos or other captive facilities (cf. Kovács 2018; Fluck 2020). However, the number of samples from captive wolves used was limited, and although we cannot completely exclude the possibility of such releases, this seems unlikely based on the observed genetic structure. Especially, that wolves are capable of dispersing over long distances (e.g. Wabakken et al. 2007; Ciucci et al. 2009; Andersen et al. 2015), thus individuals from neighbouring countries such as Slovakia, Romania or Slovenia, or e.g., the Czech Republic, Poland or Ukraine could reach Hungary in spite of the ecological barriers and unfavourable habitats. The dispersion may be facilitated by suitable habitats and ecological corridors (Köck et al. 2014), so animals originating from different populations can mix and contribute to the present genetic pool of a given population, as has been proved in several regions in Europe (e.g. Ražen et al. 2016; Hulva et al. 2018; Szewczyk et al. 2019). Thus, Hungarian wolves separating from the Slovakian and captive individuals at K = 4 in the admixture analysis may also be immigrants from other populations or their descendants.
Wolves can disperse into Hungary via least-cost paths to core areas in the Börzsöny, Bükk, Mátra and Zemplén Mountains, where most of the samples for this study were collected (see Fig. 1). A single individual was identified in the north-eastern part of the Great Plain close to the Romanian border, where the species also has a core area. Based on the least-cost path model, the animal could have reached this region from Slovakia through the North Hungarian Mountains or from Romania (Köck et al. 2014). The origin of this individual should be studied in more depth, as short-distance movements and dispersion from Slovakia would be the most obvious but longer dispersion routes from more distant Romanian Carpathian, Dinaric or even Central Europan Lowland populations could also explain its presence (Fabbri et al. 2014; Ražen et al. 2016; Hulva et al. 2018; Ericson et al. 2020). However, only Hungarian and Slovakian samples were used in this study, so we cannot determine the origin of this individual yet.
Our Hungarian wolf samples were slightly male-biased, which might reflect differences in dispersal between the sexes. The dispersal of natural grey wolf populations is usually male-biased, i.e. males seem to have a greater tendency to disperse (Stansbury et al. 2016). Nevertheless, kinship relationships found among our Hungarian samples confirm the presence of at least one reproducing pack in the Bükk Mountains, which is consistent with camera-trap records of young wolves in the area (Gombkötő, unpublished data), and reproduction has also been confirmed by genetic methods in Aggtelek National Park (Hausknecht et al. 2010).
Large carnivores present a particular set of conservation issues since one of their fundamental characteristics is that they occur at relatively low population densities, individuals tend to move over large areas (Andersen et al. 2015; Bartoń et al. 2019), and they can occur even in human-dominated areas. Many populations span international borders, thus the management and conservation of these species call for transboundary, population-level management plans (Linnell et al. 2008; Trouwborst 2010; Blanco 2012). With the technical development of animal monitoring methods (e.g. photo-trapping, GPS-satellite telemetry, isotope analysis, genetic monitoring), data on natural dispersal and long-distance movements are increasingly available, resulting in a better understanding of these processes (e.g. Kays et al. 2015). Genetic methods reveal a lot of additional information (e.g. effective population size, diversity indices, genetic structuring), which can provide valuable support for developing management and conservation measures. Future research and conservation programmes would benefit from adopting and optimizing genetic methods to acquire baseline population parameters and to enable comparison between (sub)populations (De Groot et al. 2016).
Conclusion
We studied the genetic diversity and origin of the grey wolf in Hungary using 14 microsatellite loci and a marker for sex determination. These markers are suitable for species and individual identification, as well as for the assessment of kinship relations. Our analyses revealed that the Slovakian population probably contributed to the gene pool of Hungarian wolves via natural dispersal. However, to investigate if Slovakia is the source of recolonization of Hungary or whether other wolf populations have also contributed, reference samples from additional regional populations need to be included in future analyses. Furthermore, it appears that multiple individuals have colonised Hungary, possibly multiple times. New migrants from Slovakia or possibly from other countries seem to preserve some level of gene flow, and some exchange of individuals between subpopulations can be expected based on the life history of wolves. Moreover, reconstructed kinships underpin that wolves in northern Hungary are not only dispersing or migrating individuals, but breeding packs are also becoming established in the area. Since it cannot be ruled out that populations outside Slovakia were involved in the recolonisation of Hungary, it would be necessary to further compare Hungarian individuals with reference samples from neighbouring populations. However, our analyses have found no evidence that individuals released from captivity played a role in the successful recolonisation process in Hungary.
We would recommend the use of a standardised genetic methodology to enable the comparison of results across Europe and, in particular, in the Carpathian region. This would greatly facilitate monitoring of population status and potential further expansion, and contribute to transboundary conservation and management programmes following European Union guidelines. If appropriate measures are implemented, the ongoing recovery of the wolf in northern Hungary may enable the species to resume its ecological role as an apex predator in the Pannonian Bioregion.
Availability of data and material
Not applicable.
Code availability
Not applicable.
References
Alcala N, Goudet J, Vuilleumier S (2014) On the transition of genetic differentiation from isolation to panmixia: what we can learn from Gst and D. Theor Pop Biol 93:75–84. https://doi.org/10.1016/j.tpb.2014.02.003
Andersen LW, Harms V, Caniglia R, Czarnomska SD, Fabbri E, Jędrzejewska B et al (2015) Long-distance dispersal of a wolf, Canis lupus, in northwestern Europe. Mamm Res 60:163–168. https://doi.org/10.1007/s13364-015-0228-y
Bakan J, Vukan L, Popović Z, Paule L (2014) Genetic differentiation of grey wolf population (Canis lupus L.) from Balkan and Carpathians. Balkan J Wildl Res 1:87–93. https://doi.org/10.15679/bjwr.v1i1.17
Bartoń KA, Zwijacz-Kozica T, Zięba F, Sergiel A, Selva N (2019) Bears without borders: long-distance movement in human-dominated landscapes. Glob Ecol Conserv 17:e00541. https://doi.org/10.1016/j.gecco.2019.e00541
Berger KM (2006) Carnivore–livestock conflicts: effects of subsidized predator control and economic correlates on the sheep industry. Conserv Biol 20:751–761. https://doi.org/10.1111/j.1523-1739.2006.00336.x
Blanco JC (2012) Towards a population-level approach for the management of large carnivores in Europe. Challenges and opportunities. A Large Carnivore Initiative for Europe report prepared for the European Commission (contract 070307/2012/629085/SER/B3)
Boitani L, Alvarez F, Anders O, Andrén H, Avanzinelli E, Balys V et al (2015) Key actions for large carnivore populations in Europe. Institute of Applied Ecology (Rome, Italy). Report to DG Environment, European Commission, City of Brussels. https://ec.europa.eu/environment/nature/conservation/species/carnivores/pdf/key_actions_large_carnivores_2015.pdf. Accessed June 2021
Ceballos G, Ehrlich PR (2002) Mammal population losses and the extinction crisis. Science 296:904–907. https://doi.org/10.1126/science.1069349
Chapron G, Kaczensky P, Linnell JDC, von Arx M, Huber D, Andrén H et al (2014) Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346:1517–1519. https://doi.org/10.1126/science.1257553
Cimatti M, Ranc N, Benítez-LópezA ML, Boitani L, Cagnacci F et al (2021) Large carnivore expansion in Europe is associated with human population density and landcover changes. Divers Distrib 27:602–617. https://doi.org/10.1111/ddi.13219
Ciucci P, Reggioni W, Maiorano L, Boitani L (2009) Long-distance dispersal of a rescued wolf from the Northern Apennines to the Western Alps. J Wildl Manag 73:1300–1306. https://doi.org/10.2193/2008-510
Czarnomska DS, Jedrzejewska B, Borowik T, Niedziałkowska M, Stronen AV, Nowak S et al (2013) Concordant mitochondrial and microsatellite DNA structuring between Polish lowland and Carpathian Mountain wolves. Conserv Genet 14:573–588. https://doi.org/10.1007/s10592-013-0446-2
Ðan M, Šnjegota D, Veličković N, Stefanović M, Vidaković DO, Ćirović D (2016) Genetic variability and population structure of grey wolf (Canis lupus) in Serbia. Russ J Genet 52:821–827. https://doi.org/10.1134/S1022795416080044
Dayton M, Koskinen TM, Tom KB, Mattila MA, Eric J, Halverson J et al (2009) Developmental validation of Short Tandem Repeat Reagent Kit for forensic DNA profiling of canine biological material. Croat Med J 50:268–285. https://doi.org/10.3325/cmj.2009.50.268
De Groot GA, Nowak C, Skrbinšek T, Andersen LW, Aspi J, Fumagalli L et al (2016) Decades of population genetic research reveal the need for harmonization of molecular markers: the grey wolf (Canis lupus) as a case study. Mamm Rev 46:44–59. https://doi.org/10.1111/mam.12052
Deinet S, Ieronymidou C, McRae L, Burfield IJ, Foppen RP, Collen B, Böhm M (2013) Wildlife comeback in Europe: The recovery of selected mammal and bird species. Final report to Rewilding Europe by ZSL, BirdLife International and the European Bird Census Council, London. https://rewildingeurope.com/wp-content/uploads/2013/11/Wildlife-Comeback-in-Europe-the-recovery-of-selected-mammal-and-bird-species.pdf. Accessed June 2021
Demeter A (1984) Recent records of rare or non-resident large carnivores in Hungary. Vertebr Hung 22:65–71
Duchamp C, Boyer J, Briaudet PE, Leonard Y, Moris P, Bataille A et al (2012) A dual frame survey to assess time- and space-related changes of the colonizing wolf population in France. Hystrix 23:1428. https://doi.org/10.4404/hystrix-23.1-4559
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Ericson HS, Fedorca A, Toderas I, Hegyeli Z, Plis K, Dykyy I et al (2020) Genome-wide profiles indicate wolf population connectivity within the eastern Carpathian Mountains. Genetica 148:33–39. https://doi.org/10.1007/s10709-019-00083-1
Fabbri E, Caniglia R, Kusak J, Galov A, Gomerčić T, Arbanasić H, Huber D, Randi E (2014) Genetic structure of expanding wolf (Canis lupus) populations in Italy and Croatia, and the early steps of the recolonization of the Eastern Alps. Mamm Biol 79:138–148. https://doi.org/10.1016/j.mambio.2013.10.002
Faragó S (1989) A farkas (Canis lupus LINNÉ, 1758) 1920–1985 közötti előfordulása Magyarországon. Folia Hist Nat Musei Matraiensis 14:139–164
Fluck D (2020) Észt farkasok, nem kutyadolog. In: Wallendums P (ed) Magyar Vadászlap. Virtuóz Kiadó és Nyomdaipari Kft., Budapest, pp 50–51
Frank K, Bleier N, Tóth B, Sugár L, Horn P, Barta E, Orosz L, Stéger V (2017) The presence of Balkan and Iberian red deer (Cervuselaphus) mitochondrial DNA lineages in the Carpathian Basin. Mamm Biol 86:48–55. https://doi.org/10.1016/j.mambio.2017.04.005
Goudet J (1995) Fstat (Version 1.2): a computer program to calculate F-statistics. J Hered 86:485–486. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Gula R, Hausknecht R, Kuehn R (2009) Evidence of wolf dispersal in anthropogenic habitats of the Polish Carpathian Mountains. Biodivers Conserv 18:2173–2184. https://doi.org/10.1007/s10531-009-9581-y
Hausknecht R, Szabó Á, Firmánszky G, Gula R, Kuehn R (2010) Confirmation of wolf residence in Northern Hungary by field and genetic monitoring. Mamm Biol 75:348–352. https://doi.org/10.1016/j.mambio.2009.10.001
Hindrikson M, Remm J, Pilot M, Godinho R, Stronen AV, Baltrūnaité L et al (2017) Wolf population genetics in Europe: a systematic review, meta-analysis and suggestions for conservation and management. Biol Rev 92:1601–1629. https://doi.org/10.1111/brv.12298
Hulva P, Bolfíková ČB, Woznicová V, Jindřichová M, Benešová M, Mysłajek RW et al (2018) Wolves at the crossroad: Fission–fusion range biogeography in the Western Carpathians and Central Europe. Divers Distrib 24:179–192. https://doi.org/10.1111/ddi.12676
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:1–15. https://doi.org/10.1186/1471-2156-11-94
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10:551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x
Kays R, Crofoot MC, Jetz W, Wikelski M (2015) Terrestrial animal tracking as an eye on life and planet. Science 348:aaa2478. https://doi.org/10.1126/science.aaa2478
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Köck M, Tudor P, Verghelet M, Hoffmann C, Favilli F, Elmi M, Alberton M, Meyer H, Kadlecik J, Sipos K (2014) Integrated management of biological and landscape diversity for sustainable regional development and ecological connectivity in the Carpathians. UNEP Vienna—Interim Secretariat of the Carpathian Convention (ISCC), Vienna
Kovács Z (2018) Féljünk-e a farkastól? In: Jámbor L, Bíró G, Bors R, Dénes R, Földvári A, Kovács G, Kovács Z, Rung Á, Wallendmus P (eds) Vadászévkönyv 2018. Országos Magyar Vadászkamara, Budapest, pp 128–132
Laliberte AS, Ripple WJ (2004) Range contractions of North American carnivores and ungulates. Bioscience 54:123–138. https://doi.org/10.1641/0006-3568(2004)054[0123:RCONAC]2.0.CO;2
Li YL, Liu JX (2018) StructureSelector: a web-based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Resour 18:176–177. https://doi.org/10.1111/1755-0998.12719
Linnell JDC, Cretois B (2018) Research for AGRI Committee—The revival of wolves and other large predators and its impact on farmers and their livelihood in rural regions of Europe, European Parliament, Policy Department for Structural and Cohesion Policies, Brussels
Linnell JDC, Odden J, Smith ME, Aanes R, Swenson JE (1999) Large carnivores that kill livestock: do problem individuals really exist? Wildl Soc Bull 27:698–705
Linnell JDC, Salvatori V, Boitani L (2008) Guidelines for population level management plans for large carnivores in Europe. A Large Carnivore Initiative for Europe report prepared for the European Commission (contract 070501/2005/424162/MAR/B2)
Mech LD, Boitani L (2003) Wolf social ecology. In: Mech LD, Boitani L (eds) Wolves. Behavior, ecology, and conservation. University of Chicago Press, Chicago, pp 1–34
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Pilot M, Jedrzejewski W, Branicki W, Sidorovich VE, Jedrzejewska B, Stachura K, Funk SM (2006) Ecological factors influence population genetic structure of European grey wolves. Mol Ecol 15:4533–4553. https://doi.org/10.1111/j.1365-294X.2006.03110.x
Pilot M, Branicki W, Jedrzejewski W, Goszczynski J, Jedrzejewska B, Dykyy I, Shkvyrya M, Tsingarska E (2010) Phylogeographic history of grey wolves in Europe. BMC Evol Biol 10:104. https://doi.org/10.1186/1471-2148-10-104
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Puechmaille SJ (2016) The program STRUCTURE does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Resour 16:608–627. https://doi.org/10.1111/1755-0998.12512
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org/. Accessed Sept 2020
Ražen N, Brugnoli A, Castagna C, Groff C, Kaczensky P, Kljun F, Knauer F, Kos I, Krofel M, Luštrik R, Majić A, Rauer G, Righetti D, Potočnik H (2016) Long-destance dispersal connects Dinaric-Balkan and Alpine grey wolf (Canis lupus) populations. Eur J Wildl Res 62:137–142. https://doi.org/10.1007/s10344-015-0971-z
Rigg R, Skrbinšek T, Linnell J (2014) Engaging hunters and other stakeholders in a pilot study of wolves in Slovakia using non-invasive genetic sampling. Report to DG Environment, European Commission, Bruxelles. Contract no. 07.0307/2013/654446/SER/B
Ripple WJ, Estes JA, BeschtaRL WCC, Ritchie EG, Hebblewhite M et al (2014) Status and ecological effects of the world’s largest carnivores. Science 343:6167. https://doi.org/10.1126/science.1241484
Salvatori V, Linnell J (2005) Report on the conservation status and threats for wolf (Canis lupus) in Europe. Convention on the Conservation of European Wildlife and Natural Habitats, Strasbourg
Schmitt T (2009) Biogeographical and evolutionary importance of the European high mountain systems. Front Zool 6:1–10. https://doi.org/10.1186/1742-9994-6-9
Shafer AB, Gattepaille LM, Stewart RE, Wolf JB (2015) Demographic inferences using short-read genomic data in an approximate Bayesian computation framework: in silico evaluation of power, biases and proof of concept in Atlantic walrus. Mol Ecol 24:328–345. https://doi.org/10.1111/mec.13034
Stansbury CR, Ausband DE, Zager P, Mack CM, Waits LP (2016) Identifying gray wolf packs and dispersers using noninvasive genetic samples. J Wildl Manag 80:1408–1419. https://doi.org/10.1002/jwmg.21136
Sundqvist L, Keenan K, Zackrisson M, Prodöhl P, Kleinhans D (2016) Directional genetic differentiation and relative migration. Ecol Evol 6:3461–3475. https://doi.org/10.1002/ece3.2096
Szewczyk M, Nowak S, Niedźwiecka N, Hulva P, Špinkytė-Bačkaitienė R, Demjanovičová K et al (2019) Dynamic range expansion leads to establishment of a new, genetically distinct wolf population in Central Europe. Sci Rep 9:1–16. https://doi.org/10.1038/s41598-019-55273-w
Trouwborst A (2010) Managing the carnivore comeback: international and EU species protection law and the return of lynx, wolf and bear to Western Europe. J Environ Law 22:347–372. https://doi.org/10.1093/jel/eqq013
Valiere N, Taberlet P (2000) Urine collected in the field as a source of DNA for species and individual identification. Mol Ecol 9:2150–2154. https://doi.org/10.1046/j.1365-294x.2000.11142.x
Van Oosterhout C, Hutchinson WFD, Wills DP, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Vilá C, Sundqvist AK, Flagshd A, Seddon J, Björnerfeldt S, Kojola I, Casulli A, Sand H, Wabakken P, Ellegren H (2003) Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proc R Soc Lond B Biol Sci 270:91–97. https://doi.org/10.1098/rspb.2002.2184
Wabakken P, Sand H, Kojola I, Zimmermann B, Arnemo JM, Pedersen HC, Liberg O (2007) Multistage, long-range natal dispersal by a global positioning system-collared scandinavian wolf. J Wildl Manag 71:1631–1634. https://doi.org/10.2193/2006-222
Wallendums P (2018) Helyzetjelentés. In: Jámbor L, Bíró G, Bors R, Dénes R, Földvári A, Kovács G, Kovács Z, Rung Á, Wallendums P (eds) Vadászévkönyv 2018. Országos Magyar Vadászkamara, Budapest, pp 133–138
Yan S, Bai C, Li Y, Hou J, Zhao Z, Han W (2013) Sex identification of dog by PCR based on the differences in the AMELX and AMELY genes. Anim Genet 44:606. https://doi.org/10.1111/age.12063
Acknowledgements
We are very grateful to associates of the Ranger Service of Bükk National Park Directorate, Foundation of Börzsöny, the Slovak Wildlife Society White Wilderness volunteers, Foundation for the Preservation of European Wildlife, Nagyvad Hunting Ltd. and the University of Veterinary Medicine Budapest, who participated in sample collection. We would also like to thank two anonymous reviewers, whose comments and suggestions helped to improve the manuscript.
Funding
Open access funding provided by Hungarian University of Agriculture and Life Sciences. This study was financed by the Bükki National Park Directorate (Grant no. BNPI: 8-34/1/2017; NAIK/2404-1/2017) and the Doctoral School of Animal Biotechnology and Animal Science of Hungarian University of Agriculture and Life Sciences.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Handling editor: J. Paul Grobler.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fehér, P., Frank, K., Gombkötő, P. et al. The origin and population genetics of wolves in the north Hungarian mountains. Mamm Biol 102, 1823–1833 (2022). https://doi.org/10.1007/s42991-022-00287-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42991-022-00287-7