
Abstract
While the existence of post-transcriptional modifications of RNA nucleotides has been known for decades, in most RNA species the exact positions of these modifications and their physiological function have been elusive until recently. Technological advances, such as high-throughput next-generation sequencing (NGS) methods and nanopore-based mapping technologies, have made it possible to map the position of these modifications with single nucleotide accuracy, and genetic screens have uncovered the “writer”, “reader” and “eraser” proteins that help to install, interpret and remove such modifications, respectively. These discoveries led to intensive research programmes with the aim of uncovering the roles of these modifications during diverse biological processes. In this review, we assess novel discoveries related to the role of post-transcriptional modifications during animal development, highlighting how these discoveries can affect multiple aspects of development from fertilization to differentiation in many species.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Orderly embryonic development constantly gives rise to novel cell lineages, which will ultimately lead to the appearance of numerous differentiated cell types organized in tissues and organs by the end of the developmental process. The narrowing of the developmental potential and finally the switch from proliferation to (final) differentiation in every lineage is accompanied by profound changes in the gene expression programme. Multiple levels of regulation have evolved to fine-tune the expression, stability and activity of gene products (proteins and non-coding RNAs), and the coordinated research programmes established to discern these distinct regulatory levels have led to the emergence (and proliferation) of the “omics” fields (Hasin et al. 2017).
Epitranscriptomics, the study of the biochemical modifications occurring in RNA molecules, is a relative newcomer to the family of “omics” sciences, partly due to the technological difficulties of the reliable detection of these modifications. Yet, the expansion of the biochemical toolbox coupled with the latest developments in next-generation sequencing (NGS) technologies and RNA mass-spectrometry has made it possible to study the dynamics of post-transcriptional modifications in previously unprecedented detail.
The functions of the epitranscriptomic modifications of the RNA are analogous with the well characterized epigenetic changes described in the DNA, and the number of known chemically modified nucleosides is ever increasing (about 160 at the time of writing this review, for details see: (McCown et al. 2020; Boccaletto et al. 2021). For most of these modifications, however, we have only a cursory understanding of their occurrence in living systems, whether they are actively incorporated and/or removed. Not surprisingly, therefore, the physiological relevance has been clearly demonstrated for only a handful of them.
Most epitranscriptomic modifications change the secondary structure of the transcripts and/or their stability; therefore, they will affect their interactions with other RNAs and proteins, resulting in downstream effects in the gene expression programme (Gilbert et al. 2016; Bartee et al. 2022).
The discovery of novel factors catalysing post-transcriptional modifications and the development of high-throughput detection methods that ought to detect dynamic changes in these modifications have led to an explosion of studies addressing the biological relevance of these modified nucleotides. In this mini-review, we will focus mainly on four abundant natural RNA modifications (N6-methyladenosine–m6A, 5-methylcytidine–m5C, inosine–I, pseudouridine–Ψ) that can be detected relatively robustly and accurately in the transcriptome and also discuss four sparser (N1-methyladenosine–m1A, N4-acetylcytidine–ac4C, 1-methylguanosine–m1G, N7-methylguanosine–m7G) modifications that seem to be important during animal development. As an organizing principle, we will focus on three characteristic developmental processes (maternal-zygotic transition–MZT, hematopoiesis and neural development) where the availability of multiple, high-quality datasets will allow us to compare the roles of these modifications in particular model organisms (Fig. 1).

A summary of the main post-transcriptional modifications discussed in this review. The four “standard” nucleotides are present in the middle of the figure, with the RNA modifications discussed in this review represented in an outer ring. Modifying (“writer” and “eraser”) enzymes and “reader” proteins, where known, are listed in the arrows (the direction of the arrow showing the direction of the modification). Not every developmental process is influenced by every modification and it is not always clear which particular RNA-type is responsible in particular developmental processes. The outer ring thus is just a graphical represnetation of the developmental processed that have been connected with a particlar epitranscriptomic modification
Enzymes involved in editing the epitranscriptome
In general, for programmable (and likely biologically relevant) post-transcriptional modifications, we expect to identify “writer” enzymes that can deposit them under certain conditions, “reader” proteins that can decode their presence directly (through the detection of the modified nucleotides) or changes in the RNA structure that signal their presence, and potentially “eraser” enzymes that can remove them from the transcripts (Kan et al. 2021; Zaccara et al. 2019; Wiener and Schwartz 2021). Of note, while the reversibility of epigenetic modifications is widely accepted, it is still hotly contested if epitranscriptomic modifications are themselves reversible, or indeed whether it is necessary to actively reverse them given the (relatively) short half-life of the RNA transcripts (Zaccara et al. 2019).
Bona fide erasers have been identified for N6-methyladenosine (m6A), while for other modifications, even those abundant in transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), similar enzymes have not been found (Wiener and Schwartz 2021). Indeed, some even hint that the case of m6A might be the exception and not the norm for epitranscriptomic modifications (Penning et al. 2022).
N6-methyladenosine is also the most widely studied post-transcriptional modification to date, somewhat related to the fact that besides a couple of “erasers” (such as the demethylases FTO and ALKBH5) a larger set of related “writers” and “readers” have been identified. The methyltransferases METTL3-METTL14 form the core of the m6A methyltransferase complex that also includes Wilms’ tumour 1-associating protein (WTAP), VIRMA, the E3 ubiquitin ligase HAKAI, the RNA-binding motif protein 15 (RBM15), the zinc-finger protein ZC3H13, Bcl-2-associated transcription factor 1 (BCLAF1) and thyroid hormone receptor-associated protein 3 (THRAP3) (for a comprehensive review see: (Zhang et al. 2021b). The m6A modifications in the transcriptome are recognized by YTH domain family proteins and IGF2BP1-3 and can regulate functions as diverse as RNA splicing, export, stability and translation (the role of HNRNP proteins as m6A readers is also supported by some evidence but currently is more controversial) (Yang et al. 2018).
While no erasers have been found up to date, some “writers” and some “readers” have been identified for 5-methylcytidine (m5C) as well. RNA methyltransferases of m5C belong to the Nop2/SUN (NSUN) domain family but the DNA methyltransferase (DNMT) homologue DNMT2 can also use mediate cytosine methylation of tRNAs, just as the tRNA-specific methyltransferase (TRDMT) family members (Yang et al. 2017; Heissenberger et al. 2019; Song et al. 2022). Readers of m5C include the mRNA export adaptor ALYREF and the human Y box binding protein 1 (YBX1), respectively (Yang et al. 2017, 2019; Zou et al. 2020).
In contrast, for the first discovered and most abundant RNA modification, pseudouridylation, identified seven decades ago (Cohn and Volkin 1951), no reader or eraser enzymes have been found to date. One plausible reason for the absence of “erasers” is that the C–C bond formed between the base and sugar in pseudouridine (Ψ), the C-glycoside isomer of uridine (U), is stronger than the C-N bond in uridine and therefore its formation may be irreversible. The current consensus is that the readout of Ψ may occur through a structural change in RNA based on its different biochemical properties compared to uridine (Spenkuch et al., 2014).
In humans, we know about 13 Ψ “writer” enzymes, called pseudouridine-synthases (PUSs). Although there are very few sequence similarities between them, all these enzymes share the same structural features in their catalytic domains and thus have the same catalytic activity (Purchal et al. 2022). PUSs can be classified into two classes based on their mode of action: RNA-independent or RNA-dependent. The first group includes enzymes that can catalyse the isomerization in a site-specific manner through direct RNA sequence/structure recognition. A broad set of PUSs can modify tRNAs, thereby affecting its stability, functionality, and metabolism (Levi and Arava 2021). For example, PUS1 was originally known to modify tRNAs and deleterious mutations in the coding gene are associated with mitochondrial myopathy, lactic acidosis and sideroblastic anaemia 1 (MLASA1, OMIM: 600,462), but recent data suggest that PUS1 directly recognises a specific sequence motif and perform modification in human mRNA too (Schwartz et al. 2014; Li et al. 2015b).
Pseudouridine is especially abundant in rRNA, being one of the most common epitranscriptomic modification in this RNA species besides acetylation and methylation. In rRNA, Ψ sites are established by RNA-dependent PUSs, which requires small nucleolar RNAs (snoRNAs) to direct it to the appropriate target RNAs. In humans, the dominant RNA-dependent PUS is dyskerin, encoded by DKC1. It can associate with hundreds of box H/ACA snoRNAs to generate a variety of functionally distinct ribonucleoproteins (RNPs) that catalyse the pseudouridylation of specific RNA residues. The primary target molecule of these RNPs is rRNA, with multiple evolutionarily highly conserved Ψ sites (Schwartz et al. 2014). rRNA is important for translational fidelity, fine-tunes ribosome affinity for highly structured RNAs (Eyler et al. 2019) and is also required for the assembly of the preinitiation translation complex on cap-independent translation pathway (Yoon et al. 2006). Alterations in the pseudouridylation patterns of rRNA have been widely reported in cancer and other ribosome-deficient conditions (reviewed in (Kampen et al. 2020)).
It is worth noting that changes can be stacked on top of each other, especially in the case of uridine, the most variably modified nucleoside (McCown et al. 2020). This can lead to the formation of hypermodified bases, such as the pseudouridine-based 1-methyl-pseudouridine (m1Ψ) and 1-methyl-3-α-amino-α-carboxyl-propyl pseudouridine (m1acp3Ψ), the latter being by far one of the most complex of all the epitranscriptomic modifications (Piekna-Przybylska et al. 2008). Ribosomal m1acp3Ψ is highly conserved, located in the decoding centre of the ribosome (18S:1248.U) where it is created by the concerted action of the EMG1 methyltransferase and the TSR3 aminocarboxyl propyl transferase (McCown et al. 2020).
While the exact biological function of m1acp3Ψ is poorly understood, changes in its levels affect translation and are an emerging hallmark of cancer (Babaian et al. 2020; Tan et al. 2021). Of note, while no human diseases have been associated with TSR3 mutations, the Bowen-Conradi syndrome (BWCNS, OMIM: 211180) is associated with homozygous EMG1 mutations (Armistead et al. 2009). This early lethal syndrome has been associated with numerous developmental skeletal abnormalities and Emg1 loss-of-function causes pre-implantation lethal phenotypes in mice (Wu et al. 2010); however, it is not clear if these are due to the defects in the EMG1 methyltransferase activity (Meyer et al. 2011).
“RNA editing”, the conversion of adenine (A) to I (A-to-I) was the first post-transcriptional modification that received wider attention. This modification is catalysed by adenosine deaminases acting on RNA (ADARs), specialized RNA editing enzymes that act on double-stranded regions of nuclear-encoded RNAs (dsRNAs) (Bass 2002; Savva et al. 2012). Deamination of A-to-I acts essentially as a mark for “self” RNA and is important in preventing the abnormal activation of the innate antiviral responses usually triggered by dsRNAs (Quin et al. 2021; O’Connell et al. 2015). A-to-I editing event are quite promiscuous in the human genome, thanks mainly to the abundance of Alu elements that can form readily dsRNA structures if they are close to each other in inverted orientation, which serve as templates for ADAR1 and show varying levels of editing (Bazak et al. 2014; Athanasiadis et al. 2004; Nishikura 2016). While most of these editing events are not conserved, the absence of ADAR genes can lead to characteristic neural phenotypes, suggesting that this epitranscriptomic modification is essential for normal development (Savva et al. 2012). Elegant genetic work (see below) has demonstrated that A-to-I editing is really essential for the developmental recoding of a handful of proteins related to neurotransmission and suggests that other editing events might lack physiological relevance (Chalk et al., 2019).
In addition to the possibility that the modifications may be superimposed, there may be other links between the different modifications. For example, 1-methylguanosine (m1G) is very precisely expressed in human tRNAs at position G9 and is formed by the TRMT10A writer enzyme (Krishnamohan and Jackman 2017). Deficiencies in TRMT10A are associated with microcephaly, short-stature and diabetes (Igoillo-Esteve et al. 2013). Recently, TRMT10A has been also reported to affect mRNA m6A levels by enhancing the demethylation activity and selectivity of FTO (Ontiveros et al. 2020).
N-acetyltransferase 10 (NAT10) and its orthologues are responsible for installing N4-acetylcytidine (ac4C) to rRNA, tRNA and mRNA (Ito et al. 2014; Arango et al. 2018; Sharma et al. 2015), while assisted by adaptor proteins orthologous to the yeast TAN1 (Johansson and Byström 2004; Sharma et al. 2015). The presence of ac4C promotes translational efficiency and fidelity in general, both by increasing the stability of tRNAs and mRNAs, and aiding the correct usage of tRNAs when present in the wobble position of the anticodon loop (for details see: (Jin et al. 2020).
The 5′ 7-methylguanosine (m7G) containing cap structure is a critical component of eukaryotic mRNAs and is essential for the stability and the correct translation of the transcripts (Furuichi 2015). Accordingly, during the past decades, the enzymatic pathways necessary for “capping” have been described in great details (Ghosh and Lima 2010). Lately, however, m7G was also detected in 18S rRNA (G1639) where it is installed by the Williams–Beuren syndrome critical region 22 (WBSCR22) methylase (Haag et al. 2015), and in tRNA at position G46 where the TRM8P/TRM82P heterodimer is involved in the modification (Purta et al. 2005). Due to its low levels in mRNA (~ 0.04% of all guanines), the detection of this modification with current methods at single nucleotide resolution is difficult; therefore, the location of m7Gs in mRNAs (other than the cap) is still debated (Wiener and Schwartz 2020).
N1-methyladenosine (m1A) is significantly less abundant than m6A and it is mainly present in the A58 position of tRNAs (Jin et al. 2022; Wiener and Schwartz 2020). This conserved methylation is dependent on the activity of TRMT6 and TRMT61 (Ozanick et al. 2005). A conserved m1A methylation pattern was also observed in 28S rRNA (A1309) that is catalysed by the nucleomethylin (NML) enzyme, which is essential for ribosomal assembly and function (Waku et al. 2016). The presence (and function) of m1A in mRNA is still debated (Wiener and Schwartz 2020), but the TRMT6/TRMT61A complex is thought to be responsible for the methylation of this RNA species as well, while the demethylation of both tRNA and mRNA is dependent on ALKBH3 (Jin et al. 2022; Ougland et al. 2004). YTHDF1-3 and YTHDC1 can act as “readers” of m1A (Dai et al. 2018).
Localization and dynamics of epitranscriptomic modifications
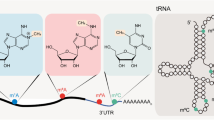
The physiological effect of post-transcriptional mRNA modifications is also dependent on their location within the transcripts. While the difference in detection methods makes direct comparisons more difficult, for more abundant modifications, such as m6A, m5C, Ψ and A-to-I editing, we have enough data to make some general observations (Fig. 2). For example, while m6A is widely distributed in mRNAs, it is most abundant in the proximity of the stop codon and in long introns (Fig. 2A). It can be also detected close to the 5’ cap (Meyer et al. 2015; Zhou et al. 2015). On the other hand, m5C sites are most abundant in coding sequence (CDS), and depleted in the UTRs of most model species (Fig. 2B), with the exception of mammals where 5’UTRs are especially enriched in this modification (Hartstock et al. 2022; Liu et al. 2022).

Schematic distribution of four abundant epitranscriptomic marks across mRNAs and the dynamics of their abundance during development. (A) m6A is enriched in the cap region and around the stop codon, present in the CDS and missing from the UTRs. (A’) The developmental dynamics of m6A is different in the organisms where data are available, but in general tissue-specific differences can be detected in adults. (B,B’) m5C marks are mostly detectable in the CDS, during early development. (C) Ψ sites are present both in the CDS and the 3’UTR. (There is not enough data to assess the developmental dynamics of these modifications. (D) I is most abundant in the 3’UTR and somewhat more enriched in the CDS than in the 5’UTR. (D’) It is present at high levels in the oocytes, but during development is removed and only detectable later in adult tissues at varying levels. (Dashed lines in A’ and B depict somewhat divergent signals detected in mammals. Funnel shapes in panels A’ and D’ mark a wide range of modification levels observed in adult tissues. For details and sources see text.)
Distribution of Ψ sites within the mRNAs is also not even and shows a heavy skew towards the CDS and 3’UTR (Fig. 2C) where more than 90% of the detected sites occur (Tavakoli et al. 2022; Carlile et al. 2014; Li et al. 2015a). The current amount of data about Ψ, however, is not sufficient to assess its dynamics during development.
For other modifications with a low stoichiometry, current mapping methods are less reliable. For example, while initially m1A was thought to be enriched in the 5’UTRs in the proximity of the start codons, later experiments demonstrated that off-target detection of m7G was responsible for this signal (Grozhik et al. 2019). The detection of m7G itself is controversial: current methods give contradictory results about its presence (or absence) and exact localization in mRNAs and small RNAs (Li et al. 2022; Enroth et al. 2019; Zhang et al. 2019). Low levels of ac4C make its detection with current immunoprecipitation-based protocols also difficult —a high ratio of aspecific signals can result in an excess false-positive results (Arango et al. 2018).
It is much more straightforward to detect adenosine to inosine (A-to-I) RNA editing catalysed by the adenosine-deaminase (ADAR) enzymes, as the reverse-transcriptase (RT) step ubiquitous in most NGS methods will detect these as miss-matches (Nishikura 2010). Most of the A-to-I edits occur in introns and often target repetitive sequences, such as Alu elements in humans and regions enriched in short and long interspersed nuclear elements (SINEs and LINEs) in rodents. This is most likely due to the occurrence of double-stranded secondary RNA structures that can attract editing enzymes with dsRNA-binding domains, such as ADAR1 and ADAR2 (Tajaddod et al. 2016).
Multiple studies have looked at the dynamic of post-transcriptional RNA modifications during development (Fig. 2A’, B’, D’). A-to-I editing appears abundant especially during early stages of vertebrate development: in humans the peak is before 8 cell stage (Shtrichman et al. 2012; Qiu et al. 2016), while in zebrafish before the maternal-zygotic transition (MZT) (Buchumenski et al. 2021). In contrast, in Drosophila A-to-I editing events can be detected from late pupal stages and become more abundant in adults (Graveley et al. 2011). This later wave of A-to-I editing might coincide with the changes in the neural transcriptome that seem to occur in most metazoan species, from cephalopods to mammals (Liscovitch-Brauer et al. 2017; Hwang et al. 2016; Behm et al. 2017; Cuddleston et al. 2021).
In contrast to A-to-I editing, m5C levels appear to be highest in the zygotes of all taxa and become progressively eliminated after (or around) MZT (Liu et al. 2022). While m6A is the most widely studied of the epitranscriptomic modifications, interestingly, the available data about its developmental dynamics is harder to interpret. While in zebrafish, m6A levels peak around MZT (Aanes et al. 2019; Zhao et al. 2017a), in Drosophila and mice we can observe the opposite dynamics and m6A levels dip around MZT before bouncing back (Fig. 2A’)(Lence et al. 2016; Wu et al. 2021; Sui et al. 2020). That said, interestingly, the number of genes affected by m6A modification is actually increased in murine embryos at MZT (Wu et al. 2021). It will be interesting to understand if these differences reflect some genuine, species-specific alterations in the physiological role(s) of m6A, especially as the pattern of distribution of this epitranscriptomic modification across the transcripts appears to be highly conserved evolutionarily.
The epitranscriptome of the maternal-zygotic transition (MZT)
Different animals employ different developmental strategies after fertilization and these strategies are reflected in the amount and composition of the maternal pool of RNAs and proteins that will guide the earliest phases of embryogenesis. Yet, regardless of the actual strategy, the time will come for the embryo to become its own master and start expressing the genes on the chromosomes it inherited from its parents (Vastenhouw et al. 2019).
The activation of the zygotic genome is a hallmark event of early development, and besides the obvious changes of the epigenetic landscape, it is usually accompanied by a tightly regulated restructuring of the transcriptome as well. Maternal transcripts are progressively eliminated as transcripts emerging from the zygotic genome become more abundant. This process is essential, as a failure to eliminate maternal transcripts will almost always lead to embryonic lethality (Giraldez et al. 2006; Liu et al. 2020).
The roles played by post-transcriptional modifications in regulating MZT have become the subject of multiple research programmes and the emerging data suggest that they play a fundamental role in the process.
In zebrafish, one of the major models in MZT research, previous studies have clearly established the zygotically expressed mir-430 as one of the key regulators of maternal transcript clearance (Liu et al. 2020; Giraldez et al. 2006). Later studies, however, have also unveiled the other, redundant processes also contribute to the gradual elimination of maternal mRNAs (Kontur et al. 2020).
One of the first epitranscriptomic modifications that was found to have a role during zebrafish MZT was m6A (Zhao et al. 2017b). Maternal transcripts are rich in m6A sites and, importantly, the removal of the Ythdf2 m6A “reader” resulted in the failed clearance of maternal mRNAs (Zhao et al. 2017b; Kontur et al. 2020). This modification appears to be also essential for oogenesis as the removal of multiple Ythdf “readers” or the Mettl3 writer resulted in oocyte maturation defects and a male bias during development (Xia et al. 2018; Kontur et al. 2020). The latter phenotype is likely due to an early failure in the formation of the bipotential gonad and the gonocytes essential for later female development (Liew and Orbán 2014; Aharon and Marlow 2022).
In contrast, the m5C modification, while also abundant in the early transcriptome, appears to have the opposite effect: the elimination of the Ybx1 “reader” results in the destabilization of maternal transcripts, suggesting that m5C-modified maternal transcripts are protected from an early degradation during MZT (Yang et al. 2019; Liu et al. 2022).
Recent studies also suggest that the early embryonic role of m6A and m5C might also be conserved during development. For example, m6A is also essential for the regulation of transcript stability both in the mouse oocyte and during MZT and the maternal depletion of YTHDF2 results in female infertility (Wu et al. 2022; Ivanova et al. 2017). The dynamics of m5C around MZT also appears to be conserved in and Drosophila embryos lacking NSUN2 fail to initiate the MZT in a timely manner (Liu et al. 2022). These observations suggest and essential and role for m5C during early development.
For other modifications, such as Ψ or ac4C, we just do not have enough data to assess their role during MZT. Of note, however, zebrafish maternal-zygotic snord13 mutants, defective in 18S rRNA ac4C acetylation do not show any morphological defects (Bortolin-Cavaillé et al. 2022), whereas knockout of dyskerin function results in larval lethality in the same species (Balogh et al. 2020; Zhang et al. 2012) and very early embryonic lethality in mice (He et al. 2002).
One possible role of Ψ during MZT may be the regulation of ribosomal function through the modulation of rRNA biogenesis and folding. For example, in zebrafish, multiple lines of evidence suggest that besides the miR-430, m6A and m5C, the process of the ribosome-driven codon-mediated mRNA decay can influence the degradation of maternal transcripts (Bazzini et al. 2016; Mishima and Tomari 2016; Mishima et al. 2022; Medina-Muñoz et al. 2021). As zebrafish development also sees a transition from the use of maternal rRNAs to the use of somatic ones (Locati et al. 2017), one hitherto unexplored possibility is the differential tuning of these ribosomal types to the different mRNA pools. As the maternal and somatic ribosomes might be differentially pseudouridylated and this might affect their specificity, rRNA pseudouridylation might be a more indirect way how post-transcriptional modifications can effect MZT.
Post-transcriptional modifications in the hematopoietic system
Hematopoiesis occurs in multiple waves during animal development and is a hallmark feature of adult forms as well. Key to this process is multipotent hematopoietic stem cells (HSCs) that form a heterogenic population of progenitors with the potential to differentiate into typical hematopoietic lineages, such as the erythroid and lymphoid lineages of vertebrates.
HSC differentiation is a tightly regulated process and a string of well characterized signalling events and accompanying epigenetic changes ensure that combinations of mature hematopoietic cells, in proportions required by the physiological status of the (developing) animal are established (Hu and Shilatifard 2016). Recent studies also highlight the importance of epitranscriptomic marks in these dynamic fate decisions.
The role of RNA methylation during the development of adult HSCs has been first recognized in zebrafish. In the absence of Mettl3, the “writer” of m6A modifications, Notch signalling levels in HSCs became abnormally elevated leading to defects in the endothelial-to-hematopoietic transition. This effect is due to an impaired decay in notch1a transcripts, normally mediated by the Ythdf2 m6A reader protein (Zhang et al. 2017). Off note, however, animals lacking functional Ythdf2 appeared phenotypically normal (Zhao et al., 2017).
Other studies also highlighted the elevated levels of Mettl3 (and Mettl14) in HSCs compared to differentiated cell types and have shown that the impairment of Mettl3 leads to a failure of stem cell self-renewal and differentiation both in mice (Yao et al. 2018; Lee et al. 2019) and humans (Vu et al. 2017; Bueno-Costa et al. 2022). Recent studies have also been able to map the dynamic changes in m6A patterns during human HSC differentiation at a single-base resolution (Hu et al. 2022), and it will be interesting to see if similar dynamic patterns of transcriptome methylation will be detected in other tissues as well.
While pseudouridylation is also essential in hematopoiesis, current datasets mostly highlight the importance of Ψ in non-coding RNAs. Pseudouridylation dependent on pseudouridine synthase 1 (PUS1) occurs in many tRNAs and at multiple positions. Several studies have reported mitochondrial myopathy, lactic acidosis and sideroblastic anaemia 1 (MLASA1 OMIM: 600462) as a result of PUS1 pathogenic mutations (Oncul et al. 2021), but interestingly Pus1 loss in mice do not results in hematological phenotype (Mangum et al. 2016).
The impairment of dyskerin-dependent pseudouridylation leads to X-linked recessive dyskeratosis congenita (DC, OMIM: 305000) that mostly affects highly regenerative tissues. The failure of primitive erythropoiesis caused by the lack of dyskerin was showed in zebrafish, mice and human cell lines. While originally this failure was attributed to the role of dyskerin in telomere biogenesis, more recent studies suggest that the impairment of pseudouridylation is at least equally important to the process (Balogh et al. 2020; Ruggero et al. 2003; Donaires et al. 2019). Additionally, the dynamic changes in snoRNA expression during hematopoiesis was already highlighted by the earlier studies (Warner et al. 2018) and it might be instrumental to the correct differentiation of HSCs, therefore.
More recently, the importance of rRNA pseudouridylation has also been highlighted by the phenotype of Ddx41-deficient mice. This enzyme is essential for the processing of snoRNAs; therefore, the impaired function will result in altered rRNA pseudouridylation. Homozygous Ddx41 mutations result in HSC differentiation, whereas monoallelic mutations sill result in age-dependent defects in the hematopoietic process (Chlon et al. 2021). In summary, both the dysfunction of dyskerin and the aberrant processing of snoRNAs lead to a failure in HSC differentiation.
A relatively new discovery also connects hematopoiesis with the recently recognized gene expression regulatory role of tRNA-derived fragments (tRFs). For a specific subtype of tRFs (mini tRFs containing a 5′ terminal oligoguanine – mTOGs), an important, Ψ-dependent role has been proposed in the inhibition of aberrant protein synthesis programmes (Lee et al. 2022; Guzzi et al. 2022).
Finally, the role of A-to-I editing in hematopoiesis has been also getting more attention recently. Multiple studies showed that mutations of ADAR1 can lead to embryonic lethality I mice and mutants show foetal liver defects coupled with impairment of erythroid differentiation (Hartner et al. 2004; Liddicoat et al. 2013). In humans, hyperactivity of ADAR1 leads to the missplicing of the GSK3B gene and results in the accumulation of β-catenin, the ectopic activation of the canonical Wnt signalling pathway and consequently a defect in HSC self-renewal (Jiang et al., 2013).
RNA editing and other post-transcriptional modifications in neural development
While the role of post-transcriptional RNA modifications has been demonstrated in many cell differentiation processes, they seem especially important in the development of the nervous system. This is because the complexity of the nervous system requires a remarkable diversity of cell types with unique gene expression patterns (Zeng 2022) and during cognitive development these expression patterns change in an activity-dependent manner. Additional mechanisms are also required for the dynamic establishment and breakdown of synaptic connections (Campbell & Wood, 2019). Accordingly, the temporal and spatial regulation of gene expression in neurons is highly complex and involves several layers. Due to its rapidity and complexity, post-transcriptional regulatory processes play a prominent role compared to other tissues, such as brain-specific miRNAs and lncRNAs (Petri et al. 2014; Ang et al. 2020). Nevertheless, it is becoming increasingly evident that RNA modifications are also required to mediate this extraordinary diversity and complexity.
Based on our current knowledge, A-to-I editing by ADAR enzymes is most prominent and essential epitranscriptomic modification in the nervous system (Tariq and Jantsch 2012). The rate of A-to-I editing increases spectacularly with neuronal complexity, which suggests that this modification is an evolutionary driver for the formation of more complex neuronal structures (Duan et al. 2022; Li and Church 2013). Consistent with this hypothesis, the developmental impact of A-to-I editing seems less pronounced in lower complexity groups.
The absence of ADAR enzymes in C. elegans results in impaired chemotaxis (Tonkin et al. 2002) and Adar knockout in Drosophila causes locomotor defects and age-dependent neurodegeneration (Sapiro et al. 2019; Deng et al. 2020). In contrast, (Chalk et al., 2019)2 deficiency in mice causes epilepsy and postnatal lethality.
In mammals, ADAR2 is expressed in all tissues and largely shares target positions with ADAR1. The severe phenotype of Adar2 mutant mice would suggest that ADAR2 has an essential role in brain development that cannot be compensated by ADAR1. Notably, however, this severe phenotype can be rescued by converting a single ADAR2 target site in the glutamate receptor ionotropic AMPA 2 (Gria2) from adenine to guanine (Chalk et al. 2019; Higuchi et al. 2000).
A previous study of human ADAR activity shows that most of the A-to-I editing occurs on Alu sequences and less than 1% of the total editing activity accounts for coding sequences (Bazak et al. 2014). Most of the human specific Alu elements are located in genes with neuronal functions and the rare recoding events can have a substantial impact on the neurodevelopmental programme (Ramaswami et al. 2013).
Increased rates of mRNA editing have also been observed in the cephalopods, which have a notoriously well-developed nervous system. In this group, A-to-I editing sites are evolutionarily conserved; those in the coding regions are clearly associated with distinct neuronal functions, but many of the sites are not neural tissue specific and located in non-coding regions, too (Albertin et al. 2022; Liscovitch-Brauer et al. 2017). Interestingly, RNA editing appears to be a major driver for generating transcriptome diversity in cephalopods, and similarly to vertebrates, it has profound effects on the physiology of voltage-gated ion channels (Liscovitch-Brauer et al. 2017).
In mammals, the third and final member of the ADAR family is the peculiar ADAR3, which has no catalytic activity and is expressed specifically in brain tissue only. The function of this protein is still largely mysterious, but overexpression of ADAR3 in glioblastoma can inhibit the editing of Gria2 transcripts by ADAR2 (Oakes et al. 2017) and the lack of its RNA-binding domain increases anxiety and reduces cognitive abilities in mice (Mladenova et al. 2018).
Regarding A-to-I editing, it is also worth highlighting that a very severe human neurodevelopmental abnormality with poor growth and dysmorphic facies (OMIM: 615,286) is caused by the loss of function of the Adenosine Deaminase tRNA-Specific 3 (ADAT3) writer enzyme, specific for tRNAs (Sharkia et al. 2019).
The epitranscriptomic modification of tRNAs seems generally important for neurodevelopmental processes. In vertebrates, the m1G modification of the tRNA G9 position located very precisely at the transition between the acceptor and D-strand. The addition of this modification is nucleo-cytoplasmically performed by the methyltransferase TRMT10A, while in the case of mitochondrial tRNAs (mtRNAs) this is driven by TRM10C (Holzmann et al., 2008). Both writer enzymes are known to have pathogenic mutations: defects in TRMT10A mainly lead to neurodegenerative pathologies and glucose degradation problems (OMIM: 616,033) (Narayanan et al. 2015; Vilardo et al. 2020; Igoillo-Esteve et al. 2013).
The function of the highly conserved Dnmt2 is mainly to modify the C38 positions of tRNAs to m5C. Depletion of Dnmt2 in zebrafish leads to abnormal neurogenesis, similar to that in human patients in which it leads to central nervous system lesions (Franke et al. 2009; Rai et al. 2007). Relatedly, the absence of NSUN3-mediated mt-tRNA C34 methylation impedes the differentiation of mouse embryonic stem cells towards the neuroectoderm lineage (Trixl et al. 2018).
Targets of the NSUN methyltransferase family, however, are not only tRNAs and expression of its members is increased in the developing brain, consistent with the fact that mRNA m5C modification levels in mice are highest in brain tissue (Chi and Delgado-Olguín 2013; Yang et al. 2017). Interestingly, when comparing mouse ESC and brain tissue the m5C signal for given transcripts is specifically present only in one or the other tissue. This differential methylation allows for the differentiation of transcripts between cell types (Amort et al. 2017).
NSUN2 deficient patients also develop an ID phenotype, characterized by retardation, microcephaly, impaired cognition and poor motor function (OMIM: 611,091) (Khan et al. 2012; Abbasi-Moheb et al. 2012). These phenotypes could be related to the observations that the depletion of NSUN2 inhibits neuroepithelial stem cell migration, whereas its overexpression contributes to tumourigenesis by stabilizing mRNAs in migration pathways (Chen et al. 2019b; Flores et al. 2017).
A link between NSUN5 rRNA methyltransferase and neuronal function has been also established, with Nsun5 loss in mice resulting in reduced cortical thickness and abnormal neuronal layer formation (Chen et al. 2019a). Deletion of NSUN5 has been linked to Williams-Beuren syndrome (OMIM: 194050), which is characterized by cognitive deficits and neuronal abnormalities (Heissenberger et al. 2019).
As for other NSUN family members, a recent preprint suggests that NSUN6 and NSUN7 m5C writers and the ALYREF reader expression in certain brain regions are significantly altered in different stages of Alzheimer's disease with NSUN6 expression being also reduced following brain injury, independent from Alzheimer's disease develops or not (Perezgrovas-Saltijeral et al. 2022).
Insufficient pseudouridylation resulting from the loss of PUS3 and PUS7 also produces neuronal phenotypes through damage to tRNA function. PUS3 catalyses the formation of tRNA Ψ at positions U38 and U39 of the anticodon stem. Accumulation of its mRNA genes has been demonstrated in neuronal tissues of mouse embryos (Diez-Roux et al. 2011). Mutations in PUS3 have been associated with neurodevelopmental disorders, including microcephaly and intellectual disability (OMIM: 617051) and shorter lifespan (Borghesi et al. 2022; Shaheen et al. 2016). The lack of functional PUS7 is also associated with intellectual disability and aggressive behavior (OMIM: 618342) and in these cases not only the tRNA Ψ13 site is absent, but pseudouridylation of some mRNAs is also affected (Martinez et al. 2022; Brouwer et al. 2018; Shaheen et al. 2019; Darvish et al. 2019). Notably, the complete loss of pus7 function in flies also leads to behavioural problems, such as aggression and disorientation (Brouwer et al. 2018). In humans, aggressive behaviour and motor impairments are only observed in patients with missense mutations in PUS7, while nonsense and frameshift mutations cause more severe forms of ID (Darvish et al. 2019).
Even though dyskerin does not modify tRNAs, high expression of its orthologous gene is observed in certain cells of mouse embryonic neural tissues and in adult mouse cerebellar and olfactory bulb neurons. (Heiss et al. 2000). It is also notable that in one of the most severe forms of DC, Hoyeraal-Hreidarsson syndrome shows a greater severity of neurological symptoms such as mental retardation, cerebellar hypoplasia and microcephaly, in addition to haematological problems (Zhang et al. 2021a). These findings demonstrate an important role for dyskerin in neural development and suggest that insufficient pseudouridylation contributes to the development of neurological problems in general.
Perhaps m6A has the richest literature in neuroscience compared to other modifications (He & He, 2021; J. Li et al., 2019; Livneh et al., 2020; Shu et al., 2021). Interestingly, the levels of m6A are low during early mouse brain development, but increases dramatically in adulthood (Meyer et al., 2012). In addition, a recent study showed that the pattern of m6A in rodents is specific to brain regions and neuronal subtypes (Chang et al., 2017).
Depletion of both METTL3 and METTL14 disrupts m6A-mediated degradation of mRNAs of several transcription factors associated with cell cycle, stem cell and neuronal differentiation functions. The defective writer complex prolongs the cell cycle of neural progenitors and delays the cortical neurogenesis, leading to incorrect neuronal layering in the mouse brain (Yoon et al., 2017). A study implicates oncogenic roles for these genes in glioblastoma, the most common brain tumour (Cui et al., 2017).
FTO knockout mice exhibits a less severe phenotype including learning difficulties and impaired memory. Consistent with this finding, the loss of FTO reduces the number of adult neural stem cells (L. Li et al., 2017).
The YTHDF reader proteins have also been associated neuronal functions. Ythdf1 conditional knockout mice have strongly reduced axon regeneration, suggesting a crucial role for m6A modification in the response to injury (Weng et al., 2018).
Dysregulation of m6A has been associated with human brain-disorders risk genes (Yoon et al., 2017). For example, the fragile X mental retardation protein, encoded by the most prominent monogenic risk gene for autism, binds RNAs m6A-dependent manner. The amount of FTO increases in Parkinson's disease models compared to wild-type rats (Qiu et al., 2020) and genetic variants of FTO have been associated with Alzheimer's disease (Keller et al., 2011). Taken together, these results suggest that the correct balance of m6A is essential for the proper development of the mammalian brain and normal neural function.
Conclusion and outlook
Post-transcriptional RNA modifications appear to have essential roles in fine-tuning several biological processes. As we have summarized in our review, different epitranscriptomic modification can affect the stability of RNAs, the efficiency of the translation process and even the composition of certain proteins. All RNA types can be targeted to differing degree, although current evidence is equivocal for the role of some modifications especially in mRNAs and lncRNAs. Nevertheless, epitranscriptomic marks appear to be indispensable in regulating the stability of maternal transcripts, the complicated hematopoietic differentiation process and multiple aspects of neural development.
As we will learn more about the RNA modifications themselves, their combinatorial and/or cumulative effects and the many functions of the proteins related to them, we will be able to get a better understanding about their potential role in pathologies as well. We will also understand if these modifications and the processes that regulate them can be used as targets of therapies.
The emerging picture suggests that different cell types might have characteristic epitranscriptome signatures and the actual “meaning” of certain post-transcriptional modifications might not only depend on their position within the transcripts, but also on their relative abundance, the abundance of the RNA molecules affected.
References
Aanes H, Engelsen D, Manaf A, Alemu EA, Vågbø CB, Martín L, Lerdrup M, Hansen K, Mathavan S, Winata C, et al. 2019. N6-methyladenosine dynamics during early vertebrate embryogenesis. Biorxiv 528422
Abbasi-Moheb L, Mertel S, Gonsior M, Nouri-Vahid L, Kahrizi K, Cirak S, Wieczorek D, Motazacker MM, Esmaeeli-Nieh S, Cremer K et al (2012) Mutations in NSUN2 cause autosomal- recessive intellectual disability. Am J Hum Genetics 90:847–855
Aharon D, Marlow FL (2022) Sexual determination in zebrafish. Cell Mol Life Sci 79:8
Albertin CB, Medina-Ruiz S, Mitros T, Schmidbaur H, Sanchez G, Wang ZY, Grimwood J, Rosenthal JJC, Ragsdale CW, Simakov O et al (2022) Genome and transcriptome mechanisms driving cephalopod evolution. Nat Commun 13:2427
Amort T, Rieder D, Wille A, Khokhlova-Cubberley D, Riml C, Trixl L, Jia X-Y, Micura R, Lusser A (2017) Distinct 5-methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol 18:1
Ang CE, Trevino AE, Chang HY (2020) Diverse lncRNA mechanisms in brain development and disease. Curr Opin Genet Dev 65:42–46
Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, Hosogane M, Sinclair WR, Nanan KK, Mandler MD et al (2018) Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175:1872-1886.e24
Armistead J, Khatkar S, Meyer B, Mark BL, Patel N, Coghlan G, Lamont RE, Liu S, Wiechert J, Cattini PA et al (2009) Mutation of a gene essential for ribosome biogenesis, EMG1, causes Bowen-Conradi syndrome. Am J Hum Genet 84:728–739
Athanasiadis A, Rich A, Maas S (2004) Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. Plos Biol 2:e391
Babaian A, Rothe K, Girodat D, Minia I, Djondovic S, Milek M, Miko SES, Wieden H-J, Landthaler M, Morin GB et al (2020) Loss of m1acp3Ψ Ribosomal RNA Modification Is a Major Feature of Cancer. Cell Rep 31:107611
Balogh E, Chandler JC, Varga M, Tahoun M, Menyhárd DK, Schay G, Goncalves T, Hamar R, Légrádi R, Szekeres Á et al (2020) Pseudouridylation defect due to DKC1 and NOP10 mutations causes nephrotic syndrome with cataracts, hearing impairment, and enterocolitis. Proc National Acad Sci 117:15137–15147
Bartee D, Nance KD, Meier JL (2022) Site-specific synthesis of N4-acetylcytidine in RNA reveals physiological duplex stabilization. J Am Chem Soc 144:3487–3496
Bass BL (2002) RNA Editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71:817–846
Bazak L, Haviv A, Barak M, Jacob-Hirsch J, Deng P, Zhang R, Isaacs FJ, Rechavi G, Li JB, Eisenberg E et al (2014) A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res 24:365–376
Bazzini AA, del Viso F, Moreno-Mateos MA, Johnstone TG, Vejnar CE, Qin Y, Yao J, Khokha MK, Giraldez AJ (2016) Codon identity regulates mRNA stability and translation efficiency during the maternal-to-zygotic transition. Embo J 35:2087–2103
Behm M, Wahlstedt H, Widmark A, Eriksson M, Öhman M (2017) Accumulation of nuclear ADAR2 regulates adenosine-to-inosine RNA editing during neuronal development. J Cell Sci 130:745–753
Boccaletto P, Stefaniak F, Ray A, Cappannini A, Mukherjee S, Purta E, Kurkowska M, Shirvanizadeh N, Destefanis E, Groza P et al (2021) MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res 50:D231–D235
Borghesi A, Plumari M, Rossi E, Viganò C, Cerbo RM, Codazzi AC, Valente EM, Gana S (2022) PUS3-related disorder: Report of a novel patient and delineation of the phenotypic spectrum. Am J Med Genet A 188:635–641
Bortolin-Cavaillé M-L, Aurélie Q, Supuni TG, Thomas JM, Sas-Chen A, Sharma S, Plisson-Chastang C, Vandel L, Blader P, Lafontaine DLJ et al (2022) Probing small ribosomal subunit RNA helix 45 acetylation across eukaryotic evolution. Nucleic Acids Res 50:6284–6299
Buchumenski I, Holler K, Appelbaum L, Eisenberg E, Junker JP, Levanon EY (2021) Systematic identification of A-to-I RNA editing in zebrafish development and adult organs. Nucleic Acids Res 49:4325–4337
Bueno-Costa A, Piñeyro D, García-Prieto CA, Ortiz-Barahona V, Martinez-Verbo L, Webster NA, Andrews B, Kol N, Avrahami C, Moshitch-Moshkovitz S et al (2022) Remodeling of the m6A RNA landscape in the conversion of acute lymphoblastic leukemia cells to macrophages. Leukemia 36:1–4
Campbell RR, Wood MA (2019) How the epigenome integrates information and reshapes the synapse. Nat Rev Neurosci 20(3):133–147
Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515:143–146
Chalk AM, Taylor S, Heraud-Farlow JE, Walkley CR (2019) The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol 20:268
Chang M, Lv H, Zhang W, Ma C, He X, Zhao S, Zhang Z-W, Zeng Y-X, Song S, Niu Y, Tong W-M (2017) Region-specific RNA m6A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol 7(9):170166
Chen P, Zhang T, Yuan Z, Shen B, Chen L (2019a) Expression of the RNA methyltransferase Nsun5 is essential for developing cerebral cortex. Mol Brain 12:74
Chen X, Li A, Sun B-F, Yang Y, Han Y-N, Yuan X, Chen R-X, Wei W-S, Liu Y, Gao C-C et al (2019b) 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat Cell Biol 21:978–990
Chi L, Delgado-Olguín P (2013) Expression of NOL1/NOP2/sun domain (Nsun) RNA methyltransferase family genes in early mouse embryogenesis. Gene Expr Patterns 13:319–327
Chlon TM, Stepanchick E, Hershberger CE, Daniels NJ, Hueneman KM, Davis AK, Choi K, Zheng Y, Gurnari C, Haferlach T et al (2021) Germline DDX41 mutations cause ineffective hematopoiesis and myelodysplasia. Cell Stem Cell 28:1966-1981.e6
Cuddleston R, Sloofman L, Liang L, Mossotto E, Fan X, Wang M, Zhang B, Wang J, Sestan N, Devlin B, et al. 2021. Expansion of RNA sequence diversity and RNA editing rates throughout human cortical development. Biorxiv 2021.06.10.447947.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang C-G, Riggs AD, He C, Shi Y (2017) m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep 18(11):2622–2634
Dai X, Wang T, Gonzalez G, Wang Y (2018) Identification of YTH domain-containing proteins as the readers for N1-methyladenosine in RNA. Anal Chem 90:6380–6384
Darvish H, Azcona LJ, Alehabib E, Jamali F, Tafakhori A, Ranji-Burachaloo S, Jen JC, Paisán-Ruiz C (2019) A novel PUS7 mutation causes intellectual disability with autistic and aggressive behaviors. Neurology Genetics 5:e356
de Brouwer APM, Jamra RA, Körtel N, Soyris C, Polla DL, Safra M, Zisso A, Powell CA, Rebelo-Guiomar P, Dinges N et al (2018) Variants in PUS7 cause intellectual disability with speech delay, microcephaly, short stature, and aggressive behavior. Am J Hum Genetics 103:1045–1052
Deng P, Khan A, Jacobson D, Sambrani N, McGurk L, Li X, Jayasree A, Hejatko J, Shohat-Ophir G, O’Connell MA et al (2020) Adar RNA editing-dependent and -independent effects are required for brain and innate immune functions in Drosophila. Nat Commun 11:1580
Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, Magen A, Canidio E, Pagani M, Peluso I et al (2011) A high-resolution anatomical atlas of the transcriptome in the mouse embryo. Plos Biol 9:e1000582
Donaires FS, Alves-Paiva RM, Gutierrez-Rodrigues F, da Silva FB, Tellechea MF, Moreira LF, Santana BA, Traina F, Dunbar CE, Winkler T et al (2019) Telomere dynamics and hematopoietic differentiation of human DKC1-mutant induced pluripotent stem cells. Stem Cell Res 40:101540
Duan Y, Tang X, Lu J (2022) Evolutionary driving forces of A-to-I editing in metazoans. Wiley Interdiscip Rev Rna 13:e1666
Edupuganti RR, Geiger S, Lindeboom RGH, Shi H, Hsu PJ, Lu Z, Wang S-Y, Baltissen MPA, Jansen PWTC, Rossa M, Müller M, Stunnenberg HG, He C, Carell T, Vermeulen M (2017) N6-methyladenosine (m6A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol 24(10):870–878
Enroth C, Poulsen LD, Iversen S, Kirpekar F, Albrechtsen A, Vinther J (2019) Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res 47:e126–e126
Eyler DE, Franco MK, Batool Z, Wu MZ, Dubuke ML, Dobosz-Bartoszek M, Jones JD, Polikanov YS, Roy B, Koutmou KS (2019) Pseudouridinylation of mRNA coding sequences alters translation. Proc National Acad Sci 116:23068–23074
Flores JV, Cordero-Espinoza L, Oeztuerk-Winder F, Andersson-Rolf A, Selmi T, Blanco S, Tailor J, Dietmann S, Frye M (2017) Cytosine-5 RNA methylation regulates neural stem cell differentiation and motility. Stem Cell Rep 8:112–124
Franke B, Vermeulen SHHM, Steegers-Theunissen RPM, Coenen MJ, Schijvenaars MMVAP, Scheffer H, den Heijer M, Blom HJ (2009) An association study of 45 folate-related genes in spina bifida: Involvement of cubilin (CUBN) and tRNA aspartic acid methyltransferase 1 (TRDMT1). Birth Defects Res Part Clin Mol Teratol 85:216–226
Furuichi Y (2015) Discovery of m7G-cap in eukaryotic mRNAs. Proc Jpn Acad Ser B 91:394–409
Ghosh A, Lima CD (2010) Enzymology of RNA cap synthesis. Wiley Interdiscip Rev Rna 1:152–172
Gilbert WV, Bell TA, Schaening C (2016) Messenger RNA modifications: Form, distribution, and function. Science 352:1408–1412
Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Dongen SV, Inoue K, Enright AJ, Schier AF (2006) Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312:75–79
Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW et al (2011) The developmental transcriptome of Drosophila melanogaster. Nature 471:473–479
Grozhik AV, Olarerin-George AO, Sindelar M, Li X, Gross SS, Jaffrey SR (2019) Antibody cross-reactivity accounts for widespread appearance of m1A in 5’UTRs. Nat Commun 10:5126
Guzzi N, Muthukumar S, Cieśla M, Todisco G, Ngoc PCT, Madej M, Munita R, Fazio S, Ekström S, Mortera-Blanco T et al (2022) Pseudouridine-modified tRNA fragments repress aberrant protein synthesis and predict leukaemic progression in myelodysplastic syndrome. Nat Cell Biol 24:299–306
Haag S, Kretschmer J, Bohnsack MT (2015) WBSCR22/Merm1 is required for late nuclear pre-ribosomal RNA processing and mediates N7-methylation of G1639 in human 18S rRNA. RNA 21:180–187
Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH (2004) Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1*. J Biol Chem 279:4894–4902
Hartstock K, Ovcharenko A, Kueck NA, Spacek P, Cornelissen NV, Hüwel S, Dieterich C, Rentmeister A. 2022. MePMe-seq: Antibody-free simultaneous m6A and m5C mapping in mRNA by metabolic propargyl labeling and sequencing. Biorxiv 2022.03.16.484494.
Hasin Y, Seldin M, Lusis A (2017) Multi-omics approaches to disease. Genome Biol 18:83
He J, Navarrete S, Jasinski M, Vulliamy T, Dokal I, Bessler M, Mason PJ (2002) Targeted disruption of Dkc1, the gene mutated in X-linked dyskeratosis congenita, causes embryonic lethality in mice. Oncogene 21:7740–7744
He PC, He C (2021) m6A RNA methylation: from mechanisms to therapeutic potential. EMBO J. https://doi.org/10.15252/embj.2020105977
Heiss NS, Bächner D, Salowsky R, Kolb A, Kioschis P, Poustka A (2000) Gene structure and expression of the mouse dyskeratosis congenita gene, Dkc1. Genomics 67:153–163
Heissenberger C, Liendl L, Nagelreiter F, Gonskikh Y, Yang G, Stelzer EM, Krammer TL, Micutkova L, Vogt S, Kreil DP et al (2019) Loss of the ribosomal RNA methyltransferase NSUN5 impairs global protein synthesis and normal growth. Nucleic Acids Res 47:11807–11825
Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH (2000) Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406:78–81
Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, Rossmanith W (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135(3):462–474
Hu L, Liu S, Peng Y, Ge R, Su R, Senevirathne C, Harada BT, Dai Q, Wei J, Zhang L et al (2022) m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat Biotechnol. https://doi.org/10.1038/s41587-022-01616-4
Hu D, Shilatifard A (2016) Epigenetics of hematopoiesis and hematological malignancies. Gene Dev 30:2021–2041
Hwang T, Park C-K, Leung AKL, Gao Y, Hyde TM, Kleinman JE, Rajpurohit A, Tao R, Shin JH, Weinberger DR (2016) Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci 19:1093–1099
Igoillo-Esteve M, Genin A, Lambert N, Désir J, Pirson I, Abdulkarim B, Simonis N, Drielsma A, Marselli L, Marchetti P et al (2013) tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. Plos Genet 9:e1003888
Ito S, Horikawa S, Suzuki T, Kawauchi H, Tanaka Y, Suzuki T, Suzuki T (2014) Human NAT10 Is an ATP-dependent RNA acetyltransferase responsible for N 4-acetylcytidine formation in 18 S ribosomal RNA (rRNA)*. J Biol Chem 289:35724–35730
Ivanova I, Much C, Giacomo MD, Azzi C, Morgan M, Moreira PN, Monahan J, Carrieri C, Enright AJ, O’Carroll D (2017) The RNA m6A reader YTHDF2 Is essential for the post-transcriptional regulation of the maternal transcriptome and oocyte competence. Mol Cell 67:1059-1067.e4
Jiang Q, Crews LA, Barrett CL, Chun H-J, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, Rusert JM, Dao K-HT, Morris SR, Goldstein LSB, Marra MA, Frazer KA, Jamieson CHM (2013) ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci 110(3):1041–1046
Jin G, Xu M, Zou M, Duan S (2020) The processing, gene regulation, biological functions, and clinical relevance of N4-acetylcytidine on RNA: a systematic review. Mol Ther Nucleic Acids 20:13–24
Jin H, Huo C, Zhou T, Xie S (2022) m1A RNA modification in gene expression regulation. Genes-Basel 13:910
Johansson MJO, Byström AS (2004) The Saccharomyces cerevisiae TAN1 gene is required for N4-acetylcytidine formation in tRNA. RNA 10:712–719
Kampen KR, Sulima SO, Vereecke S, Keersmaecker KD (2020) Hallmarks of ribosomopathies. Nucleic Acids Res 48:1013–1028
Kan L, Ott S, Joseph B, Park ES, Dai W, Kleiner RE, Claridge-Chang A, Lai EC (2021) A neural m6A/Ythdf pathway is required for learning and memory in Drosophila. Nat Commun 12:1458
Keller L, Xu W, Wang H-X, Winblad B, Fratiglioni L, Graff C (2011) The obesity related gene, FTO, interacts with APOE, and is associated with Alzheimer’s disease risk: a prospective cohort study. J Alzheimer’s Dis 23(3):461–469
Khan MA, Rafiq MA, Noor A, Hussain S, Flores JV, Rupp V, Vincent AK, Malli R, Ali G, Khan FS et al (2012) Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. Am J Hum Genetics 90:856–863
Kontur C, Jeong M, Cifuentes D, Giraldez AJ (2020) Ythdf m6A Readers Function Redundantly during Zebrafish Development. Cell Rep 33:108598
Krishnamohan A, Jackman JE (2017) Mechanistic features of the atypical tRNA m1G9 SPOUT methyltransferase, Trm10. Nucleic Acids Res 45:9019–9029
Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, Hanna JH, Ding L (2019) Stage-specific requirement for Mettl3-dependent m6A mRNA methylation during haematopoietic stem cell differentiation. Nat Cell Biol 21:700–709
Lee AK, Aifantis I, Thandapani P (2022) Emerging roles for tRNAs in hematopoiesis and hematological malignancies. Trends Immunol 43:466–477
Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, Kreim N, Andrade-Navarro MA, Poeck B, Helm M et al (2016) m6A modulates neuronal functions and sex determination in Drosophila. Nature 540:242–247
Levi O, Arava YS (2021) RNA modifications as a common denominator between tRNA and mRNA. Curr Genet 67:545–551
Li JB, Church GM (2013) Deciphering the functions and regulation of brain-enriched A-to-I RNA editing. Nat Neurosci 16:1518–1522
Li X, Zhu P, Ma S, Song J, Bai J, Sun F, Yi C (2015a) Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol 11:592–597
Li Y-H, Zhang G, Cui Q (2015b) PPUS: a web server to predict PUS-specific pseudouridine sites. Bioinformatics 31:3362–3364
Li L, Zang L, Zhang F, Chen J, Shen H, Shu L, Liang F, Feng C, Chen D, Tao H, Xu T, Li Z, Kang Y, Wu H, Tang L, Zhang P, Jin P, Shu Q, Li X (2017) Fat mass and obesity-associated (FTO) protein regulates adult neurogenesis. Hum Mol Genet 26(13):2398–2411
Li J, Yang X, Qi Z, Sang Y, Liu Y, Xu B, Liu W, Xu Z, Deng Y (2019) The role of mRNA m6A methylation in the nervous system. Cell Biosci 9(1):66
Li W, Li X, Ma X, Xiao W, Zhang J (2022) Mapping the m1A, m5C, m6A and m7G methylation atlas in zebrafish brain under hypoxic conditions by MeRIP-seq. BMC Genomics 23:105
Liddicoat B, Hartner J, Chalk A, Lu J, Orkin SH, Walkley CR (2013) ADAR1 Is essential for erythroid development. Blood 122:9–9
Liew WC, Orbán L (2014) Zebrafish sex: a complicated affair. Brief Funct Genomics 13:172–187
Liscovitch-Brauer N, Alon S, Porath HT, Elstein B, Unger R, Ziv T, Admon A, Levanon EY, Rosenthal JJC, Eisenberg E (2017) Trade-off between transcriptome plasticity and genome evolution in cephalopods. Cell 169:191-202.e11
Liu Y, Zhu Z, Ho IHT, Shi Y, Li J, Wang X, Chan MTV, Cheng CHK (2020) Genetic deletion of miR-430 disrupts maternal-zygotic transition and embryonic body plan. Frontiers Genetics 11:853
Liu J, Huang T, Chen W, Ding C, Zhao T, Zhao X, Cai B, Zhang Y, Li S, Zhang L et al (2022) Developmental mRNA m5C landscape and regulatory innovations of massive m5C modification of maternal mRNAs in animals. Nat Commun 13:2484
Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, Dominissini D (2020) The m6A epitranscriptome: transcriptome plasticity in brain development and function. Nat Rev Neurosci 21(1):36–51
Locati MD, Pagano JFB, Girard G, Ensink WA, van Olst M, van Leeuwen S, Nehrdich U, Spaink HP, Rauwerda H, Jonker MJ et al (2017) Expression of distinct maternal and somatic 5.8S, 18S, and 28S rRNA types during zebrafish development. RNA (new York, NY) 23:1188–1199
Mangum JE, Hardee JP, Fix DK, Puppa MJ, Elkes J, Altomare D, Bykhovskaya Y, Campagna DR, Schmidt PJ, Sendamarai AK et al (2016) Pseudouridine synthase 1 deficient mice, a model for Mitochondrial Myopathy with Sideroblastic Anemia, exhibit muscle morphology and physiology alterations. Sci Rep-Uk 6:26202
Martinez NM, Su A, Burns MC, Nussbacher JK, Schaening C, Sathe S, Yeo GW, Gilbert WV (2022) Pseudouridine synthases modify human pre-mRNA co-transcriptionally and affect pre-mRNA processing. Mol Cell 82:645-659.e9
McCown PJ, Ruszkowska A, Kunkler CN, Breger K, Hulewicz JP, Wang MC, Springer NA, Brown JA (2020) Naturally occurring modified ribonucleosides. Wiley Interdiscip Rev Rna 11:e1595–e1595
Medina-Muñoz SG, Kushawah G, Castellano LA, Diez M, DeVore ML, Salazar MJB, Bazzini AA (2021) Crosstalk between codon optimality and cis-regulatory elements dictates mRNA stability. Genome Biol 22:14
Meyer B, Wurm JP, Kötter P, Leisegang MS, Schilling V, Buchhaupt M, Held M, Bahr U, Karas M, Heckel A et al (2011) The Bowen-Conradi syndrome protein Nep1 (Emg1) has a dual role in eukaryotic ribosome biogenesis, as an essential assembly factor and in the methylation of Ψ1191 in yeast 18S rRNA. Nucleic Acids Res 39:1526–1537
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149(7):1635–1646
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian S-B, Jaffrey SR (2015) 5’ UTR m(6)A promotes cap-independent translation. Cell 163:999–1010
Mishima Y, Tomari Y (2016) Codon usage and 3′ UTR length determine maternal mRNA stability in zebrafish. Mol Cell 61:874–885
Mishima Y, Han P, Ishibashi K, Kimura S, Iwasaki S (2022) Ribosome slowdown triggers codon-mediated mRNA decay independently of ribosome quality control. Embo J 41:e109256
Mladenova D, Barry G, Konen LM, Pineda SS, Guennewig B, Avesson L, Zinn R, Schonrock N, Bitar M, Jonkhout N et al (2018) Adar3 Is involved in learning and memory in mice. Front Neurosci-Switz 12:243
Narayanan M, Ramsey K, Grebe T, Schrauwen I, Szelinger S, Huentelman M, Craig D, Narayanan V, Group CR (2015) Case Report: Compound heterozygous nonsense mutations in TRMT10A are associated with microcephaly, delayed development, and periventricular white matter hyperintensities. Research 4:912
Nishikura K (2010) Functions and regulation of RNA editing by ADAR deaminases. Annu Rev Biochem 79:321–349
Nishikura K (2016) A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17:83–96
O’Connell MA, Mannion NM, Keegan LP (2015) The epitranscriptome and innate immunity. Plos Genet 11:e1005687
Oakes E, Anderson A, Cohen-Gadol A, Hundley HA (2017) Adenosine deaminase that acts on RNA 3 (ADAR3) Binding to glutamate receptor subunit B Pre-mRNA inhibits RNA editing in glioblastoma*. J Biol Chem 292:4326–4335
Oncul U, Unal-Ince E, Kuloglu Z, Teber-Tiras S, Kaygusuz G, Eminoglu FT (2021) A Novel PUS1 mutation in 2 siblings with MLASA syndrome: a review of the literature. J Pediatric Hematology Oncol 43:e592–e595
Ontiveros RJ, Shen H, Stoute J, Yanas A, Cui Y, Zhang Y, Liu KF (2020) Coordination of mRNA and tRNA methylations by TRMT10A. Proc National Acad Sci 117:7782–7791
Ougland R, Zhang C-M, Liiv A, Johansen RF, Seeberg E, Hou Y-M, Remme J, Falnes PØ (2004) AlkB Restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol Cell 16:107–116
Ozanick S, Krecic A, Andersland J, Anderson JT (2005) The bipartite structure of the tRNA m1A58 methyltransferase from S. cerevisiae is conserved in humans. RNA 11:1281–1290
Penning A, Jeschke J, Fuks F (2022) Why novel mRNA modifications are so challenging and what we can do about it. Nat Rev Mol Cell Bio 23:385–386
Perezgrovas-Saltijeral A, Rajkumar AP, Knight HM. 2022. Differential expression of m5C RNA methyltransferase genes NSUN6 and NSUN7 in Alzheimer’s disease and Traumatic Brain Injury
Petri R, Malmevik J, Fasching L, Åkerblom M, Jakobsson J (2014) miRNAs in brain development. Exp Cell Res 321:84–89
Piekna-Przybylska D, Decatur WA, Fournier MJ (2008) The 3D rRNA modification maps database: with interactive tools for ribosome analysis. Nucleic Acids Res 36:D178–D183
Purchal MK, Eyler DE, Tardu M, Franco MK, Korn MM, Khan T, McNassor R, Giles R, Lev K, Sharma H et al (2022) Pseudouridine synthase 7 is an opportunistic enzyme that binds and modifies substrates with diverse sequences and structures. P Natl Acad Sci Usa 119:e2109708119
Purta E, van Vliet F, Tricot C, Bie LGD, Feder M, Skowronek K, Droogmans L, Bujnicki JM (2005) Sequence–structure–function relationships of a tRNA (m7G46) methyltransferase studied by homology modeling and site-directed mutagenesis. Proteins Struct Funct Bioinform 59:482–488
Qiu S, Li W, Xiong H, Liu D, Bai Y, Wu K, Zhang X, Yang H, Ma K, Hou Y et al (2016) Single-cell RNA sequencing reveals dynamic changes in A-to-I RNA editome during early human embryogenesis. BMC Genomics 17:766
Qiu X, He H, Huang Y, Wang J, Xiao Y (2020) Genome-wide identification of m6A-associated single-nucleotide polymorphisms in Parkinson’s disease. Neurosci Lett 737:135315
Quin J, Sedmík J, Vukić D, Khan A, Keegan LP, O’Connell MA (2021) ADAR RNA Modifications, the Epitranscriptome and Innate Immunity. Trends Biochem Sci 46:758–771
Rai K, Chidester S, Zavala CV, Manos EJ, James SR, Karpf AR, Jones DA, Cairns BR (2007) Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Gene Dev 21:261–266
Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O’Connell MA, Li JB (2013) Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10:128–132
Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, Cordon-Cardo C, Pandolfi PP (2003) Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 299:259–262
Sapiro AL, Shmueli A, Henry GL, Li Q, Shalit T, Yaron O, Paas Y, Li JB, Shohat-Ophir G (2019) Illuminating spatial A-to-I RNA editing signatures within the Drosophila brain. P Natl Acad Sci Usa 116:2318–2327
Savva YA, Rieder LE, Reenan RA (2012) The ADAR protein family. Genome Biol 13:252
Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, Engreitz JM, Guttman M, Satija R, Lander ES et al (2014) Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159:148–162
Shaheen R, Han L, Faqeih E, Ewida N, Alobeid E, Phizicky EM, Alkuraya FS (2016) A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum Genet 135:707–713
Shaheen R, Tasak M, Maddirevula S, Abdel-Salam GMH, Sayed ISM, Alazami AM, Al-Sheddi T, Alobeid E, Phizicky EM, Alkuraya FS (2019) PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum Genet 138:231–239
Sharkia R, Zalan A, Jabareen-Masri A, Zahalka H, Mahajnah M (2019) A new case confirming and expanding the phenotype spectrum of ADAT3-related intellectual disability syndrome. Eur J Med Genet 62:103549
Sharma S, Langhendries J-L, Watzinger P, Kötter P, Entian K-D, Lafontaine DLJ (2015) Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res 43:2242–2258
Shtrichman R, Germanguz I, Mandel R, Ziskind A, Nahor I, Safran M, Osenberg S, Sherf O, Rechavi G, Itskovitz-Eldor J (2012) Altered A-to-I RNA editing in human embryogenesis. PLoS ONE 7:e41576
Shu L, Huang X, Cheng X, Li X (2021) Emerging roles of N6-methyladenosine modification in neurodevelopment and neurodegeneration. Cells 10(10):2694
Song H, Zhang J, Liu B, Xu J, Cai B, Yang H, Straube J, Yu X, Ma T (2022) Biological roles of RNA m5C modification and its implications in cancer immunotherapy. Biomark Res 10:15
Spenkuch F, Motorin Y, Helm M (2014) Pseudouridine: still mysterious, but never a fake (uridine)! RNA Biol 11(12):1540–1554
Sui X, Hu Y, Ren C, Cao Q, Zhou S, Cao Y, Li M, Shu W, Huo R (2020) METTL3-mediated m6A is required for murine oocyte maturation and maternal-to-zygotic transition. Cell Cycle 19:1–14
Tajaddod M, Jantsch MF, Licht K (2016) The dynamic epitranscriptome: A to I editing modulates genetic information. Chromosoma 125:51–63
Tan K-T, Ding L-W, Wu C-S, Tenen DG, Yang H (2021) Repurposing RNA sequencing for discovery of RNA modifications in clinical cohorts. Sci Adv 7:eabd2605
Tariq A, Jantsch MF (2012) Transcript diversification in the nervous system: A to I RNA editing in CNS function and disease development. Front Neurosci-Switz 6:99
Tavakoli S, Nabizadehmashhadtoroghi M, Makhamreh A, Gamper H, Rezapour NK, Hou Y-M, Wanunu M, Rouhanifard SH. 2022. Detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct, long-read sequencing. Biorxiv 2021.11.03.467190
Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL (2002) RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. Embo J 21:6025–6035
Trixl L, Amort T, Wille A, Zinni M, Ebner S, Hechenberger C, Eichin F, Gabriel H, Schoberleitner I, Huang A et al (2018) RNA cytosine methyltransferase Nsun3 regulates embryonic stem cell differentiation by promoting mitochondrial activity. Cell Mol Life Sci 75:1483–1497
Vastenhouw NL, Cao WX, Lipshitz HD (2019) The maternal-to-zygotic transition revisited. Development 146:dev161471
Vilardo E, Amman F, Toth U, Kotter A, Helm M, Rossmanith W (2020) Functional characterization of the human tRNA methyltransferases TRMT10A and TRMT10B. Nucleic Acids Res 48:6157–6169
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M et al (2017) The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23:1369–1376
Waku T, Nakajima Y, Yokoyama W, Nomura N, Kako K, Kobayashi A, Shimizu T, Fukamizu A (2016) NML-mediated rRNA base methylation links ribosomal subunit formation to cell proliferation in a p53-dependent manner. J Cell Sci 129:2382–2393
Warner WA, Spencer DH, Trissal M, White BS, Helton N, Ley TJ, Link DC (2018) Expression profiling of snoRNAs in normal hematopoiesis and AML. Blood Adv 2:151–163
Weng Y-L, Wang X, An R, Cassin J, Vissers C, Liu Y, Liu Y, Xu T, Wang X, Wong SZH, Joseph J, Dore LC, Dong Q, Zheng W, Jin P, Wu H, Shen B, Zhuang X, He C, Ming G (2018) Epitranscriptomic m6A regulation of axon regeneration in the adult mammalian nervous system. Neuron 97(2):313-325.e6
Wiener D, Schwartz S (2020) The epitranscriptome beyond m6A. Nat Rev Genetics 22:119–131
Wiener D, Schwartz S (2021) The epitranscriptome beyond m6A. Nat Rev Genet 22:119–131
Wu X, Sandhu S, Patel N, Triggs-Raine B, Ding H (2010) EMG1 is essential for mouse pre-implantation embryo development. Bmc Dev Biol 10:99
Wu Y, Xu X, Qi M, Chen C, Li M, Yan R, Kou X, Zhao Y, Liu W, Li Y et al (2022) N6-methyladenosine regulates maternal RNA maintenance in oocytes and timely RNA decay during mouse maternal-to-zygotic transition. Nat Cell Biol 24:917–927
Wu Y, Xu X, Qi M, Chen C, Li M, Yan R, Kou X, Zhao Y, Liu W, Li Y, et al. 2021. The dual role of N6-methyladenosine on mouse maternal RNAs and 2-cell specific RNAs revealed by ULI-MeRIP sequencing. Biorxiv 2021.12.13.472368
Xia H, Zhong C, Wu X, Chen J, Tao B, Xia X, Shi M, Zhu Z, Trudeau VL, Hu W (2018) Mettl3 mutation disrupts gamete maturation and reduces fertility in zebrafish. Genetics 208:729–743
Yang X, Yang Y, Sun B-F, Chen Y-S, Xu J-W, Lai W-Y, Li A, Wang X, Bhattarai DP, Xiao W et al (2017) 5-methylcytosine promotes mRNA export — NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res 27:606–625
Yang Y, Hsu PJ, Chen Y-S, Yang Y-G (2018) Dynamic transcriptomic m6A decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 28:616–624
Yang Y, Wang L, Han X, Yang W-L, Zhang M, Ma H-L, Sun B-F, Li A, Xia J, Chen J et al (2019) RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol Cell 75:1188-1202.e11
Yao QJ, Sang L, Lin M, Yin X, Dong W, Gong Y, Zhou BO (2018) Mettl3–Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res 28:952–954
Yoon A, Peng G, Brandenburger Y, Brandenburg Y, Zollo O, Xu W, Rego E, Ruggero D (2006) Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science (new York, NY) 312:902–906
Yoon K-J, Ringeling FR, Vissers C, Jacob F, Pokrass M, Jimenez-Cyrus D, Su Y, Kim N-S, Zhu Y, Zheng L, Kim S, Wang X, Doré LC, Jin P, Regot S, Zhuang X, Canzar S, He C, Ming G, Song H (2017) Temporal control of mammalian cortical neurogenesis by m6A methylation. Cell 171(4):877-889.e17
Zaccara S, Ries RJ, Jaffrey SR (2019) Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Bio 20:608–624
Zeng H (2022) What is a cell type and how to define it? Cell 185:2739–2755
Zhang Y, Morimoto K, Danilova N, Zhang B, Lin S (2012) Zebrafish models for dyskeratosis congenita reveal critical roles of p53 activation contributing to hematopoietic defects through RNA processing. PLoS ONE 7:e30188
Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, Lv J, Heng J, Ding Y, Xue Y et al (2017) m6A modulates haematopoietic stem and progenitor cell specification. Nature 549:273–276
Zhang L-S, Liu C, Ma H, Dai Q, Sun H-L, Luo G, Zhang Z, Zhang L, Hu L, Dong X et al (2019) Transcriptome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA. Mol Cell 74:1304-1316.e8
Zhang M, Cao Y, Wu H, Li H (2021a) Brain imaging features of children with Hoyeraal-Hreidarsson syndrome. Brain Behav 11:e02079
Zhang W, Qian Y, Jia G (2021b) The detection and functions of RNA modification m6A based on m6A writers and erasers. J Biological Chem 297:100973
Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C (2017) m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542:475–478
Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C (2017a) m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542:475–478
Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C (2017b) m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542:475–478
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian S-B (2015) Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature 526:591–594
Zou F, Tu R, Duan B, Yang Z, Ping Z, Song X, Chen S, Price A, Li H, Scott A et al (2020) Drosophila YBX1 homolog YPS promotes ovarian germ line stem cell development by preferentially recognizing 5-methylcytosine RNAs. Proc National Acad Sci 117:3603–3609
Acknowledgements
Work in the laboratory of MV was supported by the Hungarian National Research, Development and Innovation Office (NRDI) Grant NRDI-FK124230 and ELTE Eötvös Loránd University Institutional Excellence Program. MV is also a János Bolyai Fellow of the Hungarian Academy of Sciences.
Funding
Open access funding provided by Eötvös Loránd University.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hamar, R., Varga, M. The role of post-transcriptional modifications during development. BIOLOGIA FUTURA 74, 45–59 (2023). https://doi.org/10.1007/s42977-022-00142-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42977-022-00142-3