
Abstract
(Dihydro)lipoamide dehydrogenase (LADH) deficiency is an autosomal recessive genetic metabolic disorder. It generally presents with an onset in the neonatal age and premature death. The clinical picture usually involves metabolic decompensation and lactic acidosis that lead to neurological, cardiological, and/or hepatological outcomes. Severity of the disease is due to the fact that LADH is a common E3 subunit to the pyruvate, alpha-ketoglutarate, alpha-ketoadipate, and branched-chain alpha-keto acid dehydrogenase complexes and is also part of the glycine cleavage system; hence, a loss in LADH activity adversely affects several central metabolic pathways simultaneously. The severe clinical manifestations, however, often do not parallel the LADH activity loss, which implies the existence of auxiliary pathological pathways; stimulated reactive oxygen species (ROS) production as well as dissociation from the relevant multienzyme complexes proved to be auxiliary exacerbating pathomechanisms for selected disease-causing LADH mutations. This review provides an overview on the therapeutic challenges of inherited metabolic diseases, structural and functional characteristics of the mitochondrial alpha-keto acid dehydrogenase complexes, molecular pathogenesis and structural basis of LADH deficiency, and relevant potential future medical perspectives.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Therapeutic challenges concerning inherited metabolic diseases
Inherited metabolic disorders may develop due to various genetic drivers including mutations in genes encoding metabolically relevant proteins–metabolic enzymes, transporters of metabolites or regulators of the former two. Loss of function of any such protein can lead to the accumulation of their substrates and depletion of their products disturbing metabolite levels that can manifest in a variety of symptoms. If the mutated protein is involved in basic catabolic pathways, even energy provision can be compromised resulting in energy deficit of the cells of either specific tissues or the entire body. Therefore, the severity of such diseases is quite variable and highly depends on the function of the affected protein. In the most severe cases, symptoms appear in early childhood and are often lethal. On the other hand, some cases are characterized by episodic onsets in response to triggers like diet, starvation, acute illnesses, and surgical interventions.
There is no generally accepted cure for most of the inherited diseases. Patients usually need to follow life-long dietary restrictions and supplementation with coenzyme precursors, while antioxidants may also be beneficial in certain cases. If a metabolic decompensation develops, appropriate medical intervention is inevitable, but that often targets symptoms only. Novel approaches to provide real long-term remedies for genetic diseases are gene therapy and enzyme replacement therapy (ERT), as they both aim at restoring the missing protein function. Gene therapy targets gene expression by a variety of mechanisms. Even though a single intervention might provide a life-long solution for monogenic disorders, gene therapy is highly expensive, a factor that might even seal the fate of the drug—despite being effective in the treatment of lipoprotein lipase deficiency (Gaudet et al. 2013), the (in)famous drug Glybera got discontinued due to its price. As of 2022, gene therapy is approved by both the FDA and EMA to treat inherited retinal dystrophy and spinal muscular dystrophy and by only EMA for adenosine deaminase deficiency (Mendell et al. 2021).
In the course of ERT, the wild-type copy of the dysfunctional protein is heterologously expressed, purified and then, administered to the patient parenterally. Internalization of the protein into cells or even distinct cell organelles is provoked by conjugating or fusing it to short peptide segments referred to as protein transduction domains (PTDs) or cell-penetrating peptides (CPPs). High purity—including complete lack of bacterial toxins, if bacterial expression was applied—is of utmost significance. Disadvantages of ERT derive from the fact that protein drugs generally exhibit a much shorter internal life-span (and also shelf-life) as compared to, e.g., DNA drugs; hence, the treatments need to be frequently and regularly repeated usually under medical supervision. There is also a quite high chance for provoking an immune response in the course of an ERT treatment. ERT has already been approved as a clinical treatment for selected metabolic diseases, like liposomal storage diseases (Desnick and Schuchman 2012). Based on promising results reported in the literature, ERT might also provide remedy for certain mitochondrial disorders in the near future (Lichtenstein et al. 2021). One of the first inherited mitochondrial diseases which were addressed via ERT is the deficiency of the (dihydro)lipoamide dehydrogenase (LADH). The laboratory of Prof. Loberboum-Galski at the Hebrew University of Jerusalem in Israel reported the first successful PDT-mediated delivery of the wild-type LADH into the mitochondria of fibroblasts derived from LADH-deficient probands (Rapoport et al. 2008) or murine model of the disease (Rapoport et al. 2011).
An additional—yet currently rather theoretical—approach is to target the mechanistic cause of the protein dysfunction by structure-based drug design. For this purpose, as a very first step, the wild-type protein and its disease-causing variant(s) need to be expressed, purified, and characterized both structurally and functionally. If the protein in question operates as a subunit of a multienzyme complex, pathogenic mutations might impair the interactions and/or proper substrate channeling with other subunits; in this case, the structures and interactions of all relevant subunits have to be also explored.
Our research group at the Semmelweis University has devoted the last decades to studying the α-ketoglutarate (or 2-oxoglutarate) dehydrogenase enzyme complex (KGDHc or OGDHc) with special focus on its LADH subunit and the molecular pathomechanism of the inherited deficiency of the human (h) LADH. In the present mini-review, we wish to guide the readers through recent structural and functional findings that, as we hope, one day might provide the basis for structure-based drug design against the rare but clinically severe hLADH deficiency.
Structural and functional characteristics of the mitochondrial α-keto acid dehydrogenase complexes
LADH is the common third subunit of the α-keto acid dehydrogenase complexes (Nemeria et al. 2018; Reed 1974; Yeaman 1989)—including the KGDHc, the pyruvate dehydrogenase complex (PDHc), the branched-chain α-keto acid dehydrogenase complex (BCKDHc), and the α-ketoadipate dehydrogenase complex (KADHc)—and also a constituent of the glycine cleavage system (GCS) (Kikuchi 1973); all the above complexes play key roles in metabolism and energy homeostasis. These complexes carry out oxidative decarboxylation on their corresponding α-keto acid substrates using specific E1 and E2 subunits (Kikuchi 1973; Reed 1974; Yeaman 1989), except for the common E2 in KGDHc and KADHc (Nemeria et al. 2018). The LADH (also referred to as the E3 subunit) oxidatively regenerates the reduced E2-bound lipoate cofactor by transferring its electrons to the NAD+ cofactor with the help of a tightly but non-covalently bound FAD prosthetic group (Scheme 1) (Massey 1960). This enables the complexes to enter a new catalytic cycle. The subunits in the GCS are designated differently, but the catalyzed reactions are analogous. Under pathological conditions, characterized here by a decreased NAD+/NADH ratio and/or mildly acidic pH, the KGDHc is capable of producing significant amounts of the reactive oxygen species (ROS) superoxide and, its spontaneous dismutation product, H2O2 by using molecular oxygen as the final electron acceptor. ROS formation can occur in both the forward and reverse catalytic directions, using α-ketoglutarate and NADH, respectively, as electron donors. The KGDHc was reported to be the principal ROS generator among the α-keto acid dehydrogenase complexes in vivo/in situ (Mailloux et al. 2016; Quinlan et al. 2014; Starkov et al. 2004), even though, for intact complexes at least, the radical formation has unequivocally been attributed to the common LADH subunit; the hPDHc produced rather significant amounts of ROS only when the complex was assembled from recombinant enzyme subunits and investigated in vitro (Ambrus et al. 2015a, b). This moonlighting activity of the KGDHc is considered to significantly contribute to the mitochondrial oxidative stress under pathologically relevant conditions (Bunik and Sievers 2002; Starkov et al. 2004; Tretter and Adam-Vizi 2004).

Forward, reverse, and ROS-generating reactions of LADH. LA, lipoic acid; DHLA, dihydrolipoic acid
The isolated LADH could also produce ROS, both superoxide and H2O2, in both the forward and reverse catalytic directions (Ambrus et al. 2009; Bando and Aki 1991; Gazaryan et al. 2002; Massey et al. 1969). The ROS-producing activity of the LADH in the reverse reaction showed pH sensitivity and could be antagonized by exogenous lipoic acid in vitro (Ambrus et al. 2009).
The rather elegant structures that support the highly coordinated operation of the subunits in each α-keto acid dehydrogenase complex have been studied for decades, and findings about their above-mentioned role in oxidative stress keep them in the spotlight. Multiple copies of the respective E2 subunits provide the core structures to which the corresponding E1 and E3 (LADH) subunits associate. Swinging arms of the E2 chains with lipoate cofactors attached to them (via Lys residues) connect the active sites of the different subunits/components in all these complexes (Reed 1974). Composition and chain stoichiometry varies not only among these cognate complexes, but also among species for the same complex. The core structures generally consist of 24 E2 subunits in an octahedral (cube-shaped, 8 × 3-type) arrangement/symmetry (see Fig. 1) (Nagy et al. 2021; Reed 1974). The eukaryotic PDHc represents an exception among the α-keto acid dehydrogenase complexes as it adopts a 60-meric pentagonal dodecahedron (20 × 3-type) assembly instead, where non-catalytic E3-binding protein (E3BP or protein X) subunits, specialized for tethering the LADH (Patel and Roche 1990; Reed 1974), are proposed to be either built in (replacing either 12 (Hiromasa et al. 2004) or 20 (Brautigam et al. 2009) copies of the E2 subunit) or coordinated (with 12 chains) (Smolle et al. 2006) on the 60-meric core. In the prokaryotic PDHc (Patel and Roche 1990) and BCKDHc (Yeaman 1989), the LADH subunits compete with the other peripheral (E1) subunits for binding to the E2 subunits (in a mutually exclusive manner), while in the mammalian KGDHc, LADH associates to the complex via the E1k (k for KGDHc) subunit (see Fig. 1) (McCartney et al. 1998; Zhou et al. 2018). In hLADH, the binding loci for the peripheral subunit-binding domains (PSBD) of the E3BP (Brautigam et al. 2006; Ciszak et al. 2006) and hE2b (b for BCKDHc) (Brautigam et al. 2011) overlap, according to co-crystallization studies. Recently, hydrogen-/deuterium-exchange mass spectrometry (HDX-MS) analysis revealed a distinct interaction hotspot for hE1k on hLADH (Zhou et al. 2018).

Structural topology of the hKGDHc. a Core structure of the hKGDHc. The 24-meric hE2 assembly adopts an 8 × 3-type (cubic) symmetry according to our cryo-EM results (PDB ID:6H05 (Nagy et al. 2021)). Flexible regions of the hE2 subunits could not experimentally be observed and therefore, are not shown here. Subunits are represented via color shades. b Model of the overall hKGDHc. E1 (pale pink, two shades represent the two monomers of a homodimer; homology model) and E3 (green, two shades represent the two monomers of a homodimer; PDB ID: 6I4Q (Szabo et al. 2019)) subunits associate with the edges and faces, respectively, of the cubic core (blue, three shades represent the three monomers of a homotrimer). The model was manually assembled in PyMOL based on results published by Nagy et al. (Nagy et al. 2021)
LADH is the subunit that has been the easiest to investigate owing to its moderate size and the lack of highly flexible domains—in contrast to the other subunits. LADH is a functional/obligate homodimer since both monomers contribute to the formation of the active sites that reside on the homodimer interface; one monomer carries the Cys45 and Cys50 residues that form a redox-active disulphide bridge (numbering refers to the human enzyme), while the other monomer provides the catalytic base, His452′ (′ indicates amino acids of the adjacent monomer) (Brautigam et al. 2005; Mattevi et al. 1991; Szabo et al. 2018, 2019). The proper conformation of His452′ is adjusted by the cis-peptide bond formed with Pro453′ and H-bonding with Glu457′ (Brautigam et al. 2005; Mattevi et al. 1991; Szabo et al. 2018, 2019); the latter residue also modulates the pKa of the catalytic base (Benen et al. 1992; Kim and Patel 1992). The two substrates bind on the two opposite faces of the isoalloxazine ring of the FAD prosthetic group: the lipoate substrate on the si, whereas NAD+/NADH on the re face (Brautigam et al. 2005; Mattevi et al. 1991). The lipoate substrate is proposed to approach the active site through a relatively narrow channel with a length of 10 Å (de Kok et al. 1993) that corresponds to the length of the lysine-bound lipoic acid (Reed 1974). De Kok et al. (1993) revealed that the lipoate-binding channel continues as an even longer and narrower channel all the way to the surface of the protein (de Kok and van Berkel 1996). This second leg of the channel was proposed to function as an outlet for H2O molecules (de Kok et al. 1993) and/or H+ (de Kok and van Berkel 1996) potentially indirectly playing role in the catalytic function (since H+ is generated in the course of the normal catalytic cycle). A recently published high-resolution structure of the hLADH revealed that the side chains of two residues, namely Glu332 and Arg460′, adopt alternative conformations and consequently modulate the geometry and polarity of the H+/H2O-channel, which in turn presumably affect the governance of the active site (Szabo et al. 2019). According to our very recent HDX-MS results, once bound, the lipoate substrate enters an interaction with a peptide from the H+/H2O channel, which also includes the flexible Glu332 (unpublished data). Hence, the previously defined boundaries of the lipoate and H+/H2O channels ought likely to be revisited.
Molecular pathogenesis of LADH deficiency
LADH deficiency is a rare genetic condition that develops due to pathogenic hLADH variants (Ambrus and Adam-Vizi 2018; Cameron et al. 2006; Quinonez et al. 2013; Quinonez and Thoene 2014). Hitherto, 14 disease-causing mutations of the hLADH protein have been reported in the clinical literature (Fig. 2) (Ambrus and Adam-Vizi 2018; Brassier et al. 2013; Cameron et al. 2006; Carrozzo et al. 2014; Cerna et al. 2001; Grafakou et al. 2003; Hong et al. 1996, 1997, 2003; Liu et al. 1993; Odievre et al. 2005; Quinonez et al. 2013; Quinonez and Thoene 2014; Quintana et al. 2010; Shaag et al. 1999; Shany et al. 1999). These amino acid substitutions all occur in functionally relevant regions of the enzyme—the active site or its close vicinity (G101del, P453L), the FAD-binding site (I12T, K37E), the NAD+/NADH-binding site (G194C, I318T, M326V, I358T), or the dimer interface (E340K, G426E, D444V, I445M, R447G, R460G).

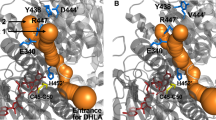
Overall structure of hLADH with the 14 disease-causing mutation sites. The two monomers of the hLADH homodimer are colored beige and gray. The active site disulphide bond, catalytic His and FAD are depicted with sticks. The two channels that lead to the active sites are computed with the Caver plug-in of PyMOL and depicted as spheres. Cα atoms of residues with known disease-causing mutations are indicated with spheres around one of the two active sites; variants whose high-resolution crystal structures have been determined (Szabo et al. 2018, 2019) are shown with red, while all the others are blue
The above disease-causing mutations lead to the dysfunction of the LADH-bearing multienzyme complexes via various independent mechanisms: (i) decreased provision of the pathogenic hLADH variant due to improper folding and/or stability, often also leading to accelerated degradation, (ii) reduced specific activity of hLADH, and (iii) impaired interactions among subunits/components. Western blot analyses revealed reduced hLADH protein levels in all the patient tissue samples investigated, except the one affected by the I445M mutation in homozygous form (Ambrus and Adam-Vizi 2018; Brassier et al. 2013; Cameron et al. 2006; Carrozzo et al. 2014; Cerna et al. 2001; Hong et al. 1996, 1997, 2003; Liu et al. 1993; Odievre et al. 2005; Quinonez and Thoene 2014; Quintana et al. 2010; Shaag et al. 1999; Shany et al. 1999) (protein expression was not assessed in two compound heterozygous patients (I318T/G101del, I358T/splice variant). Instability was so significant for the I12T-, G101del-, and M326V-hLADH variants that it even prevented implementing many of the in vitro studies. Most of the pathogenic mutations compromised the specific LADH activity relative to the wild-type hLADH to various degrees and usually in both catalytic directions, but the pattern did not display any direct correlation with the localizations of the substitutions (Ambrus et al. 2011; Babady et al. 2007; Brautigam et al. 2006; Kim 2012; Liu et al. 1995; Patel et al. 2009; Vaubel et al. 2011; Yuan et al. 2008; Yuan and Kim 2010, 2012). While the first two mechanisms, i.e., decrease in the amount and/or activity of hLADH, could theoretically affect all the cognate complexes evenly, mutations that (directly or indirectly) also impair/perturb residues involved in rather complex-specific inter-subunit interactions generally compromise the cognate complexes to different degrees, evident from residual overall complex activities in clinical samples (Brautigam et al. 2006, 2011; Zhou et al. 2018). For selected dimer interface mutants (E340K-, D444V-, R447G-, and R460G-hLADH), the K37E- and P453L-hLADH reduced affinities for the PDHc-specific E3BP were indeed reported (Brautigam et al. 2006; Patel et al. 2009); binding affinities toward other cognate subunits/components were not assessed.
Moreover, selected disease-causing mutations (P453L, G194C, E340K, and D444V) augmented the ROS-producing capacity of hLADH and its pH sensitivity, which may also exacerbate the clinical outcomes (Ambrus et al. 2011). Comprehensive in vitro analysis showed that the mutations affect the different activities of hLADH independently from each other (Ambrus et al. 2011). Representing the two extremities, the P453L substitution led to the complete loss of the physiological activity and turned hLADH into a pure ROS-producing enzyme, whereas the G194C substitution only enhanced the ROS-producing activity with no alteration in the normal LADH activity (Ambrus et al. 2011). Enhanced ROS production by the G194C mutation might explain the associated adult-onset form of LADH deficiency (Barak et al. 1998; Brassier et al. 2013; Cameron et al. 2006; Hong et al. 2003; Quinonez and Thoene 2014; Shaag et al. 1999), as the oxidative damages caused by ROS may accumulate over the lifetime.
For all the above reasons, the clinical pictures associated with the disease-causing LADH mutations are quite diverse (Ambrus and Adam-Vizi 2018; Cameron et al. 2006; Quinonez et al. 2013; Quinonez and Thoene 2014). The affected overall complexes determine which laboratory values turn abnormal, while the severity of the clinical symptoms is a function of the activity losses of these complexes; interestingly, the residual KGDHc activity correlates the best with the clinical severity (Cameron et al. 2006; Shany et al. 1999). In addition to the rather systemic metabolic decompensations (lactic acidosis, hypoglycemia, and hyperammonemia), tissues with the highest energy expenditures are principally affected. As a consequence, neurological (Cameron et al. 2006; Cerna et al. 2001; Grafakou et al. 2003; Hong et al. 1996, 1997; Odievre et al. 2005; Quinonez et al. 2013; Quinonez and Thoene 2014; Sakaguchi et al. 1986; Shaag et al. 1999; Shany et al. 1999) and cardiovascular (Odievre et al. 2005; Shany et al. 1999) symptoms are quite frequent, while hepatic complications might also develop (Barak et al. 1998; Brassier et al. 2013; Cameron et al. 2006; Hong et al. 2003; Quinonez and Thoene 2014; Shaag et al. 1999). Additionally, a myopathic phenotype of LADH deficiency with rather mild symptoms has recently been also reported (Carrozzo et al. 2014; Quintana et al. 2010).
Structural basis of LADH deficiency
Molecular pathomechanisms of the disease-causing hLADH mutants previously could only be interpreted in light of the crystal structure of hLADH (Brautigam et al. 2005). Later, analytical ultracentrifugation (Brautigam et al. 2006) and calibrated size-exclusion chromatography coupled to nanoLC-MS (Ambrus et al. 2011) revealed that, contrary to earlier proposals for the pathogenic dimer interface mutants, none of the investigated mutations cause the dissociation of the LADH homodimer. Circular dichroism (CD) spectroscopy showed no significant alterations in the folding or overall conformation of hLADH by any of the studied pathogenic mutations (Ambrus et al. 2011). Detailed structural results derive from HDX-MS (Ambrus et al. 2016) and X-ray crystallography (Szabo et al. 2018, 2019), two techniques that greatly complement one another; HDX-MS provides peptide-level structural information under physiologically relevant conditions, whereas X-ray crystallography yields atomic resolution data at cryogenic temperatures (usually 100 K). HDX-MS is currently used in our laboratory to characterize the substrate-binding and subunit-subunit interactions of the pathogenic hLADH variants.
The crystal structure of P453L-hLADH confirmed the earlier hypothesis (Brautigam et al. 2005), i.e., substitution of Pro453 with Leu indeed hinders the formation of the peptide bond with the catalytic base His452 in its characteristic cis configuration (Szabo et al. 2019). As a consequence, His452 turns to be incapable of forming a H-bond with the FAD prosthetic group and adopts a perturbed conformation in P453L-hLADH relative to hLADH (Fig. 3a). The extensive structural changes in the active site most likely prevent the lipoate substrate from binding and/or getting deprotonated by the catalytic base (Szabo et al. 2019). The enhanced superoxide production by P453L-hLADH could potentially be associated with the structural changes in the vicinity of the FAD isoalloxazine ring (Fig. 3a); the loss of specific interactions may also alter the redox potential of FAD (Szabo et al. 2019). A common feature of the G194C and G426E substitutions is that the newly incorporated side chain is polar (Glu426 is also protonatable). According to the relevant crystal structures, these new side chains both triggered only minor structural changes; they primarily altered the local charge distributions and side-chain dynamics in the vicinity of the NAD+/NADH-binding pocket (Szabo et al. 2019). The structural alterations associated with the P453L, G194C, and G426E mutations all correlate well with both the changes in the catalytic activities and severities of the disease phenotypes. On the other hand, the increased ROS-producing activity of the G194C-hLADH variant could only be explained by the HDX-MS results. The latter revealed augmented solvent accessibility/flexibility for selected peptides involved in FAD binding upon the mutation; one of these peptides also includes Lys54 (this same peptide was also perturbed in P453L-hLADH) (Ambrus et al. 2016). Certain structural alterations may not manifest at cryogenic temperatures; hence at times, complementary structure analysis methods are to be simultaneously exploited.

Structural changes induced by the disease-causing P453L and R460G mutations. a Structural changes in the active site induced by the P453L substitution are represented on the superimposed structures of the mutant (PDB ID: 6I4Z (Szabo et al. 2019); two shades of green) and wild-type hLADH (PDB ID: 6I4Q (Szabo et al. 2019); beige and gray). FAD prosthetic groups of the P453L-hLADH and hLADH are displayed as orange and gray sticks, respectively. b Changes in the H+/H2O-channel, hence accessibility of the active site, induced by the R460G substitution are displayed via computing the channels by the Caver plug-in of PyMOL in the aligned structures of the R460G-hLADH (PDB ID: 6I4R (Szabo et al. 2019); red mesh) and hLADH (PDB ID: 6I4Q (Szabo et al. 2019); pale green spheres). Residues in the mutant and wild-type structures are colored by two shades of blue and beige/gray, respectively; FAD prosthetic groups are displayed as orange and beige sticks, respectively
Molecular pathomechanisms of action of the disease-causing dimer interface variants of hLADH have been much more enigmatic than those of any other pathogenic hLADH mutants. The mutation sites are at considerable distances from the cofactor-binding regions as well as the active site, despite the fact that the latter is also at the homodimer interface. Nevertheless, all of these mutations led to significant losses in hLADH activity (Ambrus et al. 2011; Vaubel et al. 2011). Two of these mutations stimulated the ROS production by hLADH (Ambrus et al. 2011). The E340K, D444V, R447G, and R460G substitutions were reported to directly affect residues that form inter-monomeric interactions; hence, they were believed to cause the dissociation of the homodimer (Brautigam et al. 2005; Carrozzo et al. 2014; Shany et al. 1999). This hypothesis was later disproved by ultracentrifugation (Brautigam et al. 2006), calibrated size-exclusion chromatography coupled to nanoLC-MS (Ambrus et al. 2011), MD simulation (Ambrus and Adam-Vizi 2013; Ambrus et al. 2015a, b), HDX-MS (Ambrus et al. 2016), and X-ray crystallography (Szabo et al. 2018, 2019), as it was already mentioned above. Recently, all the disease-causing dimer interface variants as well as the I445M mutant got associated with the rather neglected H+/H2O-channel (Figs. 2, 3b). These mutations all perturb the channel-forming helices, while some also affect the conformation and/or flexibility of the Glu332 and/or Arg460′ residues, which are potentially important in catalysis (see above) (Szabo et al. 2018, 2019). These findings may well be relevant since no significant structural alterations could be found in the active site and the cofactor (FAD/NAD+/NADH)-binding regions in the crystal structures of the D444V-, R447G-, R460G-, and I445M-hLADH variants (crystal structure is not available for the E340K-hLADH, yet; Fig. 2) (Szabo et al. 2018, 2019). These results are also in accord with the HDX-MS findings which revealed altered dynamics/solvent accessibility for selected peptides in the H+/H2O-channel (e.g., His452-Phe468, Leu327-Gly344, and the C-terminus to a varying extent) upon these mutations (Ambrus et al. 2016). Based on all these data, we propose that the local as well as relayed structural alterations induced by these mutations may also perturb the side-chain pKa of the active site residue His452 and/or the redox potential of FAD via the dipole moments of the channel-forming helices which point toward the active site with their N-termini (Szabo et al. 2018, 2019).
Potential structure-based drug design strategies
The long-term goal of the in-depth structure-function analysis of the LADH-bearing complexes and the pathogenic hLADH variants is to establish structure-based drug design and potentially develop therapy for LADH deficiency. One potential strategy is to strengthen the inter-subunit interactions hence the assembly in the respective multienzyme complexes by adaptor molecules since dissociation of one or more of the α-keto acid dehydrogenase complexes is often implicated in human LADH deficiency (Brautigam et al. 2006; Patel et al. 2009). Molecular docking studies have already been initiated in our laboratory using commercially available ligand libraries against the crystal structure of the wild-type hLADH-E3BP subcomplex and the computed subcomplexes of the relevant pathogenic hLADH variants and the E3BP. Experiments could be also extended in the future to the hLADH-interacting subunits of the other cognate complexes; however, the availability of truly high-resolution structures currently limits these efforts.
Inhibition of the ROS generation by relevant pathogenic hLADH mutants (as well as the liberated hLADH in acidosis) would be another possibility for drug design. A selective drug candidate should potentially work only temporally and/or locally in acidosis (to protect the residual normal hLADH function between acute episodes): for instance, this molecule could be a pH-dependent drug candidate that would turn its inhibitory/binding potential on only in the course of acute episodes that involve a considerable load of lactic acidosis. Lipoic acid derivatives might be good initial molecular skeletons for such purposes since lipoic acid serves as a good substrate for hLADH only at lower pH where the pH optimum of the ROS generation by hLADH also lies, and hence, it exhibits a powerful inhibitory effect against ROS generation by hLADH (Ambrus et al. 2009); however, lipoic acid might not be as efficient or might not work at all against hLADH variants, a hypothesis to be tested.
Concluding remarks
Significant progress has recently been attained in the structural characterization of the pathogenic hLADH variants. The work is however far from completion. Structures of seven further disease-causing hLADH variants are yet to be determined (two of these structures are in fact already being refined and ought to be released/published soon). However, with all the available wild type and (disease-causing) mutant hLADH structures in hand, structure-based drug design has already become potentially feasible (against, e.g., ROS formation or dissociation from the alpha-keto acid dehydrogenase complexes).
Abbreviations
- KGDHc:
-
Alpha-ketoglutarate (also known as 2-oxoglutarate) dehydrogenase complex
- KG:
-
Alpha-ketoglutarate
- PDHc:
-
Pyruvate dehydrogenase complex
- BCKDHc:
-
Branched-chain α-keto acid dehydrogenase complex
- GCS:
-
Glycine cleavage system
- KADHc:
-
Alpha-ketoadipate dehydrogenase complex
E3: (Dihydro)lipoamide dehydrogenase, the common E3 component of the alpha-keto acid dehydrogenase complexes
- LADH:
-
(Dihydro)lipoamide dehydrogenase
- LA :
-
Lipoic acid
- DHLA:
-
Dihydrolipoic acid
- HDX-MS:
-
Hydrogen-/deuterium-exchange mass spectrometry
- FAD:
-
Flavin adenine dinucleotide
- NAD+/NADH:
-
Nicotinamide adenine dinucleotide (oxidized/reduced forms)
- ROS:
-
Reactive oxygen species (superoxide anion and hydrogen peroxide)
- wt:
-
Wild type
- h:
-
Human origin
- LC:
-
Liquid chromatography
- MD:
-
Molecular dynamics
References
Ambrus A, Adam-Vizi V (2013) Molecular dynamics study of the structural basis of dysfunction and the modulation of reactive oxygen species generation by pathogenic mutants of human dihydrolipoamide dehydrogenase. Arch Biochem Biophys 538(2):145–155. https://doi.org/10.1016/j.abb.2013.08.015
Ambrus A, Adam-Vizi V (2018) Human dihydrolipoamide dehydrogenase (E3) deficiency: novel insights into the structural basis and molecular pathomechanism. Neurochem Int 117:5–14. https://doi.org/10.1016/j.neuint.2017.05.018
Ambrus A, Tretter L, Adam-Vizi V (2009) Inhibition of the alpha-ketoglutarate dehydrogenase-mediated reactive oxygen species generation by lipoic acid. J Neurochem 109:222–229. https://doi.org/10.1111/j.1471-4159.2009.05942.x
Ambrus A, Torocsik B, Tretter L, Ozohanics O, Adam-Vizi V (2011) Stimulation of reactive oxygen species generation by disease-causing mutations of lipoamide dehydrogenase. Hum Mol Genet 20(15):2984–2995. https://doi.org/10.1093/hmg/ddr202
Ambrus A, Mizsei R, Adam-Vizi V (2015a) Structural alterations by five disease-causing mutations in the low-pH conformation of human dihydrolipoamide dehydrogenase (hLADH) analyzed by molecular dynamics – implications in functional loss and modulation of reactive oxygen species generation by pathogenic hLADH forms. Biochem Biophys Rep 2:50–56. https://doi.org/10.1016/j.bbrep.2015.04.006
Ambrus A, Nemeria NS, Torocsik B, Tretter L, Nilsson M, Jordan F, Adam-Vizi V (2015b) Formation of reactive oxygen species by human and bacterial pyruvate and 2-oxoglutarate dehydrogenase multienzyme complexes reconstituted from recombinant components. Free Radic Biol Med 89:642–650. https://doi.org/10.1016/j.freeradbiomed.2015.10.001
Ambrus A, Wang JJ, Mizsei R, Zambo Z, Torocsik B, Jordan F, Adam-Vizi V (2016) Structural alterations induced by ten disease-causing mutations of human dihydrolipoamide dehydrogenase analyzed by hydrogen/deuterium-exchange mass spectrometry: implications for the structural basis of E3 deficiency. BBA-Mol Basis Dis 1862(11):2098–2109. https://doi.org/10.1016/j.bbadis.2016.08.013
Babady NE, Pang YP, Elpeleg O, Isaya G (2007) Cryptic proteolytic activity of dihydrolipoamide dehydrogenase. Proc Natl Acad Sci USA 104(15):6158–6163. https://doi.org/10.1073/pnas.061018104
Bando Y, Aki K (1991) Mechanisms of generation of oxygen radicals and reductive mobilization of ferritin iron by lipoamide dehydrogenase. J Biochem 109(3):450–454. https://doi.org/10.1093/oxfordjournals.jbchem.a123
Barak N, Huminer D, Segal T, Ben Ari Z, Halevy J, Kaspa RT (1998) Lipoamide dehydrogenase deficiency: a newly discovered cause of acute hepatitis in adults. J Hepatol 29(3):482–484. https://doi.org/10.1016/s0168-8278(98)80069-x
Benen J, van Berkel W, Dieteren N, Arscott D, Williams C Jr, Veeger C, de Kok A (1992) Lipoamide dehydrogenase from Azotobacter vinelandii: site-directed mutagenesis of the His450-Glu455 diad. Kinetics of wild-type and mutated enzymes. Eur J Biochem 207(2):487–497. https://doi.org/10.1111/j.1432-1033.1992.tb17075.x
Brassier A, Ottolenghi C, Boutron A, Bertrand AM, Valmary-Degano S, Cervoni JP, Chretien D, Arnoux JB, Hubert L, Rabier D, Lacaille F, de Keyzer Y, Di Martino V, de Lonlay P (2013) Dihydrolipoamide dehydrogenase deficiency: a still overlooked cause of recurrent acute liver failure and Reye-like syndrome. Mol Genet Metab 109(1):28–32. https://doi.org/10.1016/j.ymgme.2013.01.017
Brautigam CA, Chuang JL, Tomchick DR, Machius M, Chuang DT (2005) Crystal structure of human dihydrolipoamide dehydrogenase: NAD(+)/NADH binding and the structural basis of disease-causing mutations. J Mol Biol 350(3):543–552. https://doi.org/10.1016/j.jmb.2005.05.014
Brautigam CA, Wynn RM, Chuang JL, Machius M, Tomchick DR, Chuang DT (2006) Structural insight into interactions between dihydrolipoamide dehydrogenase (E3) and E3 binding protein of human pyruvate dehydrogenase complex. Structure 14(3):611–621. https://doi.org/10.1016/j.str.2006.01.001
Brautigam CA, Wynn RM, Chuang JL, Chuang DT (2009) Subunit and catalytic component stoichiometries of an in vitro reconstituted human pyruvate dehydrogenase complex. J Biol Chem 284(19):13086–13098. https://doi.org/10.1074/jbc.M806563200
Brautigam CA, Wynn RM, Chuang JL, Naik MT, Young BB, Huang TH, Chuang DT (2011) Structural and thermodynamic basis for weak interactions between dihydrolipoamide dehydrogenase and subunit-binding domain of the branched-chain alpha-ketoacid dehydrogenase complex. J Biol Chem 286(26):23476–23488. https://doi.org/10.1074/jbc.M110.202960
Bunik VI, Sievers C (2002) Inactivation of the 2-oxo acid dehydrogenase complexes upon generation of intrinsic radical species. Eur J Biochem 269(20):5004–5015. https://doi.org/10.1046/j.1432-1033.2002.03204.x
Cameron JM, Levandovskiy V, MacKay N, Raiman J, Renaud DL, Clarke JTR, Feigenbaum A, Elpeleg O, Robinson BH (2006) Novel mutations in dihydrolipoamide dehydrogenase deficiency in two cousins with borderline-normal PDH complex activity. Am J Med Genet A 140(14):1542–1552. https://doi.org/10.1002/ajmg.a.31313
Carrozzo R, Torraco A, Fiermonte G, Martinelli D, Di Nottia M, Rizza T, Vozza A, Verrigni D, Diodato D, Parisi G, Maiorana A, Rizzo C, Pierri CL, Zucano S, Piemonte F, Bertini E, Dionisi-Vici C (2014) Riboflavin responsive mitochondrial myopathy is a new phenotype of dihydrolipoamide dehydrogenase deficiency. The chaperon-like effect of vitamin B2. Mitochondrion 18:49–57. https://doi.org/10.1016/j.mito.2014.09.006
Cerna L, Wenchich L, Hansiková H, Kmoch S, Peskova K, Chrastina P, Brynda J, Zeman J (2001) Novel mutations in a boy with dihydrolipoamide dehydrogenase deficiency. Med Sci Monit 7(6):1319–1325
Ciszak EM, Makal A, Hong YS, Vettaikkorumakankauv AK, Korotchkina LG, Patel MS (2006) How dihydrolipoamide dehydrogenase-binding protein binds dihydrolipoamide dehydrogenase in the human pyruvate dehydrogenase complex. J Biol Chem 281(1):648–655. https://doi.org/10.1074/jbc.M507850200
de Kok A, van Berkel WJH (1996) Lipoamide dehydrogenase. In: Patel MS, Roche TE, Harris RA (eds) Alpha-keto acid dehydrogenase complexes. Birkhäuser, Basel, pp 53–70
de Kok A, Berg A, van Berkel W, Fabisz-Kijowska A, Westphal A, van den Akker F, Mattevi A, Hol WGJ (1993) The pyruvate dehydrogenase complex from Azotobacter vinelandii. In: Yagi K (ed) Flavins and flavoproteins. Walter de Gruyter, Nagoya, Japan, pp 535–544
Desnick RJ, Schuchman EH (2012) Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genom Hum Genet 13:307–335. https://doi.org/10.1146/annurev-genom-090711-163739
Gaudet D, Méthot J, Déry S, Brisson D, Essiembre C, Tremblay G, Tremblay K, de Wal J, Twisk J, van den Bulk N, Sier-Ferreira V, van Deventer S (2013) Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther 20(4):361–369. https://doi.org/10.1038/gt.2012.43
Gazaryan IG, Krasnikov BF, Ashby GA, Thorneley RNF, Kristal BS, Brown AM (2002) Zinc is a potent inhibitor of thiol oxidoreductase activity and stimulates reactive oxygen species production by lipoamide dehydrogenase. J Biol Chem 277(12):10064–10072. https://doi.org/10.1074/jbc.M108264200
Grafakou O, Oexle K, van den Heuvel L, Smeets R, Trijbels F, Goebel HH, Bosshard N, Superti-Furga A, Steinmann B, Smeitink J (2003) Leigh syndrome due to compound heterozygosity of dihydrolipoamide dehydrogenase gene mutations. Description of the first E3 splice site mutation. Eur J Pediatr 162(10):714–718. https://doi.org/10.1007/s00431-003-1282-z
Hiromasa Y, Fujisawa T, Aso Y, Roche TE (2004) Organization of the cores of the mammalian pyruvate dehydrogenase complex formed by E2 and E2 plus the E3-binding protein and their capacities to bind the E1 and E3 components. J Biol Chem 279(8):6921–6933. https://doi.org/10.1074/jbc.M308172200
Hong YS, Kerr DS, Craigen WJ, Tan J, Pan YZ, Lusk M, Patel MS (1996) Identification of two mutations in a compound heterozygous child with dihydrolipoamide dehydrogenase deficiency. Hum Mol Genet 5(12):1925–1930. https://doi.org/10.1093/hmg/5.12.1925
Hong YS, Kerr DS, Liu TC, Lusk M, Powell BR, Patel MS (1997) Deficiency of dihydrolipoamide dehydrogenase due to two mutant alleles (E340K and G101del) - analysis of a family and prenatal testing. BBA-Mol Basis Dis 1362(2–3):160–168. https://doi.org/10.1016/S0925-4439(97)00073-2
Hong YS, Korman SH, Lee J, Ghoshal P, Qu Q, Barash V, Kang S, Oh S, Kwon M, Gutman A, Rachmel A, Patel MS (2003) Identification of a common mutation (Gly194Cys) in both Arab Moslem and Ashkenazi Jewish patients with dihydrolipoamide dehydrogenase (E3) deficiency: possible beneficial effect of vitamin therapy. J Inherit Metab Dis 26(8):816–818. https://doi.org/10.1023/b:boli.0000010004.12053.5b
Kikuchi G (1973) The glycine cleavage system: composition, reaction mechanism, and physiological significance. Mol Cell Biochem 1(2):169–187. https://doi.org/10.1007/BF01659328
Kim H (2012) Characterization of two naturally occurring mutations (Gly-101 Deletion and Glu-340 to Lys Substitution) in human dihydrolipoamide dehydrogenase of a patient with metabolic acidosis. Bull Korean Chem Soc 33(8):2477–2478. https://doi.org/10.5012/bkcs.2012.33.8.2477
Kim H, Patel MS (1992) Characterization of 2 site specifically mutated human dihydrolipoamide dehydrogenases (His-452-Gln and Glu-457-Gln). J Biol Chem 267(8):5128–5132. https://doi.org/10.0000/jbc.267/8/5128
Lichtenstein M, Zabit S, Hauser N, Farouz S, Melloul O, Hirbawi J, Lorberboum-Galski H (2021) TAT for enzyme/protein delivery to restore or destroy cell activity in human diseases. Life (basel). 11(9):924. https://doi.org/10.3390/life11090924
Liu TC, Kim H, Arizmendi C, Kitano A, Patel MS (1993) Identification of two missense mutations in a dihydrolipoamide dehydrogenase-deficient patient. Proc Natl Acad Sci USA 90(11):5186–5190. https://doi.org/10.1073/pnas.90.11.5186
Liu TC, Korotchkina LG, Hyatt SL, Vettakkorumakankav NN, Patel MS (1995) Spectroscopic studies of the characterization of recombinant human dihydrolipoamide dehydrogenase and its side-directed mutants. J Biol Chem 270(26):15545–15550. https://doi.org/10.1074/jbc.270.26.15545
Mailloux RJ, Gardiner D, O’Brien M (2016) 2-Oxoglutarate dehydrogenase is a more significant source of O2.-/H2O2 than pyruvate dehydrogenase in cardiac and liver tissue. Free Radic Biol Med 97:501–512. https://doi.org/10.1016/j.freeradbiomed.2016.06.014
Massey V (1960) The composition of the ketoglutarate dehydrogenase complex. Biochem Biophys Acta 38:447–460. https://doi.org/10.1016/0006-3002(60)91280-4
Massey V, Strickland S, Mayhew SG, Howell LG, Engel PC, Matthews RG, Schuman M, Sullivan PA (1969) The production of superoxide anion radicals in the reaction of reduced flavins and flavoproteins with molecular oxygen. Biochem Biophys Res Commun 36(6):891–897. https://doi.org/10.1016/0006-291x(69)90287-3
Mattevi A, Schierbeek AJ, Hol WG (1991) Refined crystal structure of lipoamide dehydrogenase from Azotobacter vinelandii at 2.2 Å resolution. A comparison with the structure of glutathione reductase. J Mol Biol 220(4):975–994. https://doi.org/10.1016/0022-2836(91)90367-F
McCartney RG, Rice JE, Sanderson SJ, Bunik V, Lindsay H, Lindsay JG (1998) Subunit interactions in the mammalian alpha-ketoglutarate dehydrogenase complex—evidence for direct association of the alpha-ketoglutarate dehydrogenase and dihydrolipoamide dehydrogenase components. J Biol Chem 273(37):24158–24164. https://doi.org/10.1074/jbc.273.37.24158
Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, Boye SL, Boye SE, George LA, Salabarria S, Corti M, Byrne BJ, Tremblay JP (2021) Current clinical applications of in Vivo gene therapy with AAVs. Mol Ther 29(2):464–488. https://doi.org/10.1016/j.ymthe.2020.12.007
Nagy B, Polak M, Ozohanics O, Zambo Z, Szabo E, Hubert A, Jordan F, Novaček J, Adam-Vizi V, Ambrus A (2021) Structure of the dihydrolipoamide succinyltransferase (E2) component of the human alpha-ketoglutarate dehydrogenase complex (hKGDHc) revealed by cryo-EM and cross-linking mass spectrometry: implications for the overall hKGDHc structure. Biochim Biophys Acta Gen Subj 1865(6):129889. https://doi.org/10.1016/j.bbagen.2021.129889
Nemeria NS, Gerfen G, Nareddy PR, Yang L, Zhang X, Szostak M, Jordan F (2018) The mitochondrial 2-oxoadipate and 2-oxoglutarate dehydrogenase complexes share their E2 and E3 components for their function and both generate reactive oxygen species. Free Radical Biol Med 115:136–145. https://doi.org/10.1016/j.freeradbiomed.2017.11.018
Odievre MH, Chretien D, Munnich A, Robinson BH, Dumoulin R, Masmoudi S, Kadhom N, Rötig A, Rustin P, Bonnefont JP (2005) A novel mutation in the dihydrolipoamide dehydrogenase E3 subunit gene (DLD) resulting in an atypical form of alpha-ketoglutarate dehydrogenase deficiency. Hum Mutat 25(3):323–324. https://doi.org/10.1002/humu.9319
Patel MS, Roche TE (1990) Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J 4(14):3224–3233. https://doi.org/10.1096/fasebj.4.14.2227213
Patel MS, Korotchkina LG, Sidhu S (2009) Interaction of E1 and E3 components with the core proteins of the human pyruvate dehydrogenase complex. J Mol Catal B Enzym 61(1–2):2–6. https://doi.org/10.1016/j.molcatb.2009.05.001
Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD (2014) The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. J Biol Chem 289(12):8312–8325. https://doi.org/10.1074/jbc.M113.545301
Quinonez SC, Leber SM, Martin DM, Thoene JG, Bedoyan JK (2013) Leigh syndrome in a girl with a novel DLD mutation causing E3 deficiency. Pediatr Neurol 48(1):67–72. https://doi.org/10.1016/j.pediatrneurol.2012.09.013
Quinonez SC, Thoene JG (2014) Dihydrolipoamide Dehydrogenase Deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (Eds) GeneReviews® 1993–2020. pp 1–37, University of Washington, Seattle. Retrieved from https://www.ncbi.nlm.nih.gov/books/NBK220444/
Quintana E, Pineda M, Font A, Vilaseca MA, Tort F, Ribes A, Briones P (2010) Dihydrolipoamide dehydrogenase (DLD) deficiency in a Spanish patient with myopathic presentation due to a new mutation in the interface domain. J Inherit Metab Dis 33:S315–S319. https://doi.org/10.1007/s10545-010-9169-4
Rapoport M, Saada A, Elpeleg O, Lorberboum-Galski H (2008) TAT-mediated delivery of LAD restores pyruvate dehydrogenase complex activity in the mitochondria of patients with LAD deficiency. Mol Ther 16(4):691–697. https://doi.org/10.1038/mt.2008.4
Rapoport M, Salman L, Sabag O, Patel MS, Lorberboum-Galski H (2011) Successful TAT-mediated enzyme replacement therapy in a mouse model of mitochondrial E3 deficiency. J Mol Med 89(2):161–170. https://doi.org/10.1007/s00109-010-0693-3
Reed LJ (1974) Multienzyme complexes. Acc Chem Res 7:40–46. https://doi.org/10.1021/ar50074a002
Sakaguchi Y, Yoshino M, Aramaki S, Yoshida I, Yamashita F, Kuhara T, Matsumoto I, Hayashi T (1986) Dihydrolipoyl dehydrogenase deficiency—a therapeutic trial with branched-chain amino acid restriction. Eur J Pediatr 145(4):271–274. https://doi.org/10.1007/BF00439399
Shaag A, Saada A, Berger I, Mandel H, Joseph A, Feigenbaum A, Elpeleg ON (1999) Molecular basis of lipoamide dehydrogenase deficiency in Ashkenazi Jews. Am J Med Genet 82(2):177–182. https://doi.org/10.1002/(SICI)1096-8628(19990115)82:2%3c177::AID-AJMG15%3e3.0.CO;2-9
Shany E, Saada A, Landau D, Shaag A, Hershkovitz E, Elpeleg ON (1999) Lipoamide dehydrogenase deficiency due to a novel mutation in the interface domain. Biochem Biophys Res Commun 262(1):163–166. https://doi.org/10.1006/bbrc.1999.1133
Smolle M, Prior AE, Brown AE, Cooper A, Byron O, Lindsay JG (2006) A new level of architectural complexity in the human pyruvate dehydrogenase complex. J Biol Chem 281(28):19772–19780. https://doi.org/10.1074/jbc.M601140200
Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF (2004) Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci 24(36):7779–7788. https://doi.org/10.1523/JNEUROSCI.1899-04.2004
Szabo E, Mizsei R, Wilk P, Zambo Z, Torocsik B, Weiss MS, Adam-Vizi V, Ambrus A (2018) Crystal structures of the disease-causing D444V mutant and the relevant wild type human dihydrolipoamide dehydrogenase. Free Radic Biol Med 124:214–220. https://doi.org/10.1016/j.freeradbiomed.2018.06.008
Szabo E, Wilk P, Nagy B, Zambo Z, Bui D, Weichsel A, Arjunan P, Torocsik B, Hubert A, Furey W, Montfort WR, Jordan F, Weiss MS, Adam-Vizi V, Ambrus A (2019) Underlying molecular alterations in human dihydrolipoamide dehydrogenase deficiency revealed by structural analyses of disease-causing enzyme variants. Hum Mol Genet 28(20):3339–3354. https://doi.org/10.1093/hmg/ddz177
Tretter L, Adam-Vizi V (2004) Generation of reactive oxygen species in the reaction catalyzed by alpha-ketoglutarate dehydrogenase. J Neurosci 24(36):7771–7778. https://doi.org/10.1523/JNEUROSCI.1842-04.2004
Vaubel RA, Rustin P, Isaya G (2011) Mutations in the dimer interface of dihydrolipoamide dehydrogenase promote site-specific oxidative damages in yeast and human cells. J Biol Chem 286(46):40232–40245. https://doi.org/10.1074/jbc.M111.274415
Yeaman SJ (1989) The 2-oxo acid dehydrogenase complexes: recent advances. Biochem J 257(3):625–632. https://doi.org/10.1042/bj2570625
Yuan L, Kim H (2010) Characterization of a naturally occurring mutation (Arg-460 to Gly) close to FAD in human dihydrolipoamide dehydrogenase. Bull Korean Chem Soc 31(12):3511–3512. https://doi.org/10.5012/bkcs.2010.31.12.3511
Yuan L, Kim H (2012) Characterization of a naturally occurring mutation (Ile-358 to Thr) in human dihydrolipoamide dehydrogenase of a patient with leigh syndrome. Bull Korean Chem Soc 33(5):1445–1446. https://doi.org/10.5012/bkcs.2012.33.5.1445
Yuan L, Cho YJ, Kim H (2008) Characterization of two naturally occurring mutations close to cofactors in human dihydrolipoamide dehydrogenase. Bull Korean Chem Soc 29(12):2327–2328. https://doi.org/10.5012/bkcs.2008.29.12.2327
Zhou J, Yang L, Ozohanics O, Zhang X, Wang J, Ambrus A, Arjunan P, Brukh R, Nemeria NS, Furey W, Jordan F (2018) A multipronged approach unravels unprecedented protein-protein interactions in the human 2-oxoglutarate dehydrogenase multienzyme complex. J Biol Chem 293(50):19213–19227. https://doi.org/10.1074/jbc.RA118.005432
Funding
Open access funding provided by Semmelweis University. This research was funded by the Hungarian Brain Research Program 2 (2017-1.2.1-NKP-2017-00002 grant, to Vera Adam-Vizi, Semmelweis University), Semmelweis University (STIA-OTKA-2021 grant, to A.A.), Hungarian Scientific Research Fund (OTKA grant 143627, to A.A.) and Ministry of Innovation and Technology of Hungary (TKP2021-EGA-25 grant, to A.A.). Project no. TKP2021-EGA-25 has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-EGA funding scheme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Szabó, E., Ambrus, A. Lipoamide dehydrogenase (LADH) deficiency: medical perspectives of the structural and functional characterization of LADH and its pathogenic variants. BIOLOGIA FUTURA 74, 109–118 (2023). https://doi.org/10.1007/s42977-023-00155-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42977-023-00155-6