
Abstract
Biochar (BC)-supported graphene-encapsulated zero-valent iron nanoparticle composites (BC-G@Fe0) are promising engineering nanocomposites that can be used to scavenge heavy metal from wastewater. However, the production of BC-G@Fe0 through carbothermal reduction using biomass as a carbon source remains challenging because of biomass pyrolysis complications. Here, we examined two carbothermal reduction routes for preparing BC-G@Fe0 using bamboo as the carbon source. The first route impregnated Fe ions (Fe2+/3+) into unpyrolyzed bamboo particles initially, followed by carbonization at 600–1000 °C. This process produced BC-G@Fe0 dominated by iron carbide (Fe3C), which led to low heavy metal removal efficiency (i.e., Cu2+ capacity of < 0.3 mmol g−1). In the second route, bamboo particles were pyrolyzed (600 °C) to biochar first, followed by impregnating this biochar with Fe ions, and then carbonized at 600–1000 °C. This route produces zero-valent iron nanoparticles, which resulted in high heavy metal removal capacities (i.e., 0.30, 1.58, and 1.91 mmol g−1 for Pb2+, Cu2+, and Ag+, respectively). The effects of carbonization temperature (600–1000 °C), iron source (i.e., iron nitrates, iron sulfate, ferrous chloride, and ferric chloride), and iron loading (5–40%) on the morphology, structure, and heavy metal ion aqueous uptake performance of BC-G@Fe0 were also investigated. This study revealed the formation mechanisms of BC-G@Fe0 through biomass carbothermal reduction, which could guide the application-oriented design of multifunctional iron-BC composites for water remediation.
Graphical Abstract

Highlights
-
BC-G@Fe0 nanocomposite was produced through two carbothermal reduction routes.
-
The biomass impregnating-carbonizing route produced graphene-encapsulated iron carbide (Fe3C) nanoparticles.
-
The pyrolyzing-biochar-impregnating-carbonizing route produced graphene-encapsulated nanoscale zero-valent iron particles.
-
BC-G@Fe0 nanocomposites exhibited good heavy metal ion removal performance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Heavy metal pollution is a global environmental issue that threatens 200 million people worldwide (Tchounwou et al. 2012). Heavy metals such as copper (Cu), lead (Pb), and silver (Ag) have devastating effects on humans (Wani et al. 2015). Cu2+ is a toxic heavy metal usually discharged from mining, farming, and corrosion of pipes (Briffa et al. 2020; Samaraweera et al. 2022). Pb2+ is the most toxic heavy metal originating from various sources such as leaded gasoline, water pipes, lead-based paint, battery recycling, etc. (Madduri et al. 2020). Ag+ is a rare metal that has adverse effects on human and aquatic life. The anthropogenic sources of Ag+ include emissions from smelting operations, cloud seeding, and photographic and electrical supplies (Eisler 1996).
Nanoscale zero-valent iron (nanoFe0) features a low reduction potential (E0) and high reactivity. It is a promising material for heavy metal immobilization from polluted water. Specifically, nanoFe0 can capture heavy metal ions such as Pb2+, Cu2+, and Ag+ with the E0 higher than − 0.44 eV by reduction, sequestering them as solid metal particles (Zhang et al. 2021b). Moreover, Fe2+/3+ ions released from nanoFe0 dissolution can immobilize heavy metals from wastewater solution through hydrolysis to iron hydroxide and co-precipitation (Zhang et al. 2021b). However, nanoFe0 suffers agglomeration issues because of its ferromagnetic high surface area. Agglomeration reduces the nanoFe0 surface area, which limits its mobility in polluted water, thereby reducing its water remediation performance (Huang et al. 2020; Jiao et al. 2019; Ng and Lim 2022). Depositing nanoFe0 onto a high surface area carrier can alleviate the agglomeration. Carriers including clay, zeolite, metal–organic frameworks, graphene, and biochar (BC) have been used for nanoFe0 dispersion (Asif et al. 2021; Bagheri et al. 2021; Ezzatahmadi et al. 2017; Florea et al. 2022; Li et al. 2018; Liu et al. 2019; Wang et al. 2019; Xing et al. 2020). BC is a low cost and effective candidate for dispersing nanoFe0 because of its highly porous high surface area structure and many surface functional groups (Wang et al. 2019). BC-supported nanoFe0 composite (BC-Fe0) has frequently been prepared by borohydride reduction of Fe2+/3+ containing solutions (Yang et al. 2021). However, borohydrides are toxic and expensive, making large scale borohydride reduction a non-sustainable and less appealing BC-Fe0 preparation route.
Recently, carbothermal reduction has been used to produce BC-Fe0 for water remediation (Fan et al. 2020b; Jiang et al. 2021; Ma et al. 2021). Typical carbothermal reduction process impregnates iron salts or oxides into biomass precursors and heats at high temperatures (e.g., 600–800 °C) under oxygen-limited conditions. The biomass is converted to BC while iron salts or oxides are reduced to nanoFe0 particles, which attach to biochar surfaces (Zhang et al. 2022b). Generally, both solid carbon and released pyrolysis gases (CH4, CO, H2) are the main reagents causing reduction to Fe0 (Yan et al. 2018c). However, this carbothermal reduction process is uncontrollable because of the complex biomass pyrolysis complexity. The structure, morphology, crystallinity and size of the reduced iron are hard to regulate. For instance, direct carbothermal reduction of Fe(NO3)3-impregnated rice straw at 800 °C produces BC-Fe0 dominated by nanoFe0 particles, while hydrothermally assisted carbothermal reduction yields BC with distributed nanoFe0, FeO, and Fe2O3 present (Yang et al. 2022). In contrast, the 800–1000 °C carbothermal reduction of ferric sulfate [Fe2(SO4)3]-impregnated rice husks yields Fe2O3, Fe3O4, and nanoFe0 species (Liu et al. 2020). Moreover, iron carbides (Fe3C) generated in iron-assisted biomass pyrolysis products (Mun et al. 2015; Yan et al. 2018a; Zhang et al. 2018b), have often been overlooked or misinterpreted in the carbothermal production of BC-Fe0 (Chen et al. 2021; Liu et al. 2020; Neeli and Ramsurn 2018; Yang et al. 2022). Furthermore, the role of Fe3C in water remediation is unclear. Studies have explored if Fe3C facilitates aqueous nitrate reduction (Lan et al. 2020). Fe3C is a stable compound that is not reactive to heavy metal water contaminants (Farzi et al. 2020). Additionally, the effect of the iron salts on the formation of BC-Fe0 is still unclear. For example, carbothermal reduction of iron nitrate-impregnated sawdust at 800 °C gave BC-Fe0 dominated by Fe3C (Mun et al. 2015) while carbothermal reduction of FeCl3-impregnated sawdust at the same temperature gave mainly nanoFe0 (Jiang et al. 2019).
Our previous studies revealed that graphitic carbon nanostructures usually formed during the carbothermal reduction of iron oxides/salts impregnated into biomass (Yan et al. 2018b). These nanostructures are composed of graphene nanosheets and nanoribbons that form on the surface of iron species (nanoFe0 and Fe3C) via carbon dissolution into the iron, graphene sheet formation and precipitation (Hunter et al. 2022) mechanisms. Layers of graphene sheets can encapsulate the iron particles to varying thicknesses. Recently, we have reported good heavy metal and nutrient removal from wastewater using BC-supported graphene-encapsulated nanoFe0 (BC-G@Fe0) composites synthesized via in-situ carbothermal reduction of Fe(NO3)3 impregnated lignin (Zhang et al. 2021b). BC-G@Fe0 composites were produced by the in-situ carbothermal reduction of FeCl3-impregnated sawdust and rice husk and used as persulfate activators for degrading 2,4-dichlorophenol (Zhang et al. 2021a) and acetaminophen (Zhuo et al. 2022), respectively. Biomass-derived BC-G@Fe0 composites have attracted wide research for treating wastewater, but its synthesis mechanism is still unclear because of the biomass chemical heterogeneity and pyrolysis complexity. The formation of both Fe3C and graphitic nanostructures during iron catalytic graphitization of biomass is attributed to the carbonaceous compounds released during biomass pyrolysis. Thus, it remains a challenge to regulate the formation of Fe3C nanoparticles and specific graphitic nanostructures during the BC-G@Fe0 production.
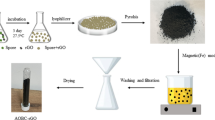
Considering these knowledge gaps, we sought to develop an effective BC-G@Fe0 composite by carbothermal reduction to remove heavy metals. Hence, two carbothermal reduction routes to generate BC-G@Fe0 were compared, i.e., the impregnating-carbonizing process and the pre-pyrolyzing-impregnating-carbonizing process (Fig. 1). The effects of the pre-pyrolyzing, carbonization temperature and iron source on the morphology, structure and heavy metal uptake performance of BC-G@Fe0 were investigated. Specifically, model heavy metal removals of BC-G@Fe0 were evaluated using aqueous Pb2+, Cu2+, and Ag+ solutions. This revealed features of the pyrolytic synthesis mechanism of biomass-derived BC-G@Fe0. This knowledge base could guide the application-oriented design of multifunctional iron-BC composites for water remediation.

Schematic illustration of two proposed carbothermal reduction routes, route I and route II, evaluated in this study for BC-G@Fe0 preparation
2 Experiments
2.1 Materials
Bamboo biomass was collected from a local farm (Starkville, MS, USA). Iron(II) chloride tetrahydrate (FeCl2·4H2O), iron(III) chloride (FeCl3), copper nitrate trihydrate [Cu(NO3)2·3H2O], lead nitrate [Pb(NO3)2] and hydrochloric acid (HCl) were purchased from Sigma Aldrich. Iron(III) nitrate nonahydrate [Fe(NO3)3·9H2O], and silver nitrate (AgNO3) were purchased from Alfa Aesar. Iron(II) sulfate heptahydrate [FeSO4·7H2O] was purchased from Acros Organics.
2.2 BC preparation
Dried bamboo strips (7% moisture content) with skin removed were ground into powders using a wood miller with a 180 µm mesh screen. Bamboo powders were pyrolyzed to BC in a tube furnace at 600 °C for 1 h at a 50 °C min−1 ramp rate under N2 (1 L min−1). After washing with 1 M HCl and distilled water to remove ash, BC was oven-dried at 100 °C overnight. The dried BC was then ground to fine particles using a pestle.
2.3 Preparation of BC-G@Fe0 by direct carbonization of Fe(NO3)3 impregnated bamboo particles (route I)
Prior to BC-G@Fe0 preparation, Fe(NO3)3 was mixed with bamboo particles through a solvent impregnation method. Briefly, 3.6 g Fe(NO3)3·9H2O was mixed with 10 g bamboo (mass ratio of iron to the oven-dried bamboo is 5/100, labeled as 5%) in a beaker, followed by dispersing the mixture in 60 mL of deionized water. The mixture was then heated on a hot plate to ~ 60 °C while stirring for 2 h until all bamboo particles were saturated with the iron salt solution and had sank to the bottom of the beaker. Then this mixture was heated for another 2–6 h with stirring to evaporate the water, followed by oven drying at 103 °C overnight.
BC-G@Fe0 was prepared through direct carbonization of Fe(NO3)3-impregnated bamboo particles at seven different targeted temperature levels, i.e., 600, 650, 675, 700, 800, 900, and 1000 °C, respectively. Specifically, 10 g of Fe(NO3)3-impregnated bamboo particles was loaded in two ceramic boats that were placed in the heating zone of a 2-inch (outer diameter) electric tubular furnace, followed by ramping (50 °C min−1) to a targeted temperature and holding for 1 h under a N2 flow (1 L min−1). After cooling the furnace to room temperature over 2 h, the sample was removed. The products from direct carbonization were labeled as BC-G@Fe0-DX, where “D” represents direct carbonization, and “X” represents the targeted carbonization temperature level used. The weights of products were measured using a digital balance (resolution 0.1 mg) giving the carbonization yield and allowing calculation of the iron content.
2.4 Preparation of BC-G@Fe0 by carbonization of Fe salt-impregnated BC (route II)
Prior to BC-G@Fe0 preparation, different iron salts were imbibed into BC (made at 600 °C, see Sect. 2.2) through a solvent impregnation method as described in Sect. 2.3. The type of iron salt used, Fe(NO3)3, FeCl2, FeCl3, and FeSO4, to impregnate BC was investigated with an iron loading of 15%. The effect of the carbonization temperature (e.g., 600, 700, 800, 900, and 1000 °C) was studied using FeCl2 to impregnate BC, employing a mass ratio of 15% iron to BC. Iron loading was evaluated using FeCl2-impregnated BCs with iron loadings of 5%, 10%, 20%, 30%, and 40%.
BC-G@Fe0 was then prepared by carbonization of Fe-impregnated BC particles at the predefined temperature using the same carbonization parameters (e.g., ramping rate and N2 flow rate) described in Sect. 2.3. The products were labeled as BC-G@Fe0-X, where “X” represents the carbonization temperature used. The product weights were measured, and the iron contents were calculated (shown in Additional file 1: Table S1).
2.5 Adsorption study
Batch sorption experiments determined the Pb2+, Cu2+, and Ag+ aqueous removal efficiency of BC-G@Fe0 samples synthesized from routes I and II. Briefly, BC-G@Fe0 samples (50 mg) were dispersed in 40 mL solutions of the heavy metal at each concentration level evaluated. The mixtures were mechanically shaken vigorously for a predefined time at 25 °C. After completing metal uptake, the adsorbents were gravity filtered. The concentrations of Pb2+ and Cu2+ were determined by atomic absorption spectrophotometry (AAS). The Ag+ concentration was determined using inductively coupled plasma mass spectrometry (ICP-MS). Copper, lead, and silver standardized solutions were prepared in the ranges of 0.5–20 mg L−1, 0.5–50 mg L−1, and 0.5–5 mg L−1, respectively. The uptake capacities of BC-G@Fe0 were calculated using Eq. 1:
where Q (mg g−1) represents the metal amount (mg) adsorbed by 1 g of adsorbent; C0 and Cf are the initial and final metal concentrations (mg L−1) in the evaluated solution, respectively; V is the metal solution volume (L) and m is the adsorbent weight (g).
2.6 Characterization
X-ray diffraction (XRD) analysis was carried out to identify sample compositions using Cu-Kα radiation over the 2θ range of 20–80° with a Rigaku Ultima III X-ray diffractometer at a scanning speed of 1° min−1 and step width of 0.05°. Field emission scanning electron microscopy (FESEM, JEOL-6500F) equipped with energy dispersive spectroscopy (EDS) was used to determine sample morphologies. BC-G@Fe0 samples were placed on a conductive carbon adhesive tab that adhere to a stub. Then a 15 nm thick layer of Pt was sputter-coated to make the sample conductive before SEM analysis. The spot diameter of the Fe0 nanoparticles in SEM images were calculated using ImageJ software. Transmission electron microscopy (TEM, JEOL-2100) equipped with EDS (Oxford X-max-80) was also used to investigate the surface morphology of BC-G@Fe0 samples. TEM specimens were prepared by mixing approximately 10 mg of BC-G@Fe0 samples with 5 mL ethanol first, followed by vortexing the suspension for 2 min. Then, a drop of the suspension was deposited onto a carbon film on a 300-mesh copper grid that was allowed to stand overnight under a vacuum before TEM analysis. BC-G@Fe0 samples for specific surface area and pore volume measurements were degassed at 130 °C for 12 h before analyses by nitrogen sorption isotherms using a TriStar II PLUS (Micrometrics Instruments). The specific surface area, micropore and mesopore volumes were obtained according to N2 adsorption isotherms using the nonlocal density functional theory (NLDFT) method by assuming a slit pore model (Zhang et al. 2022a).
The surface elemental composition of BC-G@Fe0 after heavy metal ion (Pb2+, Cu2+, and Ag+) removal tests were conducted using Thermo Scientific K-Alpha X-ray photoelectron spectroscopic system (XPS) equipped with a monochromatic X-ray source at 1486.6 eV (Al Kα line). Briefly, 100 mg of BC-G@Fe0 were added to 50 mL of heavy metal ion solution, and the mixture was then shaken mechanically (at a speed of 200 rpm min−1) for 15 min at 25 °C to characterize the surface region chemical structure of BC-G@Fe0 after the uptake of heavy metal ions. The initial solution concentration and the initial pH for each of three metal ions were 500 mg L−1 and 5, respectively. Following the removal tests, each mixture was first filtered, and the filtrate was then dried for 24 h at 80 °C under vacuum. the surface elemental composition of the dried filtrate swas then investigated using XPS, high-resolution Pb, Cu, and Ag spectra. These were deconvoluted by assigning various peak counts using the Gaussian function in OriginPro 2021 software.
3 Results and discussion
3.1 BC-G@Fe0 from direct carbonization of Fe(NO3)3 impregnated bamboo particles (route I)
The direct carbonization of iron nitrate-impregnated bamboo biomass particles yields iron-containing nanoparticles supported on bamboo-derived biochar. The decomposition of iron nitrates and biomass occurs at the temperature below 500 °C where iron oxides and carbonaceous gases compounds are produced (Eq. 2). Iron oxides conversion to metallic iron will occur at a temperature above 500 °C under a reducing environment (Crane and Scott 2012), i.e., upon exposure to carbon, CO, H2, or CH4 formed from further biomass thermal degradation (Eq. 2). Meanwhile, the dissolution of carbon atoms into iron particles above 500 °C generates iron carbides (e.g., Fe3C) (Eq. 3) (Liu et al. 2015). Both metallic iron and Fe3C nanoparticles were observed in XRD patterns of evaluated BC-G@Fe0 (Fig. 2a). Specifically, a small amount of residual Fe2O3 was found at 600 °C, while it was almost fully reduced to Fe0 above 650 °C. At 675 °C, the carbon (002) peak was observed, indicating that the formation of graphitic carbon nanostructures probably was via the precipitation or extrusion of carbon from iron-carbon solid solutions (i.e., γ-Fe and Fe3C) (Eqs. 4, 5) (Wirth et al. 2012). At 700 °C, diffraction intensities of both graphitic carbon and Fe3C increased, indicating that the formation of graphitic nanostructures was exceeded by formation of Fe3C. Further increasing the temperature from 700 to 1000 °C did not significantly change the phase composition of the products

Characterization of BC-G@Fe0 produced by route I. a XRD patterns of BC-G@Fe0 samples produced at different temperatures; b thermogravimetric analysis of iron nitrate blended into bamboo particles; c SEM images of BC-G@Fe0 produced at 1000 °C; d–f TEM images of BC-G@Fe0 that was generated at 1000 °C show the distribution of Fe-based nanoparticles in the carbon matrix (d); the encapsulation of Fe3C nanoparticle by graphitic carbon (e); the d(111) lattice structure of Fe3C (f); g, h N2 adsorption isotherms (g) and specific surface areas and pore volumes of the BC-G@Fe0 samples prepared at 600–1000 °C (h); i Cu2+ uptake capacities and iron use ratio of BC-G@Fe0
Figure 2b showed the thermogravimetric analysis of iron nitrate-impregnated bamboo particles. Three main weight loss peaks were observed on the weight derivative curve. The first peak centered at 330 °C occurs with approximately a 50% mass loss, which corresponds to the thermal degradation of bamboo. The second peak centered at 580 °C was related to the reduction of Fe2O3 to Fe0 and Fe3C nanoparticles. The third peak centered at 740 °C can be attributed to iron catalytic dehydrogenative BC carbonization and formation of graphene nanostructures. Iron-impregnated bamboo particles carbonized at 1000 °C exhibited a porous structure with many iron-based nanoparticles on their surfaces (Fig. 2c). TEM observations showed that these iron-based nanoparticles with diameters less than 200 nm were embedded in an entangled graphitic nanoribbon and nanosphere network (Fig. 2d). High magnification TEM images indicated the iron-based (e.g., Fe3C and Fe0) nanoparticles had a core–shell structure consisting mostly of an iron-based nanoparticle cores that were shelled (covered) with 5–10 nm thick graphene layered nanostructures (Fig. 2e). Fe3C, which may be formed by carbon dissolving into α-Fe (Eq. 6), was also discovered in the outer layer of these nanoparticles (Fig. 2f). Thus, we postulated that nanoG@Fe0 from direct carbonization consisted of a tertiary structure with the outer graphene surrounding layered nanostructured shell. The middle layer was Fe3C and the core was nanoFe0 (Fig. 1), as previously reported (Yan et al. 2013, 2019). Equations (2)–(6) express the general reaction features of the entire process.
The surface area and pore size of directly carbonized BC-G@Fe0 samples were determined by N2 adsorption isotherms (Fig. 2g). At 600 °C, the N2 isotherms exhibited a typical Type Ia shape with an H4 hysteresis loop, indicating their microporous structure (Thommes et al. 2015). At 700 °C, N2 isotherms showed the Type IVa shape with an H2 hysteresis loop, suggesting a developing mesoporous structure with some bottle-neck-shaped mesopores (Thommes et al. 2015). At temperatures of 700–1000 °C, the N2 isotherms of BC-G@Fe0 products showed the Type IVa shape with an H2 hysteresis loop, suggesting they all had bottle-neck-shaped mesoporous structures (Thommes et al. 2015). The microspore volume of BC-G@Fe0 samples, made from 600 to 1000 °C, decreased from 0.138 to 0.010 cm3 g−1. As a result, the specific surface area dropped from 329.1 to 102.5 m2 g−1 (Fig. 2h). However, the mesopore volume rose from 0.010 to 0.248 cm3 g−1. This microporous-to-mesoporous transaction with increasing temperature was related to the iron catalytic formation of graphene nanostructures. Specifically, the increase of temperature facilitated graphitic nanostructure (mainly mesoporous graphene nanoribbon and nanosphere) formation (Thompson et al. 2015) as indicated in XRD pattern (Fig. 2a). Moreover, the mobility of iron/iron carbide nanoparticles was increased at higher temperatures, this might also lead to reactive etching of microporous carbon and conversion of micropores to mesopore.
This BC-G@Fe0 obtained from route I was used for Cu2+ removal via batch adsorption tests, as shown in Fig. 2i. The Cu2+ removal by BC-G@Fe0 occurs mainly through a two-electron reduction reaction of Cu2+ to Cu (Eq. 7). The Cu2+ removal capacity is independent of the sample's surface area but dependent on the availability of the iron phases. As carbonization temperature rose from 600 to 700 °C, Cu2+ removal capacity (QCu) increased from 0.14 to 0.27 mmol g−1, despite 50% decrease of surface area. This took place because Fe2O3 was reduced to metallic iron nanoparticles, as illustrated in Fig. 2a. More Fe0 enhanced Cu2+ removal by reducing more Cu2+ to Cu0 by Fe0 oxidation. Increasing the carbonization temperature from 700 to 1000 °C did not change the Cu2+ uptake capacity. This agrees with XRD scans showing BC-G@Fe0 produced between 700 and 1000 °C have similar iron phase compositions. Theoretically, 1 mol of the metallic zero-valent iron nanoparticle can uptake 1 mol of Cu2+ ions from an aqueous solution, and the corresponding uptake theoretical capacity (QT-Cu) is 17.86 mmol g−1.
To evaluate the efficiency of direct carbonization of biomass/iron salt mixture (route I) for Fe0 production and its heavy metal removal efficiency, we proposed two new parameters, namely iron uptake capacity (QFe-Cu) of Cu2+ and iron use ratio. Briefly, QFe-Cu is calculated by dividing the QCu by its iron content, and Fe use ratio is calculated by QFe-Cu divided by QT-Cu. The detailed calculation steps for QT-Cu, QFe-Cu, and Fe use ratio were shown in Supporting information. As shown in Fig. 2i, as carbonization temperature rose from 600 to 700 °C, QFe-Cu increased from 0.87 to 1.69 mmol g−1 and remained at ~ 1.65 mmol g−1 between 700 and 1000 °C. Similarly, the Fe use ratio increased from 4.9 to 9.4% as the temperature increased from 600 to 700 °C and remained at ~ 9.4% thereafter. This low Fe use ratio was expected because Fe3C nanoparticles were dominant in BC-G@Fe0 products produced by direct carbonization (Fig. 2a). Only small amounts of nanoFe0 were present in BC-G@Fe0 that were tightly wrapped by both graphene layer and Fe3C shells (Fig. 2a, e). Fe3C did not reduce Cu2+.
3.2 BC-G@Fe0 from carbonization of Fe-impregnated BC (route II)
3.2.1 Effect of iron catalyst type
Fe3C lowered the Fe use ratio for Cu2+ uptake. To eliminate the formation of Fe3C and increase the nanoFe0 production in the carbonization products, bamboo particles were initially pyrolyzed at 600 °C for 1 h to make BC and remove carbonaceous gases (e.g., CH4, CO, etc.) in this first production stage without any Fe2+/3+ impregnation. The bamboo-based BC particles produced were subsequently impregnated with iron salts and then further pyrolytically carbonized at 1000 °C to generate BC-G@Fe0. As depicted in XRD analysis in Fig. 3a, this suppressed the formation of Fe3C. These BC-G@Fe0 products contained mostly metallic iron nanoparticles (i.e., α-Fe and γ-Fe). Specifically, carbonizing FeCl2 and FeCl3 impregnated in bamboo, only after it was converted to BC at 600 °C, produced mostly α-Fe nanoparticles. Carbonizing Fe(NO3)3 impregnated BC yielded both α-Fe and γ-Fe nanoparticles. However, carbonizing FeSO4 impregnated BC yielded α-Fe, γ-Fe, some Fe3C nanoparticles, as well as Fe2O3 nanoparticles that were probably generated from the air passivation of α-Fe and/or γ-Fe nanoparticles. The furnace was cooled to room temperature before the product was exposed to O2. Additionally, the carbon (002) diffraction peak was also observed in their XRD patterns (Fig. 3a).

Characterization of BC-G@Fe0 all produced from the carbonization of iron ion-impregnated-BC and subsequently carbonized at 1000 °C (route II) using different iron salts. a XRD patterns of BC-G@Fe0; b thermogravimetric analysis of BC blended with different iron salts; c Cu2+ uptake capacities and iron use ratio of BC-G@Fe0; d–k SEM images of BC-G@Fe0; l–o TEM images of BC-G@Fe0
Thermogravimetric analyses of bamboo BC are shown in Fig. 3b. Three main weight loss stages were observed. Briefly, the first stage occurred at temperatures of 50–250 °C and was related to loss of free bamboo moisture and decomposition of hydrated iron salts. The second stage occurring between 250 and 700 °C corresponded to the thermal degradation of iron salts. The third stage occurred above 700 °C and was attributed to the reduction of iron oxides and the formation of BC-G@Fe0.
Surface area and pore-size characterizations of BC-G@Fe0 performed by N2 adsorption experiments are displayed in Additional file 1: Fig S1. The specific surface areas of BC-G@Fe0 samples made by route II were only between 2 and 20 m2 g−1. This was much lower than the BC-G@Fe0 samples produced from route I (i.e., 102.5–329.1 m2 g−1). Despite their low surface areas, Cu2+ uptake capacities (QCu) of BC-G@Fe0 particles obtained from route II ranged from 0.57 to 1.01 mmol g−1 (Fig. 3c). This was significantly higher than that of BC-G@Fe0 samples obtained in route I (Fig. 2i), supporting our hypothesis that Fe3C formed at the expense of Fe0 nanoparticles reduced heavy metal uptake. FeCl2-derived BC-G@Fe0 exhibited the highest QCu of 1.00 mmol g−1, followed by FeCl3-derived BC-G@Fe0 (0.82 mmol g−1), FeSO4-derived BC-G@Fe0 (0.73 mmol g−1), and Fe(NO3)3-derived BC-G@Fe0 (0.57 mmol g−1).
To elucidate the reasons for the differences in QCu, the morphologies of BC-G@Fe0 were obtained from route II using different iron salts. Uniform distribution of iron-based nanoparticles was formed on the surfaces of all samples (Fig. 3d–k). Importantly, the sizes of iron-based nanoparticles varied with the iron salts used. Specifically, Fe(NO3)3-derived BC-G@Fe0 exhibited the largest Fe-based nanoparticles with the average diameter of 439 nm (measured by ImageJ, as shown in Additional file 1: Fig. S3). These were followed by FeSO4-derived BC-G@Fe0 (average diameter of 171 nm, Additional file 1: Fig. S3), FeCl3-derived BC-G@Fe0 (average diameter of 135 nm, Additional file 1: Fig. S3), and FeCl2-derived BC-G@Fe0 (average diameter of 97 nm, Additional file 1: Fig. S3). Generally, nanoFe0 particles with large sizes are less effective for heavy metal uptake because (1) heavy metal precipitates and (2) iron oxides formed on the surfaces. These increasingly prevent effective electron transfer from the nanoFe0 core to heavy metal ions. Thus, a larger fraction of Fe0 in the larger particles remains unoxidized. Thus, Fe(NO3)3-derived BC-G@Fe0 exhibits the lowest QCu while FeCl2-derived BC-G@Fe0 exhibits the highest QCu. In comparison with FeCl3-derived BC-G@Fe0, FeSO4-derived BC-G@Fe0 contains more Fe3C, causing a lower QCu.
The morphology and structure of G@nanoFe0 particles in BC-G@Fe0 were studied by TEM (Fig. 3l–o). Individual particles display core–shell structures composed of metallic iron nanoparticle cores (nanoFe0, dark color portion) and a graphene nanoribbon shell (light color portion and labeled by red arrows). Note that the graphene nanoribbon encapsulations on nanoFe0 surfaces are not perfect and homogeneous, i.e., some surfaces have thick graphene nanoribbons. Graphene growth on iron surfaces is crystal-structure dependent (Liu et al. 2012; Vinogradov et al. 2017), i.e., the (100) iron plane is more favorable for graphene formation. Graphene nanoribbons prevent further aggregation of nanoFe0 particles by preventing direct Fe0/Fe0 contact. These shells also increase nanoFe0 particle stability by reducing their exposed surface area to other agents.
3.2.2 Effect of carbonization temperature
The effects of temperature and iron loading during the production of BC-G@Fe0 were studied by selecting FeCl2 as the iron source and carbonizing different loadings at 600–1000 °C. Figure 4a shows the XRD patterns of FeCl2 impregnated BC, which then was carbonized at 600–1000 °C for 1 h (route II). At 600 °C, FeCl2 was partly converted to Fe2O3, but nanoFe0 was not formed. At 700 °C, FeO and α-Fe were formed due to the carbothermal reduction of Fe2O3, but FeCl2 and Fe2O3 predominated. At 800 °C, the α-Fe diffraction peak dominated in the XRD pattern, and two small peaks corresponding to graphite and γ-Fe diffractions were observed, suggesting graphene-encapsulated iron nanoparticles had formed. However, a small FeO diffraction was still visible, indicating the carbothermal reduction to Fe0 was not completed. At temperatures above 900 °C, carbonization product XRD patterns showed only α-Fe, γ-Fe, and graphite, completing the carbothermal reduction of iron oxides/chlorides. Moreover, higher γ-Fe and graphite diffraction intensities were observed in the BC-G@Fe0 produced at 1000 °C.

Characterization of BC-G@Fe0 samples produced from the carbonization of FeCl2-impregnated-BC at 600–1000 °C. a XRD patterns of BC-G@Fe0 samples; b Cu2+ uptake capacities and iron use ratio of BC-G@Fe0 samples; c–g SEM images of BC-G@Fe0 samples produced at 600 °C (c), 700 °C (d), 800 °C (e), 900 °C (f), and 1000 °C (g); h–i TEM image of BC-G@Fe0 sample produced at 1000 °C (h) and the corresponding EDS mapping results (i)
QCu values exhibited significant increases from 0.02 to 0.98 mmol g−1 upon increasing carbonization temperature from 700 to 1000 °C (Fig. 4b). The low Cu2+ uptake capacities at 600 and 700 °C resulted from the small amount of nanoFe0 formed. Carbonizing at 900 °C increased the Cu2+ uptake capacity from 0.02 to 0.44 mmol g−1, because Fe0 formation rose during the carbothermal reduction. Carbonization at 1000 °C doubled the Cu2+ uptake capacity (0.98 mmol g−1), but the XRD patterns did not show many differences between these two samples. SEM characterization found (Fig. 4c, d), nanoparticle clusters were formed on the BC surfaces at 600 and 700 °C, which were nanoparticle clusters of iron oxides/chlorides confirmed by SEM-EDS mapping (Additional file 1: Figs. S4, S5). Higher temperatures promoted the decomposition and reduction of iron oxides/chlorides to iron. The Fe/Cl and Fe/O element ratios rose. BC-G@Fe0 had a rough surface at 800 °C (Fig. 4e), attributed to etching of biochar from the carbothermal reduction. At 900 °C, nanoparticle clusters were observed on the surface of the BC (Fig. 4f). SEM–EDS mapping indicated the clusters consisted mostly of iron and carbon (Additional file 1: Fig. S6). At 1000 °C, micron-sized carbon/iron composite particles were observed in BC-G@Fe0 (Fig. 4g and Additional file 1: Fig. S7, SEM–EDS mapping). The high-resolution TEM image (Fig. 4h) and EDS mapping (Fig. 4i) revealed nanoFe0 particles were encapsulated by graphitic carbon, which agree with the XRD results. We previously reported that graphitic carbon coatings protect the core nanoFe0 from oxygen passivation and enhances its heavy metal uptake efficiency (Zhang et al. 2021b). SEM–EDS revealed that oxygen was still present in carbon/iron composites synthesized at 900 °C and 1000 °C, suggesting surface oxidation of unprotected nanoFe0 particles occurred . Specifically, the iron to oxygen atomic ratio of the 900 °C-derived BC-G@Fe0 (Fe:O = 0.9:1) was lower than those formed at 1000 °C (Fe:O = 1.5:1), implying nanoFe0 formed at 900 °C had a thinner protection layer and suffered more oxidation. This agrees with high intensity γ-Fe and graphitic XRD diffraction peaks of 1000 °C-derived BC-G@Fe0, confirming higher temperature facilitates the formation of graphitic carbon encapsulations of iron nanoparticle (Zhang et al. 2018a).
3.2.3 Effect of iron loading
The effect of iron loading on the structure, morphology and Cu2+ uptake of BC-G@Fe0 samples obtained at 1000 °C, were investigated. All XRD patterns of BC-G@Fe0 samples had α-Fe, γ-Fe, and graphite diffraction peaks (Fig. 5a). The QCu of BC-G@Fe0 increased from 0.53 to 1.14 mmol g−1 with the increase of iron loading from 5 to 40% (Fig. 5b). This confirms that nanoFe0 is responsible for the uptake of Cu2+. However, QFe-Cu and the Fe use ratio values showed a decrease as the iron loading rose. In particular, the Fe use ratio decreased from 58 to 9% as iron loading went up from 5 to 40%. To investigate this, we characterized BC-G@Fe0 samples by SEM. NanoFe0 particles were well-dispersed on the BC surfaces at a 5% iron loading (Fig. 5c). At a 10% loading, nanoFe0 clusters appeared because particle aggregation occurred. This aggregation became increasingly severe at higher loading (Fig. 5d–f), causing the Fe use ratio to drop at higher iron loadings.

Characterization of BC-G@Fe0 samples produced from FeCl2-impregnated-BC carbonization (route II) with 5–40% iron loadings. a XRD patterns of BC-G@Fe0 samples; b Cu2+ uptake capacities and iron use ratio of BC-G@Fe0 samples; c–f SEM images of BC-G@Fe0 samples produced at 5% (c), 10% (d), 30% (e), and 40% (f) of Fe added as FeCl2
3.3 Heavy metal removal study
BC-G@FE0 samples produced from FeCl2-impregnated-BC carbonization (route II) with 15% iron loading at 1000 °C were used for the heavy metal ion removal study. The uptake of Pb2+ by BC-G@Fe0 was pH-dependent. The effect of solution pH was determined within the pH range of 1–7 (Fig. 6a). The uptake capacity of Pb2+ drastically increased from 0.006 to 0.17 mmol g−1 as the solution pH increased from 1 to 3, while the capacity only slightly changed as the pH increased from 3 to 7. The removal of Pb2+ is minimized at low solution pH (i.e., pH 1 and 2) by the reaction between Fe0 and H+ resulting in oxidation of Fe0 to Fe2+ (Eq. 8). At pH 3–7, the Pb2+ removal is mainly attributed to the reduction of Pb2+ to Pb0 by Fe0 (Eq. 9).

The removal of Pb2+, Cu2+ and Ag+; a pH dependence of Pb2+ uptake (C0 = 100 mg L−1, t = 1 h), b kinetics of Pb2+ uptake (pH 6, C0 = 100 mg L−1), c isotherms of Pb2+ uptake (pH 6, t = 15 min), d pH dependence of Cu2+ uptake (C0 = 100 mg L−1, t = 1 h), e kinetics of Cu2+ uptake (pH 5, C0 = 100 mg L−1), f isotherms of Cu2+ uptake (pH 5, t = 20 min), g pH dependence of Ag+ uptake (C0 = 200 mg L−1, t = 1 h), h kinetics of Ag+ uptake (pH 5, C0 = 300 mg L−1), i isotherms of Ag+ uptake (pH 5, t = 20 min). j–l XPS spectra of BC-G@Fe0 after Pb2+ (j), Cu2+ (k), and Ag+ (l) uptake (pH 5)
XPS confirmed the presence of Pb0 on the surface of BC-G@Fe0 after Pb2+ uptake (Fig. 6j). Additionally, the presence of Pb2+ on the surface of BC-G@Fe0 can be attributed to either the oxidation of reduced Pb0 nanoparticles or precipitation of Pb2+ compounds during Fe0 dissolution (Zhang et al. 2021b).
Kinetic and isotherm studies were carried out at the initial pH of 6. The Pb2+ removal rapidly increased in the first 15 min and reached equilibrium (Fig. 6b). The data were fitted to both pseudo-first-order (1st-order) and pseudo-second-order (2nd-order) models with R2 values of 0.966 and 0.918 (Additional file 1: Table S2), respectively. The Langmuir (R2 = 0.711) and Freundlich (R2 = 0.929) models fitted to isotherm data favoring a multilayer model (Fig. 6c). The maximum Langmuir adsorption capacity was calculated to be 0.31 mmol g−1.
The uptake of Cu2+ into BC-G@Fe0 is highly pH dependent like Pb2+. The effect of solution pH was determined from pH 1–6 (Fig. 6d). The uptake capacity of Cu2+ dramatically increased from 0 to 1.04 mmol g−1 as the solution pH increased from 1 to 3. The uptake capacity slightly differs as the pH increases from 3 to 6. Like Pb2+, Cu2+ removal is low at acidic pH (i.e., pH 1 and 2) by Eq. 1, At pH 3–7, the Cu2+ removal is mainly attributed to the reduction of Cu2+ to Cu0 by Fe0 (Eq. 7). XPS of BC-G@Fe0 after Cu2+ uptake confirms the reduction of Cu2+ to Cu0 (Fig. 6k). Moreover, Cu2+ is also observed on the surface of BC-G@Fe0, which can be attributed to the oxidation of reduced Cu0 and the precipitated Cu2+ compounds during Fe0 dissolving.
Kinetic and isotherm studies were carried out at the initial pH of 5. The Cu2+ uptake rapidly increased in the first 10 min, then slowly increased between 10 and 20 min, and finally reached equilibrium after 20 min. Over 90% of ultimate Cu2+ uptake was achieved within 5 min, showing the high removal kinetics of our BC-G@Fe0 toward Cu2+. The data were fitted to both pseudo-first-order and pseudo-second-order models with R2 values of 0.803 and 0.754, respectively (Fig. 6e). Figure 6f shows concertation-dependent Cu2+ removal by BC-G@Fe0. Adsorption isotherms were drawn to illustrate the relationship between Cu2+ concentration at the equilibrium and the adsorbed Cu2+ concentration. The Langmuir (R2 = 0.984) and Freundlich (R2 = 0.904) models gave good fits to isotherm data (Additional file 1: Table S2). The maximum Langmuir adsorption capacity is calculated to be 1.58 mmol g−1.
The uptake of Ag+ by BC-G@Fe0 is also pH-dependent. The effect of solution pH is determined within the pH range of 1–6 (Fig. 6g). The uptake capacity of Ag+ increased from 0.851 to 1.345 mmol g−1 as the solution pH increased from 1 to 3. The uptake capacity becomes almost a constant value as the pH increases from 3 to 6. Like Cu2+ and Pb2+, Ag+ removal is low at acidic pH (i.e., pH 1 and 2) by Eq. 1. At pH 3–6, the Ag+ removal is mainly attributed to one electron reductions of two Ag+ to Ag0 by Fe0 (Eq. 10).
The reduction of Ag+ to Ag0 was confirmed by XPS of BC-G@Fe0 after Ag+ uptake (Fig. 6l). Moreover, the presence of Ag+ on the surface of BC-G@Fe0 can be attributed to the oxidation of reduced Ag0 and the precipitated Ag+ compounds during Fe0 dissolving.
Kinetic and isotherm studies were carried out at the initial pH of 5 (Fig. 6h, i). The Ag+ removal increased in the first 10 min and reached equilibrium after 20 min. The data were fitted to both pseudo-first-order and pseudo-second-order models with R2 values of 0.971 and 0.824 (Additional file 1: Table S2), respectively. The Langmuir (R2 = 0.99) and Freundlich (R2 = 0.74) models fitted to isotherm data favoring a monolayer model. The maximum Langmuir adsorption capacity was calculated to be 1.913 mmol g−1.
BC-G@Fe0 shows the highest uptake capacity for Ag+, followed by Cu2+ and then Pb2+. This is expected because their reduction potentials are in the following order from high to low Ag+ (0.80 V) > Cu2+ (0.34 V) > Pb2+ (− 0.13 V). In particular, the Langmuir capacity of Pb2+ is four and five times lower than that of Cu2+ and Pb2+, respectively. Such low capacity is because its reduction potential is lower than H+ (0.00 V), as a result, nanoFe0 in BC-G@Fe0 is mainly consumed by H+ during Pb2+ removal (reaction 8) (Busch et al. 2021). From stoichiometry calculations, one mole of nanoFe0 could uptake two moles of Ag+ and one mole Cu2+ via single-displacement reactions, respectively. However, the Langmuir capacity of Ag+ is only 20% higher than that of Cu2+, which is much lower than expected. Heavy metal removal by BC-G@Fe0 is a mass transfer process, where the metal ions migrate to the nanoFe0 surface and are followed by surface reduction and deposition. Despite BC-G@Fe0 showing a higher potential for Ag+ uptake than Cu2+ stoichiometrically, the exposed nanoFe0 surface area is limited. Once the exposed surfaces are occupied by the reduced heavy metal nanoparticles, the uptake is terminated. Hence, the overall Ag+ capacity is much less than the expected value.
Table 1 displays Pb2+, Cu2+ and Ag+ removal in aqueous solutions using different nanoFe0 adsorbents. Our BC-G@Fe0 shows higher uptake capacities for Pb2+, Cu2+ and Ag+ compared to some of the reported nanoFe0 adsorbents. For instance, nanoFe0 supported by reed biochar has a Pb2+ uptake capacity of 0.18 mmol g−1 (Zhu et al. 2017), which is lower than that of the present study. Additionally, BC-G@Fe0 exhibits a 13 times greater Cu2+ uptake capacity than that of amino-modified biochar supporting nanoFe0 (Yang et al. 2018). Furthermore, our BC-G@Fe0 exhibits a higher Ag+ uptake capacity of 1.91 mmol g−1 in comparison to bamboo char supported nanoFe0 using the NaBH4 method (Wang et al. 2017b). Li et al. (2020) produced hydrophilic biochar-nanoFe0, which had a higher Pb2+ uptake capacity than that of our BC-G@Fe0. (Li et al. 2020). This can be attributed to their hydrophilic biochar having a high adsorption capacity to Pb2+ because of its abundant functional groups. Similarly, expensive chitosan-stabilized nanoFe0 particles prepared by Chen et al. also exhibited a higher capacity for than this study (Chen et al. 2019) because chitosan has a high Pb2+ sorption capacity via complexation (Fan et al. 2020a). However, they have followed the NaBH4 reduction method which involves hazardous chemicals (Chen et al. 2019). Hasan et al. revealed approximately three times higher capacity for Cu2+ than this study (Hasan et al. 2020). However, they employed NaBH4 reduction which is not sustainable for scale up removal of water contaminants. Zhou et al. showed three times higher sorption capacity for Ag+ than this study. They have also used chitosan which has an additional advantage of adsorbing Ag+ through chitosan amino group complexation, hence, uplifting the uptake of heavy metal ions (Zhou et al. 2014).
4 Conclusion
This study explored BC-G@Fe0 composite synthesis by two carbothermal reduction routes and their application to remove heavy metal ions. Route I employed direct carbothermal reduction of iron salt-impregnated biomass, which generated BC-G@Fe0-containing graphene-encapsulated Fe3C nanoparticles. This route led to low heavy metal removal efficiency (i.e., < 0.3 mmol g−1 for Cu2+). Route II first pre-pyrolyzed biomass at 600 °C to biochar and then impregnated that biochar with iron salt. This was followed by carbothermal reduction. Carbonaceous gases (e.g., CH4 and CO) were responsible for Fe3C formation. Because they were removed during biomass pyrolysis, subsequent carbothermal reduction of iron salt-impregnated biochar gave BC-G@Fe0 containing graphene-encapsulated nanoFe0. This results in high heavy metal removal efficiency (i.e., 1.58 mmol g−1 for Cu). At our optimized conditions, i.e., the pre-pyrolyzing step employing FeCl2 as the iron resource, 15% iron loading, 1000 °C carbonization for 1 h, the produced BC-G@Fe0 had Pb2+, Cu2+, and Ag+ removal capacities of 0.30, 1.58, and 1.91 mmol g−1, respectively. Future studies will focus on using BC-G@Fe0 to remove heavy metals from soil.
Availability of data and materials
The authors declare that data will be made available on reasonable request.
References
Asif I, Baig N, Sher M, Ul-Hamid A, Altaf M, Mumtaz A, Sohail M (2021) MOF derived novel zero-valent iron@graphitic carbon-based nanoreactors for selective reduction of hazardous 4-nitrophenol. Clean Eng Technol 2:100081. https://doi.org/10.1016/j.clet.2021.100081
Bagheri M, Jafari SM, Eikani MH (2021) Ultrasonic-assisted production of zero-valent iron-decorated graphene oxide/activated carbon nanocomposites: chemical transformation and structural evolution. Mater Sci Eng C 118:111362. https://doi.org/10.1016/j.msec.2020.111362
Briffa J, Sinagra E, Blundell R (2020) Heavy metal pollution in the environment and their toxicological effects on humans. Heliyon 6:e04691. https://doi.org/10.1016/j.heliyon.2020.e04691
Busch M, Ahlberg E, Laasonen K (2021) From absolute potentials to a generalized computational standard hydrogen electrode for aqueous and non-aqueous solvents. Phys Chem Chem Phys 23:11727–11737. https://doi.org/10.1039/D1CP00499A
Cai M, Zeng J, Chen Y, He P, Chen F, Wang X, Liang J, Gu C, Huang D, Zhang K, Gan M, Zhu J (2021) An efficient, economical, and easy mass production biochar supported zero-valent iron composite derived from direct-reduction natural goethite for Cu(II) and Cr(VI) remove. Chemosphere 285:131539. https://doi.org/10.1016/j.chemosphere.2021.131539
Chen X-L, Li F, Xie XJ, Li Z, Chen L (2019) Nanoscale zero-valent iron and chitosan functionalized Eichhornia crassipes biochar for efficient hexavalent chromium removal. Int J Environ Res Public Health 16:3046. https://doi.org/10.3390/ijerph16173046
Chen C, Liu J, Gen C, Liu Q, Zhu X, Qi W, Wang F (2021) Synthesis of zero-valent iron/biochar by carbothermal reduction from wood waste and iron mud for removing rhodamine B. Environ Sci Pollut Res 28:48556–48568. https://doi.org/10.1007/s11356-021-13962-y
Crane RA, Scott TB (2012) Nanoscale zero-valent iron: future prospects for an emerging water treatment technology. J Hazard Mater 211–212:112–125. https://doi.org/10.1016/j.jhazmat.2011.11.073
Ezzatahmadi N, Ayoko GA, Millar GJ, Speight R, Yan C, Li J, Li S, Zhu J, Xi Y (2017) Clay-supported nanoscale zero-valent iron composite materials for the remediation of contaminated aqueous solutions: a review. Chem Eng J 312:336–350. https://doi.org/10.1016/j.cej.2016.11.154
Fan H, Ren H, Ma X, Zhou S, Huang J, Jiao W, Qi G, Liu Y (2020a) High-gravity continuous preparation of chitosan-stabilized nanoscale zero-valent iron towards Cr(VI) removal. Chem Eng J 390:124639. https://doi.org/10.1016/j.cej.2020.124639
Fan J, Chen X, Xu Z, Xu X, Zhao L, Qiu H, Cao X (2020b) One-pot synthesis of nZVI-embedded biochar for remediation of two mining arsenic-contaminated soils: arsenic immobilization associated with iron transformation. J Hazard Mater 398:122901. https://doi.org/10.1016/j.jhazmat.2020.122901
Farzi N, Hydarifard M-H, Izadi ME, Sabzyan H (2020) Investigation of iron carbide (Fe3C) corrosion in water and acidic solution using ReaxFF molecular dynamics. J Mol Liq 318:114006. https://doi.org/10.1016/j.molliq.2020.114006
Florea AF, Lu C, Hansen HCB (2022) A zero-valent iron and zeolite filter for nitrate recycling from agricultural drainage water. Chemosphere 287:131993. https://doi.org/10.1016/j.chemosphere.2021.131993
Eisler R(1996) Silver hazards to fish, wildlife, and invertebrates: a synoptic review. Contaminant Hazard Reviews Report 32; Biological Report 32 U.S. National Biological Service. https://pubs.er.usgs.gov/publication/5200169
Hasan MS, Geza M, Vasquez R, Chilkoor G, Gadhamshetty V (2020) Enhanced heavy metal removal from synthetic stormwater using nanoscale zerovalent iron-modified biochar. Water Air Soil Pollut 231:220. https://doi.org/10.1007/s11270-020-04588-w
Huang X, Zhang F, Peng K, Liu J, Lu L, Li S (2020) Effect and mechanism of graphene structured palladized zero-valent iron nanocomposite (nZVI-Pd/NG) for water denitration. Sci Rep 10:9931. https://doi.org/10.1038/s41598-020-66725-z
Hunter RD, Ramírez-Rico J, Schnepp Z (2022) Iron-catalyzed graphitization for the synthesis of nanostructured graphitic carbons. J Mater Chem A. https://doi.org/10.1039/D1TA09654K
Jiang S-F, Ling L-L, Chen W-J, Liu W-J, Li D-C, Jiang H (2019) High efficient removal of bisphenol A in a peroxymonosulfate/iron functionalized biochar system: mechanistic elucidation and quantification of the contributors. Chem Eng J 359:572–583. https://doi.org/10.1016/j.cej.2018.11.124
Jiang Q, Zhang Y, Jiang S, Wang Y, Li H, Han W, Qu J, Wang L, Hu Y (2021) Graphene-like carbon sheet-supported nZVI for efficient atrazine oxidation degradation by persulfate activation. Chem Eng J 403:126309. https://doi.org/10.1016/j.cej.2020.126309
Jiao W, Song Y, Zhang D, Chang G, Fan H, Liu Y (2019) Nanoscale zero-valent iron modified with carboxymethyl cellulose in an impinging stream-rotating packed bed for the removal of lead(II). Adv Powder Technol 30:2251–2261. https://doi.org/10.1016/j.apt.2019.07.005
Lan Y, Chen J, Zhang H, Zhang W, Yang J (2020) Fe/Fe3C nanoparticle-decorated N-doped carbon nanofibers for improving the nitrogen selectivity of electrocatalytic nitrate reduction. J Mater Chem A. https://doi.org/10.1039/D0TA02317E
Li Z, Wang L, Meng J, Liu X, Xu J, Wang F, Brookes P (2018) Zeolite-supported nanoscale zero-valent iron: new findings on simultaneous adsorption of Cd(II), Pb(II), and As(III) in aqueous solution and soil. J Hazard Mater 344:1–11. https://doi.org/10.1016/j.jhazmat.2017.09.036
Li S, Yang F, Li J, Cheng K (2020) Porous biochar-nanoscale zero-valent iron composites: synthesis, characterization and application for lead ion removal. Sci Total Environ 746:141037. https://doi.org/10.1016/j.scitotenv.2020.141037
Liu X-W, Huo C-F, Li Y-W, Wang J, Jiao H (2012) Energetics of carbon deposition on Fe(100) and Fe(110) surfaces and subsurfaces. Surf Sci 606:733–739. https://doi.org/10.1016/j.susc.2011.12.018
Liu J, Yu B, Zhang Q, Hou L, Huang Q, Song C, Wang S, Wu Y, He Y, Zou J, Huang H (2015) Synthesis and magnetic properties of Fe3C–C core–shell nanoparticles. Nanotechnology 26:085601. https://doi.org/10.1088/0957-4484/26/8/085601
Liu C-M, Diao Z-H, Huo W-Y, Kong L-J, Du J-J (2018) Simultaneous removal of Cu2+ and bisphenol A by a novel biochar-supported zero valent iron from aqueous solution: synthesis, reactivity and mechanism. Environ Pollut 239:698–705. https://doi.org/10.1016/j.envpol.2018.04.084
Liu T, Zhang Z, Wang Z, Wang Z-L, Bush R (2019) Highly efficient and rapid removal of arsenic(III) from aqueous solutions by nanoscale zero-valent iron supported on a zirconium 1,4-dicarboxybenzene metal–organic framework (UiO-66 MOF). RSC Adv 9:39475–39487. https://doi.org/10.1039/C9RA08595E
Liu X, Yang L, Zhao H, Wang W (2020) Pyrolytic production of zerovalent iron nanoparticles supported on rice husk-derived biochar: simple, in situ synthesis and use for remediation of Cr(VI)-polluted soils. Sci Total Environ 708:134479. https://doi.org/10.1016/j.scitotenv.2019.134479
Ma D, Yang Y, Liu B, Xie G, Chen C, Ren N, Xing D (2021) Zero-valent iron and biochar composite with high specific surface area via K2FeO4 fabrication enhances sulfadiazine removal by persulfate activation. Chem Eng J 408:127992. https://doi.org/10.1016/j.cej.2020.127992
Madduri S, Elsayed I, Hassan EB (2020) Novel oxone treated hydrochar for the removal of Pb(II) and methylene blue (MB) dye from aqueous solutions. Chemosphere 260:127683. https://doi.org/10.1016/j.chemosphere.2020.127683
Mun SP, Cai Z, Zhang J (2015) Preparation of Fe-cored carbon nanomaterials from mountain pine beetle-killed pine wood. Mater Lett 142:45–48. https://doi.org/10.1016/j.matlet.2014.11.053
Neeli ST, Ramsurn H (2018) Synthesis and formation mechanism of iron nanoparticles in graphitized carbon matrices using biochar from biomass model compounds as a support. Carbon 134:480–490. https://doi.org/10.1016/j.carbon.2018.03.079
Ng WM, Lim JK (2022) Complex interplay between colloidal stability, transport, chemical reactivity and magnetic separability of polyelectrolyte-functionalized nanoscale zero-valent iron particles (nZVI) toward their environmental engineering application. Colloid Interface Sci Commun 46:100582. https://doi.org/10.1016/j.colcom.2021.100582
Park MH, Jeong S, Lee G, Park H, Kim JY (2019) Removal of aqueous-phase Pb(II), Cd(II), As(III), and As(V) by nanoscale zero-valent iron supported on exhausted coffee grounds. Waste Manag 92:49–58. https://doi.org/10.1016/j.wasman.2019.05.017
Prabu D, Parthiban R, Ponnusamy SK, Anbalagan S, John R, Titus T (2017) Sorption of Cu(II) ions by nano-scale zero valent iron supported on rubber seed shell. IET Nanobiotechnol 11:714–724. https://doi.org/10.1049/iet-nbt.2016.0224
Rama Chandraiah M (2016) Facile synthesis of zero valent iron magnetic biochar composites for Pb(II) removal from the aqueous medium. Alex Eng J 55:619–625. https://doi.org/10.1016/j.aej.2015.12.015
Samaraweera H, Nawalage S, Nayanathara RMO, Peiris C, Karunaratne TN, Gunatilake SR, Thirumalai RVKG, Zhang J, Zhang X, Mlsna T (2022) In situ synthesis of zero-valent iron-decorated lignite carbon for aqueous heavy metal remediation. Processes 10:1659. https://doi.org/10.3390/pr10081659
Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ (2012) Heavy metal toxicity and the environment. In: Luch A (ed) Molecular, clinical and environmental toxicology, experientia supplementum. Springer, Basel, pp 133–164. https://doi.org/10.1007/978-3-7643-8340-4_6
Thommes M, Kaneko K, Neimark AV, Olivier JP, Rodriguez-Reinoso F, Rouquerol J, Sing KSW (2015) Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC technical report). Pure Appl Chem 87:1051–1069. https://doi.org/10.1515/pac-2014-1117
Thompson E, Danks AE, Bourgeois L, Schnepp Z (2015) Iron-catalyzed graphitization of biomass. Green Chem 17:551–556. https://doi.org/10.1039/C4GC01673D
Vinogradov NA, Simonov KA, Generalov AV, Drnec J, Carlà F, Vinogradov AS, Preobrajenski AB, Mårtensson N, Felici R (2017) The structural evolution of graphene/Fe(110) systems upon annealing. Carbon 111:113–120. https://doi.org/10.1016/j.carbon.2016.09.058
Wang S, Zhou Y, Gao B, Wang X, Yin X, Feng K, Wang J (2017a) The sorptive and reductive capacities of biochar supported nanoscaled zero-valent iron (nZVI) in relation to its crystallite size. Chemosphere 186:495–500. https://doi.org/10.1016/j.chemosphere.2017.08.014
Wang S, Zhao M, Zhao Y, Wang N, Bai J, Feng K, Zhou Y, Chen W, Wen F, Wang S, Wang X, Wang J (2017b) Pyrogenic temperature affects the particle size of biochar-supported nanoscaled zero valent iron (nZVI) and its silver removal capacity. Chem Speciat Bioavailab 29:179–185. https://doi.org/10.1080/09542299.2017.1395712
Wang S, Zhao M, Zhou M, Li YC, Wang J, Gao B, Sato S, Feng K, Yin W, Igalavithana AD, Oleszczuk P, Wang X, Ok YS (2019) Biochar-supported nZVI (nZVI/BC) for contaminant removal from soil and water: a critical review. J Hazard Mater 373:820–834. https://doi.org/10.1016/j.jhazmat.2019.03.080
Wang Z, Wu X, Luo S, Wang Y, Tong Z, Deng Q (2020) Shell biomass material supported nano-zero valent iron to remove Pb2+ and Cd2+ in water. R Soc Open Sci 7:201192. https://doi.org/10.1098/rsos.201192
Wani AL, Ara A, Usmani JA (2015) Lead toxicity: a review. Interdiscip Toxicol 8:55–64. https://doi.org/10.1515/intox-2015-0009
Wirth CT, Bayer BC, Gamalski AD, Esconjauregui S, Weatherup RS, Ducati C, Baehtz C, Robertson J, Hofmann S (2012) The phase of iron catalyst nanoparticles during carbon nanotube growth. Chem Mater 24:4633–4640. https://doi.org/10.1021/cm301402g
Xing R, He J, Hao P, Zhou W (2020) Graphene oxide-supported nanoscale zero-valent iron composites for the removal of atrazine from aqueous solution. Colloids Surf Physicochem Eng Asp 589:124466. https://doi.org/10.1016/j.colsurfa.2020.124466
Xu H, Li X, Gao M, Hu X, Zhang X, Li Y, Xu X, Hu J, Tang C, Hu Xi (2022) Chitosan and biochar synergize the efficient elimination of lead from wastewater by sulfidised nano-zero-valent iron. J Environ Chem Eng 10:107101. https://doi.org/10.1016/j.jece.2021.107101
Yan Q, Wan C, Liu J, Gao J, Yu F, Zhang J, Cai Z (2013) Iron nanoparticles in situ encapsulated in biochar-based carbon as an effective catalyst for the conversion of biomass-derived syngas to liquid hydrocarbons. Green Chem 15:1631–1640. https://doi.org/10.1039/C3GC37107G
Yan Q, Li J, Zhang X, Zhang J, Cai Z (2018a) Synthetic bio-graphene based nanomaterials through different iron catalysts. Nanomaterials 8:840. https://doi.org/10.3390/nano8100840
Yan Q, Li J, Zhang X, Zhang J, Cai Z (2018b) In situ formation of graphene-encapsulated iron nanoparticles in carbon frames through catalytic graphitization of kraft lignin. Nanomater Nanotechnol 8:1–12. https://doi.org/10.1177/1847980418818955
Yan Q, Zhang X, Li J, Hassan EB, Wang C, Zhang J, Cai Z (2018c) Catalytic conversion of kraft lignin to bio-multilayer graphene materials under different atmospheres. J Mater Sci 53:8020–8029. https://doi.org/10.1007/s10853-018-2172-0
Yan Q, Li J, Zhang X, Zhang J, Cai Z (2019) Mass production of graphene materials from solid carbon sources using a molecular cracking and welding method. J Mater Chem A 7:13978–13985. https://doi.org/10.1039/C9TA01332F
Yang J, Ma T, Li X, Tu J, Dang Z, Yang C (2018) Removal of heavy metals and metalloids by amino-modified biochar supporting nanoscale zero-valent iron. J Environ Qual 47:1196–1204. https://doi.org/10.2134/jeq2017.08.0320
Yang K, Xu J, Zhang M, Lin D (2021) Re-recognizing micro locations of nanoscale zero-valent iron in biochar using C-TEM technique. Sci Rep 11:5037. https://doi.org/10.1038/s41598-021-84685-w
Yang L, Shen J, Zhang W, Wu W, Wei Z, Chen M, Yan J, Qian L, Han L, Li J, Gu M (2022) Hydrothermally assisted synthesis of nano zero-valent iron encapsulated in biomass-derived carbon for peroxymonosulfate activation: the performance and mechanisms for efficient degradation of monochlorobenzene. Sci Total Environ 829:154645. https://doi.org/10.1016/j.scitotenv.2022.154645
Zhang X, Yan Q, Li J, Chu I-W, Toghiani H, Cai Z, Zhang J (2018a) Carbon-based nanomaterials from biopolymer lignin via catalytic thermal treatment at 700 to 1000 °C. Polymers 10:183. https://doi.org/10.3390/polym10020183
Zhang X, Yan Q, Li J, Zhang J, Cai Z (2018b) Effects of physical and chemical states of iron-based catalysts on formation of carbon-encapsulated iron nanoparticles from kraft lignin. Materials 11:139. https://doi.org/10.3390/ma11010139
Zhang C, Tang J, Gao F, Yu C, Li S, Lyu H, Sun H (2021a) Tetrahydrofuran aided self-assembly synthesis of nZVI@gBC composite as persulfate activator for degradation of 2,4-dichlorophenol. Chem Eng J. https://doi.org/10.1016/j.cej.2021.134063
Zhang X, Navarathna CM, Leng W, Karunaratne T, Thirumalai RVKG, Kim Y, Pittman CU Jr, Mlsna T, Cai Z, Zhang J (2021b) Lignin-based few-layered graphene-encapsulated iron nanoparticles for water remediation. Chem Eng J 417:129199. https://doi.org/10.1016/j.cej.2021.129199
Zhang X, Elsayed I, Nayanathara RMO, Song X, Shmulsky R, Hassan EB (2022a) Biobased hierarchically porous carbon featuring micron-sized honeycomb architecture for CO2 capture and water remediation. J Environ Chem Eng 10:107460. https://doi.org/10.1016/j.jece.2022.107460
Zhang X, Karunaratne T, Navarathna C, Zhang J, Pittman CU (2022b) Nanoscale zero-valent iron-decorated biochar for aqueous contaminant removal. In: Mohan D, Pittman CU Jr, Mlsna TE (eds) Sustainable biochar for water and wastewater treatment. Elsevier, Amsterdam, pp 611–641. https://doi.org/10.1016/B978-0-12-822225-6.00001-4
Zhou Y, Gao B, Zimmerman AR, Cao X (2014) Biochar-supported zerovalent iron reclaims silver from aqueous solution to form antimicrobial nanocomposite. Chemosphere 117:801–805. https://doi.org/10.1016/j.chemosphere.2014.10.057
Zhu S, Ho S-H, Huang X, Wang D, Yang F, Wang L, Wang C, Cao X, Ma F (2017) Magnetic nanoscale zerovalent iron assisted biochar: interfacial chemical behaviors and heavy metals remediation performance. ACS Sustain Chem Eng 5:9673–9682. https://doi.org/10.1021/acssuschemeng.7b00542
Zhuo S-N, Ren H-Y, Cao G-L, Xie G-J, Xing D-F, Ren N-Q, Liu B-F (2022) Highly efficient activation of persulfate by encapsulated nano-Fe0 biochar for acetaminophen degradation: rich electron environment and dominant effect of superoxide radical. Chem Eng J 440:135947. https://doi.org/10.1016/j.cej.2022.135947
Acknowledgements
The authors thank USDA-NIFA provide the funding support for this project. The authors also the Institute of Imaging and Analytical Technology, Mississippi State University (MSU) for providing access to XRD, SEM, and TEM characterizations. This manuscript is publication #SB1077 of the Department of Sustainable Bioproducts, MSU. This publication is a contribution of the Forest and Wildlife Research Center, MSU.
Funding
This work was supported by the USDA National Institute of Food and Agriculture (NIFA) (Grant No. 2020-65210-30763).
Author information
Authors and Affiliations
Contributions
TNK: Investigation, Methodology, Data analysis, Writing original draft, Review & editing. RMON: Investigation, Methodology, Data analysis. CMN: Investigation, Methodology. PMR: Methodology, Data analysis. RVKGT: Methodology. CUPJr: Scientific discussion, Review & editing. YK: Funding acquisition, Scientific discussion. TM: Funding acquisition, Methodology, Scientific discussion; JZ: Funding acquisition, Scientific discussion, Review & editing. XZ: Funding acquisition, Project administration, Conceptualization, Investigation, Data analysis, Writing original draft, Review & editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary Information
Additional file 1: Figure S1.
Gas adsorption study of iron salts-derived BC-G@Fe0 (a) N2 adsorption isotherms of iron salts-derived BC-G@Fe0. (b) Specific surface area and pore volume results. Figure S2. Gas adsorption study of BC-G@Fe0-X (a) N2 adsorption isotherms of BC-G@Fe0-600-BC-G@Fe0-1000 (b) Specific surface area and pore volume results. Figure S3. Spot diameter distribution of the Fe0 nanoparticles made with different impregnated iron salts (route II) shown in the SEM images (Fig. 3h–k). Figure S4. SEM image and EDS elemental mapping of BC-G@Fe0-600. Figure S5. SEM image and EDS elemental mapping of BC-G@Fe0-700. Figure S6. SEM image and EDS elemental mapping of BC-G@Fe0-900. Figure S7. SEM image and EDS elemental mapping of BC-G@Fe0-1000. Table S1. Fe content in all BC-G@Fe0 products prepared by route II. Table S2. Kinetic and Isotherm Parameters for Pb2+, Cu2+ and Ag+ Adsorptions onto BC-G@Fe0
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Karunaratne, T.N., Nayanathara, R.M.O., Navarathna, C.M. et al. Pyrolytic synthesis of graphene-encapsulated zero-valent iron nanoparticles supported on biochar for heavy metal removal. Biochar 4, 70 (2022). https://doi.org/10.1007/s42773-022-00196-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42773-022-00196-5