
Abstract
SIRT7 plays critical roles in tumorigenesis and tumor progression; however, the underlying mechanisms are poorly understood. Here, we aimed to identify downstream targets of SIRT7 to help delineate its precise function. In this study, we demonstrate that SIRT7 is essential to regulate IDH1 expression in various cancer cell types. Interestingly, both SIRT7 and IDH1 levels are downregulated in breast cancer lung metastases and are useful for predicting disease progression and prognosis. Mechanistically, SIRT7 enhances IDH1 transcription, and this process is mediated by SREBP1. SIRT7 insufficiency reduces cellular α-ketoglutarate, a metabolite product of IDH1, and suppresses lipogenesis and gluconeogenesis. Moreover, α-ketoglutarate decline increases HIF1α protein levels and, thus, promotes glycolysis. This effect permits cancer cells to facilitate Warburg effect and undergo fast proliferation. Overall, the SIRT7–IDH1 axis regulates cancer cell metabolic reprogramming and, thus, might serve as a point of therapeutic intervention.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The essence of a tumor can be characterized as uncontrolled cell proliferation (Hanahan and Weinberg 2000). To support that, cancer cells frequently adjust their energy metabolism to respond to cell growth and division signals (Xie and Simon 2017). Via a process known as the Warburg effect, most cancer cells utilize glycolysis to generate energy and metabolites, even when sufficient oxygen is present for the oxidative phosphorylation pathway (Warburg 1956; Liberti and Locasale 2016). To support such neoplastic metabolic reprogramming, several metabolic enzymes exhibit dysregulated functions or gain unexpected mutations (Sreedhar and Zhao 2018). The isocitrate dehydrogenase enzyme, IDH1, is one such enzyme that frequently carries a gain-of-function mutation resulting in abnormal expression in certain cancers (Suzuki et al. 2015; Yan et al. 2009). IDH1 localizes in the cytoplasm and peroxisomes, catalyzing the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) and NADPH (Geisbrecht and Gould 1999; D'Adamo and Haft 1965). IDH1 reversibly converts α-KG into isocitrate and NADP+ via reductive carboxylation (Ochoa 1948) and participates in lipid biosynthesis from glutamine in some cancers (Filipp et al. 2012; Metallo et al. 2011). As a co-factor, α-KG is indispensable for the enzymatic activity of (α-KG)-dependent dioxygenases, such as histone demethylases, prolyl hydroxylases (PHDs), collagen prolyl-4-hydroxylases, and the TET (ten-eleven translocation) family of 5-methlycytosine (5mC) hydroxylases (Chowdhury et al. 2011; Tsukada et al. 2006; Tahiliani et al. 2009). Moreover, IDH1 regulates cellular histone methylation to manipulate tumorigenesis. A somatic heterozygous mutation, IDH1R132, leads to the generation of d-2-hydroxygluarate (2-HG), which competes with α-KG and subsequently blocks (α-KG)-dependent dioxygenases, resulting in global DNA or histone hypermethylation and hypoxia-inducible factor (HIF1α) stabilization; this effect can accelerate glioma and leukemia development (Clark et al. 2016).
IDH1 mutations, albeit at low frequency, have been reported in other solid cancers, and emerging evidence suggests that non-mutated IDH1 also has important biological functions and clinical effects in breast cancers and some gliomas (Calvert et al. 2017; Liu et al. 2015, 2018). IDH1 mRNA and protein levels are elevated in primary glioblastomas (GBMs), promoting growth and therapeutic resistance (Calvert et al. 2017). IDH1 loss during breast cancer progression promotes metastasis via SLUG upregulation and HIF1α stabilization (Liu et al. 2018). However, the mechanisms of non-mutated IDH1 regulation remain elusive in cancers.
SIRT7 is a member of the sirtuin family of proteins that have NAD+-dependent deacetylase activity (Blank and Grummt 2017). Recent findings suggest that SIRT7 is involved in ribosomal RNA (rRNA) transcription (Ford et al. 2006), chromatin remodeling (Vazquez et al. 2016), DNA repair and cell survival (Tang et al. 2017a, b), lipid metabolism (Tang 2015), and cancer metastasis (Li et al. 2018; Tang et al. 2017a, b). Here, we aimed to understand the roles of SIRT7 in regulation of tumor associated metabolism reprogramming. To do so, we analyzed the link between SIRT7 and metabolism-related enzymes and identified IDH1 as a new downstream target of SIRT7, which modulates cancer lipogenesis, gluconeogenesis and glycolysis via α-KG.
Materials and methods
Cell cultures
Human normal hepatic L-O2 cells; human hepatocellular carcinoma HepG2 and CRL-8024 cells; human glioma U-118 MG, U87, and U251 cells; and human breast cancer MCF-7 and MDA-MB-231 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, high glucose) (Thermo Fisher, USA) supplemented with 10% fetal bovine serum (FBS, PAN-Biotech GmbH, Germany), 100 U/ml penicillin, and 100 g/ml streptomycin (Thermo Fisher), at 37 °C under a 5% CO2 atmosphere.
Constructs, siRNAs, and transfection
The human IDH1 promoter was amplified from genomic DNA extracted from HEK293 cells, with the following primer sequence. Forward primer: 5′-CCGCTCGAGCCGTTTTCTAAGGCTTCACATC-3′; reverse primer: 5′-GGAAGATCTTCCGATGATATGCTGGCGAAGAG-3′. Then, the promoter region was cloned into the luciferase reporter system vector pGL-4.20 (Promega, USA). To mutate the SREBP1-recognizing motif on IDH1 promoter, site mutagenesis was performed with the following primers. Forward primer: 5′-CGGGGCTGGGGGAGGTTTCGTGAGGAGGCGGGGCCT-3′; reverse primer: 5′-AGGCCCCGCCTCCTCACGAAACCTCCCCCAGCCCCG-3′. For gene silencing, we synthesized specific custom siRNAs purchased from GenePharma Company (Shanghai, China). All oligo RNA sequences used in this manuscript are listed below: human SIRT7 siRNA-1, 5′-CUCACCGUAUUUCUACUACUAdTdT-3′ and.
human SREBP1 siRNA-1, 5′-GCUCCUCUCUUGAAGCCUUTT-3′. For plasmid and oligo RNA transient transfections, we employed Lipo3000 reagent (Thermo Fisher), following the operational guidelines.
RNA isolation and qRT-PCR
Whole RNA extracts were isolated using Trizol reagent RNAiso Plus (TaKaRa, Japan). To transcribe RNA into cDNAs, 1000 ng fresh RNA was incubated with the Primescript RT Master Mix (TaKaRa), according to the manufacturer’s instructions. To quantify gene expression, qRT-PCR was performed using 2 × SYBR Green Mix (TaKaRa, RR064) in the Bio-Rad detection system. All primers are shown in Supplementary Table 1.
Cellular α-KG measurement
HepG2 and L-O2 cells with indicated treatments were cultured in 6-well-plates to 70% confluence. Then, the cells were washed twice with cold PBS and lysed with the indicated elusion buffer. The levels of α-KG were measured using a commercial kit (K677-100, BioVision, USA), according to the manufacturer’s instructions.
Dual luciferase reporter assays
HEK293 cells were cultured in 24-well plates to 60% confluence. The indicated plasmids were cotransfected with Lipo3000 reagent. After 48 h, luminescence was measured using a Dual-Luciferase Reporter Assay System (Promega), according to the manufacturer’s instructions. The relative luciferase activity was calculated as the ratio of firefly to Renilla luciferase activity (Fluc/Rluc).
Protein extraction and immunoblotting
To prepare protein samples, cells were lysed in RIPA buffer. Then, protein extracts were resolved in Laemmli sample buffer, separated using SDS-PAGE, transferred to a PVDF membrane (Millipore), blocked with 5% non-fat milk, and probed with the respective primary antibodies. Immunoblotting images were acquired using a ChemiDoc system (Bio-Rad, USA). All antibodies used are described in Supplementary Table 2.
Immunoprecipitation (IP)
The indicated samples were prepared in IP lysis buffer (200 mM NaCl, 20 mM Tris–HCl (pH 7.9), 5 mM MgCl2, 10% glycerol, 0.2 mM EDTA, and 0.1% NP-40) supplemented with protease inhibitors (Sigma, complete™ Protease Inhibitor Cocktail and PhosSTOP™). Clarified cell supernatants were incubated with the respective antibodies for 4 h at 4 °C. After the beads were washed with IP lysis buffer and eluted in Laemmli sample buffer, the immunoprecipitates were subjected to western blotting analysis.
Kaplan–Meier survival analysis of patients with breast cancer
The online tool, PROGgene V2, was employed to analyze correlations between levels of IDH1 with clinical outcomes (relapse-free survival (RFS) or overall survival (OS) of breast cancers. All Kaplan–Meier survival diagrams shown in the manuscript were analyzed according to previously published methods (Goswami and Nakshatri 2014).
Statistical analysis
All results are presented as means ± SEM, based on at least three independent experiments. Data were analyzed by the Student’s t test using Prism 7.0 software (GraphPad Prism). A p value < 0.05 was considered to be statistically significant.
Results
SIRT7 positively regulates IDH1 expression in breast cancers
In our first analyses, we determined the IDH1 protein and mRNA expression profile in various breast cancer cell lines. We found that, consistent with previous reports (Liu et al. 2018), IDH1 was decreased at the mRNA and protein level, in invasive breast cancer cells (Fig. 1a, b). These findings suggest that IDH1 is downregulated at the level of its transcriptional regulation. We previously have found that SIRT7 expression is significantly downregulated in breast cancer lung metastases (Tang et al. 2017a, b). Interestingly, similarly to SIRT7, we found that IDH1 protein expression was remarkably decreased in invasive breast cancer cells compared to the non-invasive ones (Fig. 1a). The mRNA levels of SIRT7 and IDH1 also displayed consistent tendencies as the protein expression levels (Fig. 1b), suggesting the transcriptional regulation. To explore whether SIRT7 mediates IDH1 expression, we knocked down SIRT7 expression in human breast cancer MCF-7 cells, which have high endogenous SIRT7 and IDH1 levels. SIRT7 knockdown significantly downregulated IDH1 expression, both in terms of protein and mRNA levels (Fig. 1c, d). By contrast, other isocitrate dehydrogenase enzymes, including IDH2, IDH3α, IDH3β and IDH3γ, were affected little by SIRT7 knockdown (Supplementary Figure 1). We, thus, randomly employed IDH3α as a negative control in the following experiments. Next, we overexpressed SIRT7 in human breast cancer MDA-MB-231 cells (MDA-231), which usually express low levels of SIRT7 and IDH1. Boosting SIRT7 expression increased IDH1 transcription and protein levels (Fig. 1e, f). Taken together, our results indicate that SIRT7 might positively regulate IDH1 expression in breast cancer cells.

SIRT7 positively regulates IDH1 expression in multiple cancers. Immunoblotting (a) and qRT-PCR (b) analysis of IDH1 and SIRT7 levels in various breast cancer cells classified into non-invasive and invasive groups. Immunoblotting (c) and qRT-PCR (d) analysis of IDH1 expression in human breast cancer MCF-7 cells with or without SIRT7 knockdown (siSIRT7). Scram, scrambled siRNAs taken as a negative control. IDH3 is an isoenzyme of IDH1 and here was considered to be a control. Immunoblotting (e) and qRT-PCR (f) analysis of IDH1 expression in human breast cancer MDA-MB-231 cells with or without overexpression of ectopic SIRT7. EV (empty vector), cells were transfected with empty vector plasmid as a control. (g, i Immunoblotting (upper panel) and qRT-PCR (lower panel) analysis of IDH1 levels in human normal hepatic L-O2 cells and human liver cancer HepG2 and 8024 cells, with or without knockdown of SIRT7. (j) Immunoblotting analysis of IDH1 levels in human glioma U-118 MG, U87, and U251 cells, with or without knockdown of SIRT7. (k) Idh1 protein levels were detected in liver tissues of Sirt7+/+ or Sirt7−/− mice. Representative data are shown as means ± SEM. *p < 0.05; Student’s t test
SIRT7 regulates IDH1 expression in multiple cancers
To date, there have been few reports describing the regulation of IDH1 expression. We, thus, assessed whether the modulation of IDH1 by SIRT7 was dependent on the cell or the cancer context. To this end, we analyzed IDH1 expression changes with or without SIRT7 knockdown in L-O2 human normal hepatic cells; human hepatocellular carcinoma HepG2 and CRL-8024 cells; and human glioma U-118 MG, U87, and U251 cells. As shown, SIRT7 knockdown impaired IDH1 protein and mRNA levels, regardless of the cellular context (Fig. 1g, j). SIRT7 regulates lipogenesis and is abundant in liver tissues (Yoshizawa et al. 2014). Consistently, we found that Idh1 was markedly decreased in the liver lysate of Sirt7−/− mice (Fig. 1k). Together, these results indicate that SIRT7 likely regulates IDH1 expression in multiple cancers and tissues.
SIRT7 is required to sustain IDH1 transcription
Based on our findings thus far, we assumed that SIRT7 might regulate IDH1 transcription. To test this hypothesis, we cloned the IDH1 promoter into the pGL-4.20 luciferase reporter system (pIDH1-Pro, Fig. 2a), as previously described (Shechter et al. 2003). We saw that IDH1 promoter activity was significantly decreased in SIRT7 knockdown cells (Fig. 2b). By contrast, SIRT7 overexpression seemed to facilitate IDH1 transcription (Fig. 2c). These results confirm that SIRT7 sustains IDH1 transcription.

SIRT7 positively regulates IDH1 transcription. a A schematic diagram of the IDH1 promoter (pIDH1-Pro). b A luciferase reporter assay showing IDH1 promoter activity (upper panel) in HEK293 cells transfected with SIRT7 siRNAs (siSIRT7) or the control siRNAs (Scram). The lower panel shows SIRT7 protein levels. c A luciferase reporter assay detecting IDH1 promoter activity (upper panel) in HEK293 cells expressing ectopic SIRT7 (OESIRT7) or empty vector plasmid (EV). SIRT7 levels were monitored by immunoblotting, shown in the lower panel. d Immunoblotting analysis of IDH1 expression in MCF-7 cells treated or not treated with NAM. NAM, nicotinamide (10 mM). e A luciferase reporter assay showing IDH1 promoter activity (upper panel) in HEK293 cells transfected with wild-type (WT) SIRT7 or catalytically inactive mutant (H187Y). Representative data are shown as means ± SEM. **p < 0.001, ***p < 0.001; Student’s t test
As SIRT7 possesses deacetylase activity, we next asked whether such regulation of IDH1 relies on its enzymatic activity. Surprisingly, compared to the untreated control only with solvent, nicotinamide (NAM; a pan-sirtuin inhibitor) enhanced rather than suppressed the level of IDH1 protein, implying that SIRT7 deacetylase activity had a less profound impact on IDH1 transcriptional activity (Fig. 2d). We presumed that this NAM-induced increase in IDH1 levels could be attributed to elevated SIRT7 levels (Fig. 2d, the middle lane), as SIRT7 knockdown reversed the effects of NAM on IDH1 (Fig. 2d, the last lane). To exclude any non-specific effects of NAM, we evaluated whether a SIRT7 catalytically inactive mutant (H187Y) was sufficient to promote IDH1 transcription. Wild-type (WT) SIRT7 and H187Y had similar effects on IDH1 promoter activity: both causing ~ 2.5-fold increase of IDH1 promoter activity (Fig. 2e). Additionally, we observed that overexpression of either wild-type SIRT7 or SIRT7–H187Y sufficiently increased endogenous IDH1 protein levels (Supplementary Figure 2). These findings support that SIRT7 promotes IDH1 transcription in a deacetylase-independent manner.
SREBP1 underlies SIRT7-mediated transcriptional regulation of IDH1
Previous research has shown that the sterol regulatory element-binding protein 1 (SREBP1) also regulates IDH1 transcription (Ricoult et al. 2016; Shechter et al. 2003). We, thus, decided to test whether SIRT7 worked with SREBP1 to regulate IDH1 expression. Interestingly, we observed an apparent protein interaction between SIRT7 and SREBP1 (Fig. 3a). We, thus, investigated whether SIRT7-mediated IDH1 transcription is SREBP1 dependent. Indeed, the SIRT7 overexpression-induced increase in IDH1 transcription was significantly attenuated by SREBP1 knockdown (Fig. 3b). To gain direct evidence of such SREBP1-dependent regulation, we mutated a SREBP1-recognizing motif, GTGGGCTGAG (pIDH1-Pro-WT), to GTTTCGTGAG (pIDH1-Pro-Mut) in the IDH1 promoter (Fig. 3c). As shown by the luciferase reporter assay, pIDH1-Pro-mut significantly attenuated luciferase activity compared with pIDH1-Pro-WT and failed to respond to SIRT7 overexpression (Fig. 3d). Together, these results support that SIRT7 regulates IDH1 transcription via SREBP1.

SREBP1 underlies SIRT7-induced IDH1 transcription. a Immunoblotting analysis of HA-SIRT7 in anti-Flag-SREBP1 immunoprecipitates derived from HEK293 cells expressing the indicated plasmids. b Analysis of IDH1 promoter activity in HEK293 cells after SREBP1 knockdown or SIRT7 overexpression (upper panel). SREBP1 and SIRT7 levels were analyzed by immunoblotting (lower panel). c A schematic diagram of the SREBP1-recognizing motif within the IDH1 promoter (pIDH1-Pro-WT). Blue indicates the mutated sequence (pIDH1-Pro-Mut) to disrupt SREBP1 binding. d Analysis of IDH1 promoter activity in HEK293 cells transfected with the indicated plasmids (upper panel). Immunoblotting showing the expression of ectopic SIRT7 (lower panel). Data are presented as means ± SEM, n = 3, **p < 0.001, ***p < 0.001; Student’s t test
SIRT7 regulates lipogenesis and gluconeogenesis
IDH1 produces non-mitochondrial α-KG (Bogdanovic 2015), and as a result, an IDH1 deficiency attenuates lipogenesis and gluconeogenesis via α-KG (Shechter et al. 2003; Ye et al. 2017). We, thus, studied whether SIRT7 affected cellular α-KG levels. We found that SIRT7 knockdown significantly decreased cellular α-KG levels in human hepatocellular carcinoma HepG2 cells (Fig. 4a, b). We, thus, investigated whether SIRT7 is involved in lipid biosynthesis in cancer cells. SIRT7 knockdown in HepG2 and L-O2 cells resulted in the downregulation of FASN, Acly, GPT1, and GOT1 mRNA levels, suggesting suppression of gluconeogenesis and lipogenesis (Fig. 4c, f). Furthermore, reduced SIRT7 expression impaired lipid droplet generation in L-O2 cells, indicative of inhibited lipogenesis (Fig. 4g). Notably, FBP1 and FBP2 were upregulated (Fig. 4c, f), likely because of the feedback from decreasing glucogenic substrates giving rise to the inhibition of lipid biosynthesis (Fig. 4h). Together, these results indicate that SIRT7–IDH1 axis regulates lipogenesis and gluconeogenesis in cancer cells via modulating α-KG production.

SIRT7 regulates lipogenesis and gluconeogenesis. a Immunoblotting analysis of IDH1 levels in scramble-(Scram) and siSIRT7-treated human hepatocellular carcinoma HepG2 cells. b Quantitative analysis of α-KG levels in (a). c qRT-PCR analysis of gene expression related to lipogenesis and gluconeogenesis in (a). d Immunoblotting analysis of IDH1 levels in scramble- (Scram) and siSIRT7-treated human hepatic L-O2 cells. e Quantitative analysis of α-KG levels in (d). f qRT-PCR analysis of genes related to lipogenesis and gluconeogenesis in (d). g Representative images showing Oil Red O staining in L-O2 cells with or without SIRT7 knockdown. h A schematic diagram illustrating changes in metabolic flow in response to SIRT7 deficiency. Green: downregulation; red: upregulation. Representative data are shown as means ± SEM. *p < 0.05, ***p < 0.001; Student’s t test
SIRT7 regulates HIF1α stability and glycolysis
Most cancer cells utilize glycolysis to support fast proliferation rate and HIF1α plays pivotal roles in glycolytic metabolism (Xie and Simon 2017; Liberti and Locasale 2016). Prolyl hydroxylases (PHDs) which target HIF1α for degradation are α-KG-dependent dioxygenases (Selak et al. 2005). Given that SIRT7 regulates IDH1 and cellular α-KG levels, we asked whether SIRT7 affects HIF1α expression in cancer cells. As shown, SIRT7 knockdown elicited a prominent increase of HIF1α protein in breast cancer MCF-7 cells, while IDH1 restoration nearly reversed such effect (Fig. 5a). Moreover, SIRT7 knockdown facilitated cancer cell glycolysis, which was evidenced by the generation of apparent acidic medium during culture (Fig. 5b). Together, our results indicate that the SIRT7–IDH1 axis contributes to metabolic alterations in cancer cells.

SIRT7 regulates HIF1α expression via IDH1. a Immunoblotting analysis of HIF1α protein levels in human breast cancer MCF-7 cells subjected into SIRT7 knockdown or (and) IDH1 restoration. b Representative images showing the culture medium of MCF-7 cells in (a), noting that the medium showing yellow indicated a higher glycolytic rate
The SIRT7–IDH1 axis predicts breast cancer progression
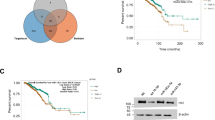
Finally, we explored the clinical relevance of the SIRT7–IDH1 axis. First, IDH1 expression was significantly downregulated in patients with breast cancer who had low SIRT7 levels (Fig. 6a). Second, supporting the SIRT7–IDH1 axis, the level of IDH1 was notably decreased in lung metastasis tumors compared with its level in paired primary tumors, reflecting the decline in the level of SIRT7 (Fig. 6b). Importantly, low IDH1 levels were associated with poor prognosis in breast cancers (Fig. 6c, d), which was similar to findings for SIRT7 (Tang et al. 2017a, b). Taken together, these results indicate that the SIRT7–IDH1 axis might be useful for predicting breast cancer progression.

IDH1–SIRT7 axis is associated with breast cancer prognosis. a Analysis of IDH1 expression in breast cancer patients stratified by SIRT7-Low (n = 316) and SIRT7-High (n = 269). Data were collected from The Cancer Genome Atlas (TCGA) database. b Representative images showing immunohistochemistry staining of SIRT7 and IDH1 in primary and paired lung metastasized tumors. Scale bar, 100 µm. c, d Low IDH1 expression predicted poor prognosis in breast cancers. Data were collected from two independent cohorts, as described in the “Materials and methods” section
Discussion
SIRT7 seems to assume various roles during tumorigenesis that are most likely dependent on the cancer context. On the one hand, SIRT7 is aberrantly increased in colorectal cancers (CRCs), hepatocellular carcinomas (HCCs), and certain stages of thyroid cancers (Wu et al. 2018). Here, SIRT7 serves as an oncogenic regulator that promotes tumor growth and ensures tumor survival (Barber et al. 2012; Wu et al. 2018). However, SIRT7 can also act as a tumor suppressor; for example, SIRT7 antagonizes metastasis in breast cancers and oral squamous cell carcinomas (Li et al. 2018; Tang et al. 2017a, b). SIRT7 acts as a tumor growth suppressor through the deacetylation of WDR77 (Qi et al. 2018), and the loss of SIRT7 is considered to protect breast cancer cells from chemotherapy-induced death via AKT activation (Yu et al. 2017). Sirt7 knockout mice exhibit accelerated development of colon cancer, highlighting its tumor suppressor characteristics (Liu et al. 2020). Here, we have shown that SIRT7 insufficiency downregulates IDH1 expression, resulting in a decline in cellular α-KG, and, thus, suppression of lipogenesis and contributed to HIF1α stabilization. We conclude that the SIRT7–IDH1 axis regulates metabolic reprogramming during cancer progression. Future studies should consider whether the SIRT7–IDH1 axis has distinct functions when cells undergo various oncogenic signals.
The recent published article unravels a novel finding that SIRT2 directly deacetylates IDH1 and inhibits colorectal cancers (Wang et al. 2020). Both SIRT7 and SIRT2 belong to Sirtuin family and are expected to have the NAD+-dependent deacetylase activity. Thus, to explore whether SIRT7 has similar roles for IDH1 deacetylation is interesting. However, we found that SIRT7 was hardly detected in the anti-IDH1 immunoprecipitates (Supplementary Fig. 3), suggesting the lack of direct interaction between SIRT7 and IDH1. Additionally, unlike SIRT2 which majorly locates at cytosol, SIRT7 is considered as a nuclear protein. Therefore, IDH1 as a cytoplasmic enzyme was less likely deacetylated by SIRT7. Altogether, our findings highlight the novel mechanism that SIRT7 regulates IDH1 expression in a transcriptional-dependent manner.
SIRT7 possesses deacetylase activity, yet here we found that SIRT7 regulates IDH1 transcription via SREBP1 in a deacetylase-independent manner, suggesting the existence of unexplored mechanisms. Indeed, when compared with other intimate nuclear-localized sirtuins (SIRT1 and SIRT6), SIRT7 exhibits much weaker deacetylase activity (Michishita et al. 2005). Furthermore, SIRT7 generally tends to form complexes with other effectors to participate in physiological activity. For instance, SIRT7 is recruited by ETS-like transcription factor 4 to deacetylate H3K18 (Barber et al. 2012). SIRT7 interacts with SIRT1 to prevent SIRT1 autodeacetylation and manipulate adipogenesis (Fang et al. 2017). Interestingly, SIRT1 directly deacetylates SREBP1 and, thus, inhibits SREBP-1C activity in regulation of hepatic lipid metabolism (Ponugoti et al. 2010). Considering that IDH1 expression is dependent on SREBP1 (Shechter et al. 2003), we propose the model that SIRT7 recruited by SREBP1 might interact with SIRT1 to inhibit the SIRT1 autocatalytic activation, which impairs the deacetylation of SREBP1 by SIRT1. In the future study, it would be interesting to explore the detailed mechanisms about SIRT7/SREBP1-mediated IDH1 regulation.
IDH1 is an isocitrate dehydrogenase, and belongs to a family that also includes IDH2 and IDH3 (Yan et al. 2009). We observed no apparent change in IDH2 or IDH3 expression following the knockdown of SIRT7, suggesting that SIRT7 uniquely regulates IDH1. Much work has focused on the functional relevance of an IDH1 mutation (R132C/H), which produces d-2-hydroxyglutarate (d-2-HG), a competitor of α-KG, and contributes to tumorigenesis via the suppression of Fe (II)/α-KG-dependent dioxygenases in low-grade gliomas and secondary GBMs (Yang et al. 2012; Yan et al. 2009). However, comparatively less is known about the regulation of non-mutated IDH1. Indeed, non-mutated IDH1 also has important roles in tumorigenesis. For example, in GBMs IDH1 is upregulated to support macromolecular synthesis, aggressive growth, and therapy resistance (Calvert et al. 2017); conversely, breast cancers that express low levels of IDH1 have invasive phenotypes and undergo epithelial-to-mesenchymal (EMT) transition (Liu et al. 2018). Additionally, dioxygenases such as JmjC domain-containing histone demethylases (KDMs) require α-KG as co-substrate to catalyze reactions (Tsukada et al. 2006). Given that SIRT7 regulates IDH1 and cellular α-KG levels, SIRT7 might affect epigenetic modifications, e.g., histone methylation, in cancer cells. Future studies to test that possibility would suggest novel roles of SIRT7 in epigenetic reprogramming besides its H3K18 deacetylation activity (Barber et al. 2012).
HIF1α plays critical roles in most of solid tumors. In this study, we did not provide many more results to show that SIRT7 regulates HIF1α protein stability. Actually, SIRT7 regulating the HIF1α expression is not novel and has been well addressed, i.e., SIRT7 promoting HIF1/2α degradation is independent of its deacetylase activity (Hubbi et al. 2013). However, the relevant mechanism is not clearly elucidated. Our findings that SIRT7 sustains IDH1 expression and maintains α-KG production somehow address that issue, i.e., the α-KG linking to PHDs activity contributing to HIF1/2α degradation is at least partially dependent on SIRT7.
Overall, our data highlight the potential of SIRT7 to modulate metabolic reprogramming in cancer via IDH1-mediated α-KG and provide a novel point to evaluate the role of non-mutated IDH1 during tumorigenesis and progression.
Availability of data and material
Any data and material in this study are available upon reasonable request.
References
Barber, M. F., Michishita-Kioi, E., Xi, Y., Tasselli, L., Kioi, M., Moqtaderi, Z., et al. (2012). SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature, 487(7405), 114–118. https://doi.org/10.1038/nature11043.
Blank, M. F., & Grummt, I. (2017). The seven faces of SIRT7. Transcription, 8(2), 67–74. https://doi.org/10.1080/21541264.2016.1276658.
Bogdanovic, E. (2015). IDH1, lipid metabolism and cancer: Shedding new light on old ideas. Biochimica et Biophysica Acta, 1850(9), 1781–1785. https://doi.org/10.1016/j.bbagen.2015.04.014.
Calvert, A. E., Chalastanis, A., Wu, Y., Hurley, L. A., Kouri, F. M., Bi, Y., et al. (2017). Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Reports, 19(9), 1858–1873.
Chowdhury, R., Yeoh, K. K., Tian, Y. M., Hillringhaus, L., Bagg, E. A., Rose, N. R., et al. (2011). The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Reports, 12(5), 463–469. https://doi.org/10.1038/embor.2011.43.
Clark, O., Yen, K., & Mellinghoff, I. K. (2016). Molecular pathways: Isocitrate dehydrogenase mutations in cancer. Clinical Cancer Research, 22(8), 1837–1842. https://doi.org/10.1158/1078-0432.CCR-13-1333.
D’Adamo, A. F., & Haft, D. E. (1965). An alternate pathway of α-ketoglutarate catabolism in the isolated, perfused rat liver. I. Studies with dl-glutamate-2-and-5-14C. Journal of Biological Chemistry, 240(2), 613–617.
Fang, J., Ianni, A., Smolka, C., Vakhrusheva, O., Nolte, H., Kruger, M., et al. (2017). Sirt7 promotes adipogenesis in the mouse by inhibiting autocatalytic activation of Sirt1. Proceedings of the National Academy of Sciences United State of America, 114(40), E8352–E8361. https://doi.org/10.1073/pnas.1706945114.
Filipp, F. V., Scott, D. A., Ronai, Z. A., Osterman, A. L., & Smith, J. W. (2012). Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell & Melanoma Research, 25(3), 375–383. https://doi.org/10.1111/j.1755-148X.2012.00989.x.
Ford, E., Voit, R., Liszt, G., Magin, C., Grummt, I., & Guarente, L. (2006). Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes & Development, 20(9), 1075–1080. https://doi.org/10.1101/gad.1399706.
Geisbrecht, B. V., & Gould, S. J. (1999). The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. Journal of Biological Chemistry, 274(43), 30527–30533. https://doi.org/10.1074/jbc.274.43.30527.
Goswami, C. P., & Nakshatri, H. (2014). PROGgeneV2: Enhancements on the existing database. BMC Cancer, 14, 970. https://doi.org/10.1186/1471-2407-14-970.
Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100(1), 57–70. https://doi.org/10.1016/s0092-8674(00)81683-9.
Hubbi, M. E., Hu, H., Kshitiz, G., & D. M., & Semenza, G. L. . (2013). Sirtuin-7 inhibits the activity of hypoxia-inducible factors. Journal of Biological Chemistry, 288(29), 20768–20775. https://doi.org/10.1074/jbc.M113.476903.
Li, W., Zhu, D., & Qin, S. (2018). SIRT7 suppresses the epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis by promoting SMAD4 deacetylation. Journal of Experimental & Clinical Cancer Research, 37(1), 148. https://doi.org/10.1186/s13046-018-0819-y.
Liberti, M. V., & Locasale, J. W. (2016). The warburg effect: How does it benefit cancer cells? Trends in Biochemical Sciences, 41(3), 211–218. https://doi.org/10.1016/j.tibs.2015.12.001.
Liu, S., Wang, H., Zhang, L., Tang, C., Jones, L., Ye, H., et al. (2015). Rapid detection of genetic mutations in individual breast cancer patients by next-generation DNA sequencing. Human Genomics, 9, 2. https://doi.org/10.1186/s40246-015-0024-4.
Liu, W. S., Chan, S. H., Chang, H. T., Li, G. C., Tu, Y. T., Tseng, H. H., et al. (2018). Isocitrate dehydrogenase 1-snail axis dysfunction significantly correlates with breast cancer prognosis and regulates cell invasion ability. Breast Cancer Research, 20(1), 25. https://doi.org/10.1186/s13058-018-0953-7.
Liu, X., Li, C., Li, Q., Chang, H. C., & Tang, Y. C. (2020). SIRT7 Facilitates CENP-A nucleosome assembly and suppresses intestinal tumorigenesis. iScience, 23(9), 101461. https://doi.org/10.1016/j.isci.2020.101461.
Metallo, C. M., Gameiro, P. A., Bell, E. L., Mattaini, K. R., Yang, J., Hiller, K., et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature, 481(7381), 380–384. https://doi.org/10.1038/nature10602.
Michishita, E., Park, J. Y., Burneskis, J. M., Barrett, J. C., & Horikawa, I. (2005). Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Molecular Biology of the Cell, 16(10), 4623–4635. https://doi.org/10.1091/mbc.e05-01-0033.
Ochoa, S. (1948). Biosynthesis of tricarboxylic acids by carbon dioxide fixation; enzymatic mechanisms. Journal of Biological Chemistry, 174(1), 133–157.
Ponugoti, B., Kim, D. H., Xiao, Z., Smith, Z., Miao, J., Zang, M., et al. (2010). SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. Journal of Biological Chemistry, 285(44), 33959–33970. https://doi.org/10.1074/jbc.M110.122978.
Qi, H., Shi, X., Yu, M., Liu, B., Liu, M., Song, S., et al. (2018). Sirtuin 7-mediated deacetylation of WD repeat domain 77 (WDR77) suppresses cancer cell growth by reducing WDR77/PRMT5 transmethylase complex activity. Journal of Biological Chemistry, 293(46), 17769–17779. https://doi.org/10.1074/jbc.RA118.003629.
Ricoult, S. J., Dibble, C. C., Asara, J. M., & Manning, B. D. (2016). Sterol regulatory element binding protein regulates the expression and metabolic functions of wild-type and oncogenic IDH1. Molecular and Cellular Biology, 36(18), 2384–2395. https://doi.org/10.1128/MCB.00163-16.
Selak, M. A., Armour, S. M., MacKenzie, E. D., Boulahbel, H., Watson, D. G., Mansfield, K. D., et al. (2005). Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell, 7(1), 77–85. https://doi.org/10.1016/j.ccr.2004.11.022.
Shechter, I., Dai, P., Huo, L., & Guan, G. (2003). IDH1 gene transcription is sterol regulated and activated by SREBP-1a and SREBP-2 in human hepatoma HepG2 cells: evidence that IDH1 may regulate lipogenesis in hepatic cells. Journal of Lipid Research, 44(11), 2169–2180. https://doi.org/10.1194/jlr.M300285-JLR200.
Sreedhar, A., & Zhao, Y. (2018). Dysregulated metabolic enzymes and metabolic reprogramming in cancer cells. Biomedical Reports, 8(1), 3–10. https://doi.org/10.3892/br.2017.1022.
Suzuki, H., Aoki, K., Chiba, K., Sato, Y., Shiozawa, Y., Shiraishi, Y., et al. (2015). Mutational landscape and clonal architecture in grade II and III gliomas. Nature Genetics, 47(5), 458–468. https://doi.org/10.1038/ng.3273.
Tahiliani, M., Koh, K. P., Shen, Y., Pastor, W. A., Bandukwala, H., Brudno, Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science, 324(5929), 930–935. https://doi.org/10.1126/science.1170116.
Tang, B. L. (2015). SIRT7 and hepatic lipid metabolism. Frontiers in Cell and Developmental Biology, 3, 1. https://doi.org/10.3389/fcell.2015.00001.
Tang, M., Lu, X., Zhang, C., Du, C., Cao, L., Hou, T., et al. (2017a). Downregulation of SIRT7 by 5-fluorouracil induces radiosensitivity in human colorectal cancer. Theranostics, 7(5), 1346–1359. https://doi.org/10.7150/thno.18804.
Tang, X., Shi, L., Xie, N., Liu, Z., Qian, M., Meng, F., et al. (2017b). SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis. Nature Communication, 8(1), 318. https://doi.org/10.1038/s41467-017-00396-9.
Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., et al. (2006). Histone demethylation by a family of JmjC domain-containing proteins. Nature, 439(7078), 811–816. https://doi.org/10.1038/nature04433.
Vazquez, B. N., Thackray, J. K., Simonet, N. G., Kane-Goldsmith, N., Martinez-Redondo, P., Nguyen, T., et al. (2016). SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO Journal, 35(14), 1488–1503. https://doi.org/10.15252/embj.201593499.
Wang, B., Ye, Y., Yang, X., Liu, B., Wang, Z., Chen, S., et al. (2020). SIRT2-dependent IDH1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Reports, 21(4), e48183. https://doi.org/10.15252/embr.201948183.
Warburg, O. (1956). On respiratory impairment in cancer cells. Science, 124(3215), 269–270.
Wu, D., Li, Y., Zhu, K. S., Wang, H., & Zhu, W. G. (2018). Advances in cellular characterization of the sirtuin isoform, SIRT7. Frontiers in Endocrinology (Lausanne), 9, 652. https://doi.org/10.3389/fendo.2018.00652.
Xie, H., & Simon, M. C. (2017). Oxygen availability and metabolic reprogramming in cancer. Journal of Biological Chemistry, 292(41), 16825–16832. https://doi.org/10.1074/jbc.R117.799973.
Yan, H., Parsons, D. W., Jin, G., McLendon, R., Rasheed, B. A., Yuan, W., et al. (2009). IDH1 and IDH2 mutations in gliomas. New England Journal of Medicine, 360(8), 765–773. https://doi.org/10.1056/NEJMoa0808710.
Yang, H., Ye, D., Guan, K. L., & Xiong, Y. (2012). IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clinical Cancer Research, 18(20), 5562–5571. https://doi.org/10.1158/1078-0432.CCR-12-1773.
Ye, J., Gu, Y., Zhang, F., Zhao, Y., Yuan, Y., Hao, Z., et al. (2017). IDH1 deficiency attenuates gluconeogenesis in mouse liver by impairing amino acid utilization. Proceedings of the National Academy of Sciences United State of America, 114(2), 292–297. https://doi.org/10.1073/pnas.1618605114.
Yoshizawa, T., Karim, M. F., Sato, Y., Senokuchi, T., Miyata, K., Fukuda, T., et al. (2014). SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin-proteasome pathway. Cell Metabolism, 19(4), 712–721. https://doi.org/10.1016/j.cmet.2014.03.006.
Yu, J., Qin, B., Wu, F., Qin, S., Nowsheen, S., Shan, S., et al. (2017). Regulation of serine-threonine kinase Akt activation by NAD(+)-dependent deacetylase SIRT7. Cell Reports, 18(5), 1229–1240. https://doi.org/10.1016/j.celrep.2017.01.009.
Acknowledgements
The authors would like to thank Dr. Jessica Tamanini (Shenzhen University and ETediting) for editing the manuscript prior to submission.
Funding
This study was supported by grants from the National Key R&D Program of China (2017YFA0503900), the National Natural Science Foundation of China (Grant nos. 91849208, 81972602, 81702909), the Science and Technology Program of Guangdong Province (Grant nos. 2017B030301016 and 2019B030301009), the Shenzhen Municipal Commission of Science and Technology Innovation (Grant nos. ZDSYS20190902093401689, KQJSCX20180328093403969 and JCYJ20180507182044945), and the German Research Council (GRK 1715).
Author information
Authors and Affiliations
Contributions
FS, XT and GL carried out most of experiments and collected data; BL and XT conceived the study and wrote the manuscript. KA contributed to editing of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no potential conflicts of interest.
Consent for publication
All authors have approved the final submission and consented to publication.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, F., Tang, X., Li, G. et al. SIRT7–SREBP1 restrains cancer cell metabolic reprogramming by upregulating IDH1. GENOME INSTAB. DIS. 2, 126–137 (2021). https://doi.org/10.1007/s42764-021-00031-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42764-021-00031-4