
Abstract
Kaolins and clays are important raw materials for production of supplementary cementitious materials and geopolymer precursors through thermal activation by calcination beyond dehydroxylation (DHX). Both types of clay contain different polytypes and disordered structures of kaolinite but little is known about the impact of the layer stacking of dioctahedral 1:1 layer silicates on optimum thermal activation conditions and following reactivity with alkaline solutions. The objective of the present study was to improve understanding of the impact of layer stacking in dioctahedral 1:1 layer silicates on the thermal activation by investigating the atomic structure after dehydroxylation. Heating experiments by simultaneous thermal analysis (STA) followed by characterization of the dehydroxylated materials by nuclear magnetic resonance spectroscopy (NMR) and scanning electron microscopy (SEM) together with first-principles calculations were performed. Density functional theory (DFT) was utilized for correlation of geometry-optimized structures to thermodynamic stability. The resulting volumes of unit cells were compared with data from dilatometry studies. The local structure changes were correlated with experimental results of increasing DHX temperature in the following order: disordered kaolinite, kaolinite, and dickite, whereupon dickite showed two dehydroxylation steps. Intermediate structures were found that were thermodynamically stable and partially dehydroxylated to a degree of DHX of 75% for kaolinite, 25% for disordered kaolinite, and 50% for dickite. These thermodynamically stable, partially dehydroxylated intermediates contained AlV while metakaolinite and metadickite contained only AlIV with a strongly distorted coordination shell. These results indicate strongly the necessity for characterization of the structure of dioctahedral 1:1 layer silicates in kaolins and clays as a key parameter to predict optimized calcination conditions and resulting reactivity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Worldwide, several million tons of metakaolin are manufactured every year by calcination of kaolin for various applications, e.g. as concrete additive, supplementary cementitious material, and geopolymer precursor but also as fillers and coating for specialty paper, or as a paint extender.
The behavior of clay minerals during calcination determines optimal activation of pozzolanic reactivity or reactivity as a geopolymer precursor. Activation is achieved upon dehydroxylation of the octahedral sheet. Thereby surface reactivity is improved. Overheating results in particle agglomeration and crystallization of inactive high-temperature phases. The temperatures at which dehydroxylation and recrystallization occur are determined by the clay mineral structure (Snellings et al. 2012). The dehydroxylation process has to be understood in detail for each clay mineral structure to tune structures of dehydroxylates (meta clay minerals) and to extend application fields.
During calcination of kaolin, its main mineral phase – kaolinite – is dehydroxylated into metakaolinite (Brindley and Nakahira 1959) according to
with Or representing residual oxygen.
Dehydroxylation of kaolinite commonly occurs between 400 and 700°C, but the dehydroxylation temperature depends on: (1) heating conditions (Bellotto et al. 1995; de Ligny and Navrotsky 1999; Ptáček et al. 2014); (2) morphology and size of kaolinite mineral particles (Kaloumenou et al. 1999); and (3) structural layer stacking order. Thereby, unknown series of stacking faults are very often described as ‘low-crystallinity’ or a result of ‘disorder of kaolinites.’ So-called disordered kaolinites commonly dehydroxylate at lower temperatures than ordered kaolinites (Smykatz-Kloss 1974).
In any case, a broad temperature gap between dehydroxylation and recrystallization favors the occurrence of metakaolinite and, if calcination is terminated prior to the formation of a defect, Al-Si spinel or even mullite at higher temperatures (Brown et al. 1985). The metakaolinite is highly reactive in alkaline solutions. This reactivity is explained by the transition of octahedrally coordinated Al (AlVI) to 5-fold coordination (AlV), established experimentally by the occurrence of a peak at ~30 ppm in 27Al nuclear magnetic resonance (NMR) spectra (Rocha and Klinowski 1990a, 1990b).
On the other hand, the octahedral Al in a fully dehydroxylated kaolinite must end up as 4-fold coordinated (AlIV) and doubts about the assignment of peaks at 30 ppm in 27Al NMR spectra to AlV are well founded. The occurrence of this resonance is more likely related to the presence of Al in much distorted tetrahedral sites (MacKenzie and Smith 2002).
While disordered kaolinites dehydroxylate at lower temperatures than well ordered kaolinites, controversial results exist about the reactivity in alkaline solutions. Some studies found that dehydroxylated disordered kaolinites show a better reactivity in alkaline solutions (Kakali et al. 2001; Bich et al. 2009; Snellings et al. 2012; Hollanders et al. 2016) while other authors reported contrary findings (e.g. Tironi et al. 2014). These observations need deeper investigation to understand the relation between the dehydroxylation behavior, the structure of metakaolinite, and reactivity.
Well ordered kaolinites are very rare, and kaolinites as main constituents of kaolins show a large variability in the degree of their structural disorder. Traditionally, this variability has been evaluated by empirical indices of “crystallinity,” i.e. by the Hinckley Index (Hinckley 1963) calculated from peak height of certain hkl reflections in XRD patterns, the P0 and P2 ratio of the OH-stretching bands of FTIR spectra, or the slope ratio of the endothermal DSC or DTA dehydroxylation peak of kaolinites (Bich et al. 2009).
Such indices cannot be related directly to the type of disorder but are used commonly to compare varieties in a qualitative manner. Drits and Tchoubar (1990) discussed several models of stacking faults, e.g. ±b/3 translations, ±120° rotations, displacement of octahedral vacancies, and models containing several enantiomorphic B-(vacant) and C-(vacant) layers. The latter model has been proved to be the most probable one and the diffraction patterns of many natural kaolinites can be described successfully based on this general principle (e.g. Bookin et al. 1989; Ufer et al. 2015; Sakharov et al. 2016). Based on this approach, ideal kaolinite consists of regularly stacked B-layers and the ideal dickite polytype is characterized by regularly stacked layers with vacant B and C octahedral positions (Fig. 1) and a translation of the layers of a/3.

Octahedral sheet of dioctahedral 1:1 layers with B vacancies (left) and with C vacancies (right)
The dehydroxylation temperature (TDHX) of dickite is ~100 K higher than TDHX of kaolinite (de Ligny and Navrotsky 1999) and most strikingly, dilatometry measurements revealed that kaolinite shrinks while dickite expands with dehydroxylation (Schomburg and Störr 1984). This observation also needs a more systematic investigation at the atomic level and must be considered in terms of current understanding of the ideal thermal activation of various kaolin raw materials.
Previous studies using density functional theory (DFT) by White et al. (2010a, 2010b) or molecular dynamics (MD) by Sperinck et al. (2011) focused on the dehydroxylation of kaolinite into metakaolinite and little attention has been given to dehydroxylation of disordered kaolinite and dickite.
The present study focused on the experimental dehydroxylation of kaolinite, disordered kaolinite, and dickite; and the differences at the atomistic level of the formation of metakaolinite from well-ordered and disordered kaolinite; and of the formation of metadickite by DFT.
The first objective of the present study was to evaluate the impact of layer stacking and disorder in dioctahedral 2:1 layer silicates on the TDHX, the number of dehydroxylation steps, and the volume changes of the minerals after DHX. The second was to elucidate the nature of apparent five-fold coordinated Al in the metamaterials.
MATERIALS AND METHODS
Materials
The kaolinite samples used in the present study were KBE-1 and KGa-2. The kaolinite KBE-1 (D) was provided by Amberger Kaolinwerke Eduard Kick GmbH & Co. KG (Hirschau, Germany) and originates from a residual kaolin deposit. Such kaolins typically contain quite well ordered kaolinites. The kaolinite KGa-2 is a sedimentary kaolin from a mine site in Warren County, Georgia (USA) obtained from the Source Clays Repository of The Clay Minerals Society. KGa-2 has been examined extensively in several studies (e.g. Ufer et al. 2015; Sakharov et al. 2016) and represents a disordered variety.
The <2 μm fraction of KBE-1 was separated by sedimentation to reduce the amount of accessory minerals, especially mica. The sample was called KBE1_M2. KGa-2 was not separated because the amount of mica was very small.
Dickite samples were from Altenberg (Saxony, Germany) and from Kohlendorf (Poland). They were used for STA measurement.
Experimental Procedures
The kaolinite content and accessory minerals were identified and quantified by X-ray diffraction (XRD) analysis of powdered samples (diameter 20 mm, top loading). A Bruker D8 Advance A25 diffractometer (Bruker AXS GmbH, Karlsruhe, Germany) equipped with a LYNXEYE XE Detector (opening degree 2.94° and 192 channels). Patterns were recorded between 5 and 80°2θ with CuKα radiation with a counting time of 3 s and a step size of 0.02°2θ, an automatic slit (primary side), Soller collimator of 2.5° (primary and secondary side), and an automatic knife edge above the specimen holder. The Rietveld software AUTOQUAN (Version 2.8.0; GE Inspection Technologies GmbH, Ahrensburg, Germany) was utilized for quantitative analysis. The XRD pattern in the range 19–24°2θ CuKα was used to calculate the Hinckley Index (Hinckley 1963) which mirrors the crystallinity of the kaolinite.
The DHX behavior was studied by simultaneous thermal analysis (STA). The measurements were performed on a STA 449 C Jupiter (Netzsch-Gerätebau GmbH, Selb, Germany) equipped with a thermogravimetry/differential scanning calorimetry (TG/DSC) sample holder. The STA is connected to a quadrupole mass spectrometer (MS) 403 C Aëolos (InProcess Instruments (IPI)/NETZSCH-Gerätebau GmbH, Selb, Germany) by a heated quartz glass capillary (T = 230°C). Samples were heated at 10°C/min from 35 to 1100°C in 50 mL/min streaming dry air (SynA; 79 mass% N2/21 mass% O2) mixed with 20 mL/min N2 (protective gas). Pt/Rh crucibles (diameter 5 mm and height 5 mm) with a punched lid were filled with 100 mg of material. An empty Pt/Rh crucible with lid served as an inert reference sample. The mass loss during thermal reactions was determined from the thermogravimetric (TG) curve. Any mass loss after dehydration (DHD) was normalized to the dry weight after DHD.
Peak decomposition and determination of peak areas of the MS curves of evolved water (m/z = 18) of the dickites was executed by means of the PeakFit software (Jandel Scientific, Version 4.12; SeaSolve Software, Framingham, Massachusetts, USA).
The STA was also used to prepare several thermally treated kaolinite and dickite samples. The calcination temperatures were between 200 and 900°C; and were selected to achieve full DHD, partial DHX, full DHX, and structural decomposition before recrystallization of high-temperature phases. A cooling rate of 20°C/min was applied.
The Al coordination and the structural changes were studied by 27Al magic angle spinning nuclear magnetic resonance (MAS NMR) spectroscopy. 27Al MAS NMR measurements were performed at room temperature on a Bruker Avance spectrometer 400 ultrashield with a magnetic field of 9.4 T. The 27Al MAS NMR spectra were obtained at a frequency of 104.28 MHz. The MAS NMR measurements were done using a 4 mm zirconium rotor at a spinning speed of 12 kHz in a dry nitrogen atmosphere. The chemical shift (δ) for 27Al was referenced to Al(NO3)3·9 H2O (solid, δ = 0 ppm).
The software DMFit (2011), developed by Dominique Massiot, Orléans, France, (http://nmr.cemhti.cnrs-orleans.fr/Dmfit/Howto/CSA/CSA_MAS.aspx) was used to evaluate MAS NMR spectra (Massiot et al. 2002).
Particle morphologies of the raw and heated samples were examined using a Philips XL 30 FEG environmental scanning electron microscope (ESEM; FEI Europe, Eindhoven, The Netherlands). Without further pre-treatment small amounts of sample powder were glued onto aluminum SEM-holders using conductive tape (Leit-C, Plano GmbH, Wetzlar, Germany). To improve the image quality, the samples were sputtered with a thin conductive layer (5 nm Au/Pd 80/20) and were investigated using an acceleration voltage of 15 kV.
Computational Methods
In the present study, Density Functional Theory (DFT) modeling was employed for better understanding of the process of formation of metakaolinite, metadiskaolinite, and metadickite during dehydroxylation; and of their resulting properties as defined in the Vienna ab initio simulation package (VASP) (Kresse and Hafner 1993; Kresse and Furthmuller 1996) for the calculation of the electronic structure (Giraudo et al. 2015). The electron–ion interaction was described within the projector-augmented wave (PAW) scheme (Kresse and Joubert 1999). The electronic wave functions were enlarged into plane waves up to a kinetic energy cutoff of 500 eV. The Perdew-Burke-Ernzerhof (PBE) functional was used to characterize electron exchange and correlation energy within the Generalized Gradient Approximation (GGA) (Perdew et al. 1996). The optimization of atomic coordinates was implemented via a conjugate gradient, which uses the total energy and the Hellmann-Feynman forces on the atoms. The structures were presumed to be entirely relaxed when the forces on the ions were <0.01 eV/Å. The open-source XcrySDen software has been used to indicate the crystalline structure and electron densities (Kokalj 1999).
The initial periodic 1×1×3 ideal kaolinite supercell (Fig. 2 left) has been simulated with a unit cell of a = 5.15 Å, b = 8.94 Å, c = 7.39 Å, α = 91.92°, β = 105.04°, γ = 89.79°, and B-vacant 1:1 layers (Bish 1993). Of course, a real kaolinite containing some stacking faults caused by layer shifts or twinning of left/right handed layers may not be described by a size-limited super cell suitable for DFT modeling. As a proxy for a disordered variety, a 1×1×3 supercell has been derived by b/3 translation of the B-vacant 1:1 layers (Fig. 2 middle). The initial periodic 1×1×6 dickite supercell has been simulated with a unit cell of a = 5.1444 Å, b = 8.9334 Å, c = 14.3896 Å, α = 90.0000°, β = 96.5440°, and γ = 90.0000° (Rocha et al. 2018); and alternating B-vacant and C-vacant 1:1 layers that are shifted by a/3 (Figs 1 and 2 right). The crystal structure and chemical bonding for kaolinite, disordered kaolinite, and dickite in each layer remained identical; the lengths of hydrogen bonding in kaolinite, disordered kaolinite, and dickite are different, however, due to different configurations of the layers.

Super cells of kaolinite (left), a disordered kaolinite (middle), and dickite (right)
RESULTS AND DISSCUSION
X-ray Diffraction Analysis
Measurements by XRD revealed that both KBE1_M2 and KGa-2 contained >90% kaolinite and small amounts of accessory minerals (Table 1). The Hinckley Index of KBE1_M2 was 1.63, characteristic of a well-ordered kaolinite (Komarneni et al. 1985a; Plançon et al. 1989). The small Hinckley Index of KGa-2 of 0.5 was in accordance with published values of 0.24–0.41 (Metz and Ganor 2001; Du et al. 2010; Ndlovu et al. 2015) and confirmed a high degree of structural disorder.
KBE1_M2 consists of 46–47 mass% ordered kaolinite and 50–51 mass% disordered kaolinite. The disordered kaolinite is characterized by 93% BB/7% BC stacking sequences. 88% of BB sequences have no additional ~b/3 stacking errors and, thus, the kaolinite is low b-axis error-ordered. KGa-2 is characterized by 86% BB stacking sequences with a strong tendency for additional ~b/3 translations (Ufer et al. 2015) and a low abundance of BC stacking. Thus, KGa-2 is intermediate disordered.
Both dickites are also quite pure and well ordered. The dickite from Altenberg (dickite AB) actually consisted of 100% dickite while the dickite from Kohlendorf (dickite KD) contained 4% accessory minerals.
Simultaneous Thermal Analysis
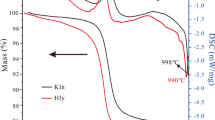
Simultaneous thermal analysis of the two natural, air-dry kaolinites (Fig. 3) showed one endothermic peak in the region between 400 and 800°C, which was associated with a maximum in the MS curve of evolved water (m/z = 18) at 562°C (KBE1_M2) and at 546°C (KGa-2) due to the dehydroxylation (DHX) of kaolinite. The normalized mass loss of 13.25 mass% and 13.24 mass% for KBE1_M2 and KGa-2, respectively, reflected perfectly the kaolinite content in both materials of ~95% determined by XRD. The DHX of KGa-2 occurred at a slightly lower temperature than did DHX of the KBE1_M2 (Figs 3 and 4).

DSC curve (green colors) and MS curve (m/z =18) of evolved water (blue colors) for both kaolinites (left) and DSC curve (red colors) and MS curve (m/z =18) of evolved water (blue colors) for both dickites (right)

Extent of DHX as a percentage of stoichiometric mass loss of 13.95% of kaolinites (left) and dickite (right)
At temperatures >950°C, two further peaks were visible in the DSC curve of both kaolinites. Neither peak is associated with any gas release and can be assigned to the first and second recrystallization of kaolinite into a spinel and mullite (Mackenzie et al. 1985).
The DHX of both dickites occurred at between 500 and 900°C, in two steps. A two-step DHX of dickites was also observed in previous studies (Frost and Vassallo 1996; Franco and Ruiz Cruz 2006). The first DHX of dickite KD is very broad while dickite AB shows a sharp peak at ~650°C. The maximum of the second endothermal peak and the second peak in the MS curve of evolved water (m/z = 18) was at ~679°C (Fig. 3). The degree of DHX of dickite KD was greater, at temperatures between 400 and 650°C, before full DHX is reached for both dickites (Fig. 4). The normalized mass loss of 13.95% (dickite AB) and 13.43% (dickite KD) corresponds to the dickite content determined by XRD.
The ratio of peak areas with a peak temperature below and above 665°C was ~40:60 for both dickites (Fig. 5).

MS curve (m/z =18) of evolved water during DHX of dickite KD (left) and dickite AB (right) with decomposed peaks
NMR Measurements
The 27Al MAS NMR spectra (Fig. 6) of the two unheated kaolinite samples showed one main resonance signal at ~0 ppm, which revealed octahedrally coordinated Al (AlVI). Both show an additional minor resonance signal at ~75 ppm, which indicated tetrahedrally (AlIV) coordinated Al, confirming the presence of dioctahedral mica with Al substitutions for Si in the tetrahedral sheet.

27Al MAS NMR spectra of the two untreated kaolinite samples (* ssb = spinning side bands)
The 27Al MAS NMR spectra of KBE1_M2 showed an additional signal between 20 and 40 ppm. Broad 27Al signals at ~20 to 40 ppm were observed for poorly crystalline aluminosilicates, X-ray amorphous aluminosilicate glasses, or gels (MacKenzie 2000); or for minerals with 5-fold coordinated Al (AlV) such as andalusite (Lippmaa et al. 1986), grandidierite (Smith and Steuernagel 1992), or augelite (Bleam et al. 1989). Neither amorphous material nor the above-mentioned minerals could be detected in KBE1_M2.
With increasing temperature, the overall intensity of the 27Al MAS NMR signals of both samples decreased drastically (Fig. 7). This indicates that only a small part of the remaining short-range order is reflected, as metakaolinite is characterized by a complex amorphous structure (Brindley and Nakahira 1959; Bellotto et al. 1995).

27Al MAS NMR spectra of KBE1_M2 and KGa-2 heated to indicated temperatures without isothermal annealing (* ssb = spinning side bands)
The recrystallization products of kaolinite are spinel and mullite. Calculations based on the structural formulaa of spinel (Verwey 1935; Low and McPherson 1988; Rozita et al. 2010) and mullite (Angel et al. 1991; Lee et al. 1997; Schneider et al. 2008) showed that the amount of AlIV ranges between 25–37.5% and 56–58%, respectively (Fig. 8). Accordingly, the AlVI is in the range 62.5–75% for spinel and 42–44% for mullite (Fig. 8). The measured AlVI and AlIV ratios match the ratio in the defect spinel/inverse spinel at ~1100°C and the ratio in the mullite at ~1400°C.

Amount of AlIV in the kaolinite–mullite reaction
Both samples showed a decreasing AlVI signal and an increasing AlIV signal as well as an increasing signal between 28 and 40 ppm with progressive DHX (Figs. 7 and 9). Thereby, the chemical shift of 28–40 ppm for the new signal is greater than published data of 20–35 ppm (Komarneni et al. 1985b; Rocha et al. 1990; He et al. 2003; Fabbri et al. 2013) and is closer to common chemical shifts of AlIV (50–80 ppm) in clay minerals. With incipient recrystallization after DHX the AlVI signal increased again and the signal between 28–40 ppm decreased (Fig. 7). The 27Al MAS NMR spectra of both kaolinites at 1100°C showed one signal for AlVI and one for AlIV.

Fractions of different Al coordinations vs. calcination temperature of KBE1_M2 and KGa-2
With increasing temperature, the quadrupole coupling constant (QCC) for all three signals in the 27 Al NMR spectra increased to values >4 MHz (Fig. 10), characteristic of heavily distorted coordination shells of Al (Fyfe et al. 2000, 2001; van Bokhoven et al. 2000; Gore et al. 2002; Omegna et al. 2003).

Quadrupole coupling constants (QCC) for KBE-1_M2 (left) and KGa-2 (right)
First-Principles Calculations
The transformation of kaolinite, disordered kaolinite, and dickite into metakaolinite, metadiskaolinite, and metadickite via several steps through dehydroxylation was investigated using DFT. First, the thermodynamic grand canonical potential was calculated for the identification of stable phases for the desorption process of water. The desorption energy alone does not allow conclusions about the stability of a specific structure, however. Rather, one has to take into account the chemical potentials μ(Ai) of the surface constituents, Ai, in order to compare energetically the interfaces with different stoichiometries. The ground state of the surface is determined by the minimum of the thermodynamic grand canonical potential Ω:
where F = E – TS is the surface free energy. Here, F is approximated by the total surface energy, E, assuming similar entropy contributions, S, for different adsorption configurations. In fact, the differences in vibrational free energy and electronic entropy are typically several orders of magnitude smaller than adsorption energies resulting from chemical-bond formation as found in the present case. The last term on the right-hand side accounts for the energy changes due to a possible surface charge, q, which creates a dependence on the chemical potential of electrons given here by the Fermi level (EF) measured relative to the valence-band maximum (EVBM).
The transformation of kaolinite into the Al12Si12O39(OH)6 phase occurred at the third step of water removal when the chemical potential of water was –1.93 eV (Fig. 11 left). The final product was a metakaolinite at a water chemical potential of –2.67 eV. A phase which had the chemical formula of Al12Si12O39(OH)6 was produced. Other phases are unstable and do not exist thermodynamically. The phase transformation from disordered kaolinite into Al12Si12O33(OH)18 happens at the first step of water removal and when the chemical potential of water is –2.03 eV (Fig. 12 left). The final product was metadiskaolinite when the chemical potential of water was –2.319 eV. The intersection of the three lines of purple, red, and green (Fig. 13 left) for the phase transformation from dickite into metadickite is not very clear but was much more evident from Fig. 14. The dehydroxylation of dickite to Al24Si24O72(OH)24 took place at the second step of water removal at a water chemical potential of –2.717 eV (Fig. 14). The final product was metadickite at the chemical potential of water of –2.82 eV.

Calculated phase diagram of kaolinite–metakaolinite transformation as a function of the water chemical potential (left) and p,T-phase diagram (right)

Calculated phase diagram of disordered kaolinite–metadiskaolinite transformation as a function of the water chemical potential (left) and p,T-phase diagram (right)

Calculated phase diagram of the dickite–metadickite transformation as a function of the water chemical potential (left) and p,T phase diagram (right)

Transition of dickite (purple) to Al24Si24O72(OH)24 (red) and to final product of metadickite Al24Si24O84 (green) at the chemical potentials of –2.717 eV and –2.82 eV, respectively
The resulting phase diagrams as a function of partial pressure of water and temperature may be compared with experimental data. Dashed lines (Figs 11, 12, and 13) indicate an excellent example for the partial pressure of H2O equal to 10 Pa vs. temperature. The chemical potential can be related directly to experimental conditions. The pressure- and temperature-dependent deviation, Δμ(H2O), from the zero-temperature value obtained from the DFT calculations can be estimated within the approximation of a polyatomic ideal gas by
where kB is the Boltzmann constant, p is the pressure, T is the temperature, and λ is the de Broglie thermal wavelength of the water molecule
where m represents the molecular mass and ℏ is Planck’s constant divided by 2.
and
The first stable phase of Al12Si12O39(OH)6 was formed at the third step at 425°C as a result of releasing nine hydroxyl groups (Fig. 11, right). As expected, Al12Si12O39(OH)6 required heating to 655°C to release the three remaining hydroxyl groups resulting in the formation of metakaolinite as the final product. The first stable phase of Al12Si12O33(OH)18 happened at the first step of water removal with a heating temperature of 457°C as a result of releasing three hydroxyl groups (Fig. 12, right). To reach complete dehydroxylation, Al12Si12O33(OH)18 must be heated to 547°C for the final production of metadiskaolinite. The formation of the first stable phase of Al24Si24O72(OH)24 happened at the second step of water removal and at 665°C as a result of releasing six hydroxyl groups. The final product was metadickite (Fig. 13 right), which needed little more than 698°C for complete DHX.
Water molecules may form when hydroxyl groups are close enough to each other upon heating. During DHX the coordination of aluminum atoms changed. The first stable phase of Al12Si12O39(OH)6 (corresponding to a degree of DHX of 75%) from kaolinite is characterized by the ratio of 1:1:0 for AlIV:AlV:AlVI (Fig. 15, middle). In contrast, the ratio is 0:1:1 for AlIV:AlV:AlVI of the first stable dehydroxylated phase of Al24Si24O72(OH)24 (corresponding to a degree of DHX of 50%), observed for dickite (Fig. 17, middle).

Stable, partially dehydroxylated kaolinite, Al12Si12O30(Or)9(OH)6 (left); metakaolinite, Al12Si12O30(Or)12 (middle); and SEM images of ordered kaolinite KBE1_M2 (upper right) and metakaolinite KBE1_M2 700°C (lower right); scale bar: 2 μm
In contrast to Mackenzie et al. (1985) and White et al. (2010a), no residual OH groups were necessary to maintain the layering of the metamaterials of any of the three 1:1 layer polytypes studied (Figs. 15, 16, and 17). The SEM images of the metamaterials confirmed the remaining platelet-like morphology (Figs. 15, 16, and 17 left).

Stable, partially dehydroxylated disordered kaolinite, Al12Si12O30(Or)3(OH)18 (left); metadiskaolinite, Al12Si12O30(Or)12 (middle); and SEM images of disordered kaolinite KGa-2 (upper right) and metadiskaolinite KGa-2 615°C (lower right); scale bar: 2 μm

Stable, partially dehydroxylated dickite, Al24Si24O60(Or)12(OH)24 (left); metadickite, Al24Si24O60(Or)24 (middle); and SEM images of dickite AB (upper right) and dickite AB 700°C (lower right); scale bar: 5 μm
The DHX of kaolinite into metakaolinite, disordered kaolinite into metadiskaolinite, and dickite into metadickite must be implemented only at the stationary point by use of the Birch-Murnaghan diagram (Bleam et al. 1989). Hence, expansion or contraction of the simulation super cells in order to compute the optimized structure at the stationary point is required. The unit cells of metakaolinite and metadiskaolinite expanded after complete dehydroxylation at the stationary points (Fig. 18). The most striking finding was that the super cells of metakaolinite and metadiskaolinite at the stationary points expanded by roughly 4% and 8% in comparison with kaolinite and disordered kaolinite, respectively. In contrast, no significant changing in the super-cell volume after complete dehydroxylation of dickite was observed.

Birch-Murnaghan diagram of kaolinite-to-metakaolinite, disordered kaolinite-to-metadiskaolinite, and dickite-to-metadickite for calculation of the optimized structure at the stationary point
The volume of kaolinite decreased after DHX by ~0.8 to 2%, determined by dilatometer measurements of kaolins, and the volume of dickite increased after DHX by ~6.5 to 7% (Schomburg and Störr 1984). While the calculated values from the Birch-Murnaghan equation of state predict the changes of the material at the atomic level, a direct correlation with measurements performed by optical dilatometer might by difficult.
One of the most important differences in polytype structures concerning the material in the present study is the differing orientation of OH groups. From a thermodynamic point of view, the hydrogen bonding of dickite is more stable than the zigzag hydrogen bonding of kaolinite between layers. This finally affects the dehydroxylation temperatures and volume changes of the super cells. TDHX,disKao < TDHX,Kao < TDHX,Dic was found experimentally and confirmed results from previous studies (e.g. Stoch and Wacławska 1981a, 1981b).
H-bonding plays a considerable role in binding layers together. To discover the strongest clay mineral, the total H-bonding energy between two immediate layers was summed and divided by the area influenced. The bond length of OH determines the calculation of the H-bonding energy (Jones et al. 2006). From DFT calculations, dickite is considered to be the strongest material with an average energy of –0.032 eV/Å2, which is in accordance with experiments. Kaolinite and disordered kaolinite were specified as the second and third strongest materials after kaolinite with average energies of –0.031 eV/Å2 and –0.02 eV/Å2, respectively.
Furthermore, two different energies (–0.03 eV/Å2 and –0.032 eV/Å2) for dickite were found, correlating to the B-C-B-C stacking sequence of layers. In contrast to that, kaolinite and metakaolinite contain only a simple B-B-B sequence of layers, which leads in the end to only one sticking energy for each material. The number of sticking energies correlated with the number of peaks that were found in the STA experiment instead of with stepwise release of inner and outer hydroxyl groups of the octahedral sheet (Frost and Vassallo 1996).
The TDHX of disordered KGa-2 is less than that of well-ordered KBE1_M2. The disorder in KGa-2 is caused by b/3 translation. For dickites, no structural models are available to describe disorder by stacking faults. In any case, the two different vacancies in the dioctahedral 1:1 layers cause a two-step dehydroxylation and the a/3 translation of the 1:1 layers results in a higher dehydroxylation temperature.
The deviation for the results between experimental and simulation studies is negligible. Two main reasons are elucidated briefly here. First, results from the computational study were limited to the thermodynamic point of view. Some of the processes might be kinetically driven or kinetically hindered. Both would mean that a thermodynamic sequence of structures is not representative. Second, as White et al. (2010b) reported, more than 3×1084 possible transformation paths exist to obtain metakaolinite from kaolinite. Thus, the results here demonstrated a possible DHX path correctly, as calculation of all possible paths is impossible.
SUMMARY AND CONCLUSIONS
Calcination of kaolin as an activation technique to enhance its reactivity with alkaline solutions is an important industrial process. Kaolin commonly contains various polytypes of dioctahedral 1:1 layer silicates that dehydroxylate during calcination. TDHX,disKao < TDHX,Kao < TDHX,Dic was confirmed experimentally and by DFT calculations. Dehydroxylation of dickite occurred, thereby, in two steps and the measured amount of evolved water corresponded to 40% of the constituent hydroxyl groups during the first step of dehydroxylation. The calculated degree of DHX for the thermodynamically stable, partially dehydroxylated intermediate of dickite was 50%. Thermodynamically stable, partially dehydroxylated intermediates of kaolinite and dickite contain AlV, while metakaolinite and metadickite contain only AlIV. The observed QCC > 4 MHz confirmed that the chemical shift at ~20–40 ppm observed for metakaolinite after full DHX corresponds to a strongly distorted coordination shell of AlIV instead of AlV.
The experimentally observed decreasing volume of kaolinite and increasing volume of dickite could not be confirmed by means of first-principles calculations, which indicates a kinetic influence on the DHX and removal of the evolved water.
The results of the present study have a significant impact on industry because they depict the material which consumes the smallest amount of energy for activation. In the future, the chemical reactivity must also be supported by further theoretical calculations incorporating the electronic structure of the material.
Change history
23 June 2021
A Correction to this paper has been published: https://doi.org/10.1007/s42860-021-00136-7
REFERENCES
Angel, R. J., McMullan, R. K., & Prewitt, C. T. (1991). Substructure and superstructure of mullite by neutron-diffraction. American Mineralogist, 76, 332–342.
Bellotto, M., Gualtieri, A., Artioli, G., & Clark, S. M. (1995). Kinetic study of the kaolinite-mullite reaction sequence. Part 1: Kaolinite dehydroxylation. Physics and Chemistry of Minerals, 22, 207–214.
Bich, C., Ambroise, J., & Pera, J. (2009). Influence of degree of dehydroxylation on the pozzolanic activity of metakaolin. Applied Clay Science, 44, 194–200.
Bish, D. L. (1993). Rietveld refinement of the kaolinite structure at 1.5 K. Clays and Clay Minerals, 41, 738–744.
Bleam, W. F., Dec, S. F., & Frye, J. S. (1989). 27Al Solid-state nuclear magnetic-resonance study of 5-coordinate aluminum in augelite and senegalite. Physics and Chemistry of Minerals, 16, 817–820.
Bookin, A. S., Drits, V. A., Plançon, A., & Tchoubar, C. (1989). Stacking faults in kaolin-group minerals in the light of real structural features. Clays and Clay Minerals, 37, 297–307.
Brindley, G. W., & Nakahira, M. (1959). The Kaolinite-Mullite Reaction Series: II, Metakaolin. Journal of the American Ceramic Society, 42, 314–318.
Brown, I. W. M., Mackenzie, K. J. D., Bowden, M. E., & Meinhold, R. H. (1985). Outstanding problems in the kaolinite-mullite reaction sequence investigated by 29Si and 27Al solid-state nuclear magnetic resonance. 2. High-temperature transformations of metakaolinite. Journal of the American Ceramic Society, 68, 298–301.
De Ligny, D., & Navrotsky, A. (1999). Energetics of kaolin polymorphs. American Mineralogist, 84, 506–516.
Drits, V. A., & Tchoubar, C. (1990). The modelization method in the determination of the structural characteristics of some layer silicates: Internal structure of the layers, nature and distribution of stacking faults. X-Ray Diffraction by Disordered Lamellar Structures (pp. 233-303). Berlin: Springer-Verlag.
Du, J. H., Morris, G., Pushkarova, R. A., & Smart, R. S. (2010). Effect of Surface Structure of Kaolinite on Aggregation, Settling Rate, and Bed Density. Langmuir, 26, 13227–13235.
Fabbri, B., Gualtieri, S., & Leonardi, C. (2013). Modifications induced by the thermal treatment of kaolin and determination of reactivity of metakaolin. Applied Clay Science, 73, 2–10.
Franco, F., & Ruiz Cruz, M. D. (2006). A comparative study of the dehydroxylation process in untreated and hydrazine-deintercalated dickite. Journal of Thermal Analysis and Calorimetry, 85, 369–375.
Frost, R. L., & Vassallo, A. M. (1996). The dehydroxylation of the kaolinite clay minerals using Infrared Emission Spectroscopy. Clays and Clay Minerals, 44, 635–651.
Fyfe, C. A., Bretherton, J. L., & Lam, L. Y. (2000). Detection of the 'invisible aluminium' and characterisation of the multiple aluminium environments in zeolite USY by high-field solid-state NMR. Chemical Communications, 17, 1575–1576.
Fyfe, C. A., Bretherton, J. L., & Lam, L. Y. (2001). Solid-state NMR detection, characterization, and quantification of the multiple aluminum environments in US-Y catalysts by Al-27 MAS and MQMAS experiments at very high field. Journal of the American Chemical Society, 123, 5285–5291.
Giraudo, N., Krolla-Sidenstein, P., Bergdolt, S., Heinle, M., Gliemann, H., Messerschmidt, F., Bruner, P., & Thissen, P. (2015). Early Stage Hydration of Wollastonite: Kinetic Aspects of the Metal-Proton Exchange Reaction. Journal of Physical Chemistry C, 119, 10493–10499.
Gore, K. U., Abraham, A., Hegde, S. G., Kumar, R., Amoureux, J. P., & Ganapathy, S. (2002). 29Si and 27Al MAS/3Q-MAS NMR studies of high silica USY zeolites. Journal of Physical Chemistry B, 106, 6115–6120.
He, H. P., Guo, J. G., Zhu, J. X., & Hu, C. (2003). 29Si and 27Al MAS NMR study of the thermal transformations of kaolinite from North China. Clay Minerals, 38, 551–559.
Hinckley, D. N. (1963). Variability in "crystallinity" values among the kaolin deposits of the coastal plain of Georgia and South Carolina. Clays and Clay Minerals, 11, 229–235.
Hollanders, S., Adriaens, R., Skibsted, J., Cizer, O., & Elsen, J. (2016). Pozzolanic reactivity of pure calcined clays. Applied Clay Science, 132, 552–560.
Jones, G., Jenkins, S. J., & King, D. A. (2006). Hydrogen bonds at metal surfaces: Universal scaling and quantification of substrate effects. Surface Science, 600, L224–L228.
Kakali, G., Perraki, T., Tsivilis, S., & Badogiannis, E. (2001). Thermal treatment of kaolin: the effect of mineralogy on the pozzolanic activity. Applied Clay Science, 20, 73–80.
Kaloumenou, M., Badogiannis, E., Tsivilis, S., & Kakali, G. (1999). Effect of the kaolin particle size on the pozzolanic behaviour of the metakaolinite produced. Journal of Thermal Analysis and Calorimetry, 56, 901–907.
Kokalj, A. (1999). XCrySDen – a new program for displaying crystalline structures and electron densities. Journal of Molecular Graphics & Modelling, 17, 176–179.
Komarneni, S., Fyfe, C. A., & Kennedy, G. J. (1985a). Order-disorder in 1:1 type clay minerals by solid-state 27Al and 29Si magic angle spinning NMR spectroscopy. Clay Minerals, 20, 327–334.
Komarneni, S., Roy, R., Roy, D. M., Fyfe, C. A., Kennedy, G. J., Bother-by, A. A., Dadok, J., & Chesnick, A. S. (1985b). 27Al and29Si magic angle spinning nuclear magnetic resonance spectroscopy of Al-substituted tobermorites. Journal of Materials Science, 20, 4209–4214.
Kresse, G., & Furthmuller, J. (1996). Efficient iterative schemes for ab initio total–energy calculations using a plane-wave basis set. Physical Review B, 54, 11169–11186.
Kresse, G., & Hafner, J. (1993). Ab initio molecular dynamics for liquid metals. Physical Review B, 47, 558–561.
Kresse, G., & Joubert, D. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Physical Review B, 59, 1758–1775.
Lee, M. H., Cheng, C. F., Heine, V., & Klinowski, J. (1997). Distribution of tetrahedral and octahedral Al sites in gamma alumina. Chemical Physics Letters, 265, 673–676.
Lippmaa, E., Samoson, A., & Magi, M. (1986). High resolution 27Al NMR of aluminosilicates. Journal of the American Chemical Society, 108, 1730–1735.
Low, I. M., & McPherson, R. R. (1988). The structure and composition of Al-Si spinel. Journal of Materials Science Letters, 7, 1196–1198.
MacKenzie, K. J. D. (2000). Applications of solid state NMR spectroscopy to ceramic research. British Ceramic Transactions, 99, 231–240.
MacKenzie, J. D., & Smith, M. E. (2002). Multinuclear Solid-state NMR of Inorganic Materials. Pergamon Materials Series, 6. Elsevier, Amsterdam.
Mackenzie, K. J. D., Brown, I. W. M., Meinhold, R. H., & Bowden, M. E. (1985). Outstanding problems in the kaolinite–mullite reaction sequence investigated by 29Si and 27Al solid state nuclear magnetic resonance. Part 1: Metakaolinite. Journal of the American Ceramic Society, 68, 293–297.
Massiot, D., Fayon, F., Capron, M., King, I., Le Calve, S., Alonso, B., Durand, J. O., Bujoli, B., Gan, Z. H., & Hoatson, G. (2002). Modelling one- and two-dimensional solid-state NMR spectra. Magnetic Resonance in Chemistry, 40, 70–76.
Metz, V., & Ganor, J. (2001). Stirring effect on kaolinite dissolution rate. Geochimica et Cosmochimica Acta, 65, 3475–3490.
Ndlovu, B., Farrokhpay, S., Forbes, E., & Bradshaw, D. (2015). Characterisation of kaolinite colloidal and flow behaviour via crystallinity measurements. Powder Technology, 269, 505–512.
Omegna, A., van Bokhoven, J. A., & Prins, R. (2003). Flexible aluminum coordination in alumino-silicates. Structure of zeolite H-USY and amorphous silica-alumina. Journal of Physical Chemistry B, 107, 8854–8860.
Perdew, J. P., Burke, K., & Ernzerhof, M. (1996). Generalized gradient approximation made simple. Physical Review Letters, 77, 3865–3868.
Plançon, A., Giese, R. F., Snyder, R., Drits, V. A., & Bookin, A. S. (1989). Stacking faults in the kaolin-group minerals – defect structures of kaolinite. Clays and Clay Minerals, 37, 203–210.
Ptáček, P., Frajkorova, F., Soukal, F., & Opravil, T. (2014). Kinetics and mechanism of three stages of thermal transformation of kaolinite to metakaolinite. Powder Technology, 264, 439–445.
Rocha, J., & Klinowski, J. (1990a). 29Si and 27Al magic angle spinning NMR studies of the thermal transformation of kaolinite. Physics and Chemistry of Minerals, 17, 179–186.
Rocha, J., & Klinowski, J. (1990b). Solid-state NMR studies of the structure and reactivity of metakaolinite. Angewandte Chemie – International Edition in English, 29, 553–554.
Rocha, J., Adams, J. M., & Klinowski, J. (1990). The rehydration of metakaolinite to kaolinite – evidence from solid-state NMR and cognate techniques. Journal of Solid State Chemistry, 89, 260–274.
Rocha, J., Paz, F. A. A., Sardo, M., & Mafra, L. (2018). Revisiting the crystal structure of dickite: X-ray diffraction, solid-state NMR, and DFT calculations study. American Mineralogist, 103, 812–818.
Rozita, Y., Brydson, R., & Scott, A. J. (2010). An investigation of commercial gamma-Al2O3 nanoparticles. Paper presented at the Electron Microscopy and Analysis Group Conference (EMAG 2009). Journal of Physics: Conference Series, 241, 012096.
Sakharov, B. A., Drits, V. A., McCarty, D. K., & Walker, G. M. (2016). Modeling powder X-ray diffraction patterns of the Clay Minerals Society kaolinite standards: KGa-1, KGa-1b, and KGa-2. Clays and Clay Minerals, 64, 314–333.
Schneider, H., Schreuer, J., & Hildmann, B. (2008). Structure and properties of mullite – A review. Journal of the European Ceramic Society, 28, 329–344.
Schomburg, J., & Störr, M. (1984). Dilatometerkurvenatlas der Tonmineralrohstoffe. Berlin: Akademie-Verlag.
Smith, M. E., & Steuernagel, S. (1992). A multinuclear magnetic resonance examination of the mineral grandidierite. Identification of a 27Al resonance from a well-defined AlO5 site. Solid State Nuclear Magnetic Resonance, 1, 175–183.
Smykatz-Kloss, W. (1974). Differential Thermal Analysis (Vol. 11). Heidelberg: Springer-Verlag.
Snellings, R., Mertens, G., & Elsen, J. (2012). Supplementary cementitious materials. In: Applied Mineralogy of Cement and Concrete (M.A.T.M. Broekmans and H. Pöllmans, editors). Reviews in Mineralogy and Geochemistry, 74, Virginia, USA, Mineralogical Society of America.
Sperinck, S., Raiteri, P., Marks, N., & Wright, K. (2011). Dehydroxylation of kaolinite to metakaolin-a molecular dynamics study. Journal of Materials Chemistry, 21, 2118–2125.
Stoch, L., & Wacławska, I. (1981a). Dehydroxylation of kaolinite group minerals. I. Kinetics of dehydroxylation of kaolinite and halloysite. Journal of Thermal Analysis, 20, 291–304.
Stoch, L., & Wacławska, I. (1981b). Dehydroxylation of kaolinite group minerals. II. Kinetics of dickite dehydroxylation. Journal of Thermal Analysis, 20, 305–310.
Tironi, A., Tressa, M. A., Scian, A. N., & Irassar, E. F. (2014). Thermal analysis to assess pozzolanic activity of calcined kaolinitic clays. Journal of Thermal Analysis and Calorimetry, 117, 547–556.
Ufer, K., Kleeberg, R., & Monecke, T. (2015). Quantification of stacking disordered Si-Al layer silicates by the Rietveld method: application to exploration for high-sulphidation epithermal gold deposits. Powder Diffraction, 30, 111–118.
van Bokhoven, J. A., Koningsberger, D. C., Kunkeler, P., van Bekkum, H., & Kentgens, A. P. M. (2000). Stepwise dealumination of zeolite beta at specific T-sites observed with 27Al MAS and 27Al MQ MAS NMR. Journal of the American Chemical Society, 122, 12842–12847.
Verwey, E. J. W. (1935). The structure of the electrolytical oxide layer on aluminium. Zeitschrift für Kristallographie – Crystalline Materials, 91, 317.
White, C. E., Provis, J. L., Proffen, T., Riley, D. P., & van Deventer, J. S. J. (2010a). Combining density functional theory (DFT) and pair distribution function (PDF) analysis to solve the structure of metastable materials: the case of metakaolin. Journal of Physical Chemistry Chemical Physics, 12, 3239–3245.
White, C. E., Provis, J. L., Proffen, T., Riley, D. P., & van Deventer, J. S. J. (2010b). Density functional modeling of the local structure of kaolinite subjected to thermal dehydroxylation. Journal of Physical Chemistry A, 114, 4988–4996.
ACKNOWLEDGMENTS
The authors thank G. Guthausen (MVM, KIT) for introduction to NMR measurements, M. Bertmer (University of Leipzig) for help with evaluation of NMR spectra, N. Werling (CMM, KIT) for preparation of SEM samples, and K. Ufer (BGR Hannover) for discussion of crystal structures. Deutsche Forschungsgemeinschaft (DFG) funded parts of the work under EM79/8-1.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
(Received 8 January 2020; revised 18 May 2020: AE: Luyi Sun)
The original online version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Izadifar, M., Thissen, P., Steudel, A. et al. COMPREHENSIVE EXAMINATION OF DEHYDROXYLATION OF KAOLINITE, DISORDERED KAOLINITE, AND DICKITE: EXPERIMENTAL STUDIES AND DENSITY FUNCTIONAL THEORY. Clays Clay Miner. 68, 319–333 (2020). https://doi.org/10.1007/s42860-020-00082-w
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42860-020-00082-w