
Abstract
The SARS-Cov-2 virus, which is evolving continuously and causing adverse effects throughout the world, needs an effective drug molecule for its treatment. There are several receptors of SARS Cov-2 which are targeted for its inhibition by many lead molecules both in-vitro and in-vivo. Papain like Protease (PLpro) is one of the two SARS-Cov-2 proteases that can be used as a drug target for SARS Cov-2. It is a coronavirus enzyme that plays a role in the cleavage and maturation of viral polyproteins, assembly of the replicase-transcriptase complex and disruption of host responses. PLpro has also been linked to the cleavage of proteinaceous post translational modifications on host proteins as a means of evading antiviral immune responses. Structure-based drug discovery can be one of the effective methods to screen for various molecules against the target receptors. In this study, PLpro of SARS CoV-2 was chosen as the target for docking. Forty phytochemicals from various plant sources and four synthetic drugs have been screened for their inhibitory potential against PLpro using AutoDock Vina. Phytochemicals such as Tinosponone, Rhoifolin, Rosmanol, Berberin, Nimbin and two other existing drugs Elbasvir and Declatasvir showed higher inhibitory potential in terms of higher binding affinities. ADME and toxicity analysis were also performed to predict the pharmacokinetics and drug likeliness properties. It was concluded from the study that Tinosponone possesss potential inhibitor property of papain-like proteases (PLpro) of SARS CoV-2. Tinosponone from the plant Tinospora cordifolia had a binding affinity of − 9.3 kcal/mol and obeyed the Lipinski rules, making it an effective lead molecule for treating SARS CoV-2. Molecular Dynamics simulation of Tinosponone with PLpro has proved the stability and validity of the binding with RMSD value in range of 0.2 nm when it was run for 50 ns using GROMACS. Therefore, Tinosponone could be considered as a potential inhibitor of PLpro of SARS CoV-2.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Severe Acute Respiratory Syndrome (SARS) Coronavirus, a novel coronavirus (2019-nCoV), also known as SARS-CoV-2, posed a huge threat to the world since 2019. The Centre for Disease Control and Prevention (CDC) tracked the outbreak of SARS-CoV-2 in Wuhan, China. The virus spread across the globe and leads to a pandemic situation with significant mortality rates that differed from country to country. People with heart disease, diabetes, lung disease, HIV, and pregnant women are at an increased risk of developing a serious illness or contracting COVID-19. The virus does not travel through the air, but rather through the respiratory droplets or aerosols of an infected individual (Dwarka et al. 2020). SARS CoV-2 is a member of the Coronaviridae family of the Nidovirales order. It is classified into four genera and SARS CoV-2 is closely related to the Betacoronavirus genus. Genome sequencing revealed higher similarity with the previous SARS CoV (2003 outbreak). Both viruses share the same membrane-bound exopeptidase Angiotensin converting enzymes (ACE2) as the host receptor to enter the cells (Chakraborty and Member 2020; Silva et al. 2020). Different structural proteins such as spike proteins play the main role in attachment to host receptors. Apart from the structural proteins, different proteases also play a major role in viral replication and immune evasion of the host. Different cysteine proteases: chymotrpsin-like proteases or main proteases (3CLpro), papain-like proteases (PLpro) are important non-structural proteins. The 3CL-like protease (3CLpro) hydrolyzes viral polyproteins to create functional proteins, which are required for coronavirus replication.Papain-like protease is essential for the release of NSP 1–16 from open reading frames ORF1a and ORF 1ab, which forms large polyproteins ppa1a and ppa1ab (Li et al. 2020). Papain-like protease (PLpro) is a SARS CoV-2 enzyme that is required for digesting viral polyproteins to generate a functional replicase complex that allows the virus to spread. It negatively regulates the host immune response by its deubiquitinating and deISGylating effect (Shah et al. 2020; Shin et al. 2020; Mohanraj et al. 2018).
Computer aided drug design tools such as molecular docking and molecular dynamic simulation help us to find the potential inhibitors against various viral targets in lesser time (El-hoshoudy 2020; Shawk et al. 2020; Meng et al. 2020). Plant-based immune modulatory compounds could reduce the effect of inflammatory response and cytokine storm (Abdellatiif et al. 2021; Pall et al. 2020; Vardhan and Sahoo 2020). Several antiviral inhibitors have been found to be more effective against the SARS CoV-2 virus (Alfaroa et al. 2020). Phytochemicals boost immunity and protect against illness (Zhu et al. 2020). This study discusses the results of docking studies of various phytochemicals against SARS CoV-2's papain-like protease (PLpro).
Material and methods
Preparation of protein
The crystal structure of papain-like protease of severe acute respiratory virus -2 (SARS-CoV-2) PDB ID 6W9C was retrieved from PDB database https://doi.org/10.2210/pdb6W9C/pdb and it was used in the present study. It has a resolution of 2.70 Å and molecular weight of 107.81 kDa. It has three chains A, B and C with a total sequence length of 317 amino acids. Energy minimization of the retrieved structure was performed using the SWISS PDB viewer. It helps to repair the distorted geometrics by moving atoms to release internal constraints and is used to minimise the protein energy (Kodchakorn et al. 2020; Tripathi et al. 2011; Trott and Olson 2010).
Preparation of ligand
Antiviral compounds derived from plant resources with variety of traditional applications were screened from a variety of phytochemical databases such as IMPPAT https://cb.imsc.res.in/imppat/ (Indian Medicinal Plants, Phytochemicals and Therapeutics) (Ni et al. 2020; Swain et al. 2020). Compounds with anti-viral properties were screened based on ADME and drug-likeliness properties using SWISS ADME server. (http://www.swissadme.ch/) (Meyer-Almes 2020; Garg et al. 2020). Based on the results, forty phytoligands were chosen for the study. As a control, five commonly recommended synthetic antiviral drugs such as Elbasvir, Cefpiramide, Cefpiramide, Daclatasvir and Delamanid were also docked with PLpro (Sharma et al. 2020; Swain et al. 2020). The structure of phytoligands and drugs was obtained from Pubchem database (https://pubchem.ncbi.nlm.nih.gov/) in SDF format. The phytoligands were then converted from SDF to PDB format using the online SMILES converter. (https://cactus.nci.nih.gov/translate/). ChemSketch was used to convert the 2D structure of certain ligands into a 3D format. (https://www.acdlabs.com/resources/free-chemistry-software-apps/chemsketch-freeware/#chemsketch_modal) (Ferreira et al. 2015). Various phytoligands and synthetic drugs were docked with the target protein, Papain-like protease of SARS CoV-2 using Autodock Vina in blind docking mode (Venkateshana et al. 2020).
Molecular dynamic simulation of Tinosponone
The complex of Tinosponone with Papain-like protease was prepared for Molecular Dynamic simulation and run for 50 ns using GROMACS (version 2022.1) through Google Colab pro (Salsbury 2010; Dutt and Roy 2020). Topology generation and energy minimization of ligand were performed and energy minimization of ligand was performed using the Google colab pro GPU server.
Results and discussion
Molecular docking analysis
The binding energies of forty phytoligands against SARS-CoV-2 papain-like protease (6W9C) ranged from − 9.3 kcal/mol (Tinosponone) to − 2.5 kcal/mol (Methyl Rosmarinate), while synthetic anti-viral drugs ranged from − 9.1 kcal/mol (Bictegravir) to − 13.0 kcal/mol (Elbasvir). Tinosponone and Hespiridin had the highest binding affinity for the PLpro. Because the latter's drug-likeliness had some flaws, Tinosponone from Tinospora cordifolia was further subjected to MD simulation to study its binding stability. The list of various phytochemicals, their sources and their docking score were given in Table 1
The pharmacokinetic properties of the selected ligands with preferred docking scores is given in Table 2. Out of five ligands and a drug, Nimbin and Rhoifolin did not meet the required properties as Nimbin’s molecular weight was greater than 500 Da and Rhoifolin’s number of hydrogen acceptors was greater than 10. Tinosponone, Berberine, Rosamanol and Elbasvir satisfied all rules of Lipinski.
Prediction of the active site of PLpro
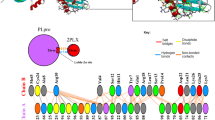
The probable active sites or binding pockets of the four viral proteins were identified with PyMOL (Bharadwaj et al. 2020; Yuan et al. 2017). Five phytochemical molecules were analyzed for the identification of the active site of the target molecule which is listed in Table 3. The target protein PLpro contains three chains A, B and C. The binding complex of Tinosponone with the active site of PLpro of SARS CoV-2 is shown in Fig. 1.

Active site predicted using PyMOL for the docked structure of Tinosponone with PLpro of SARS CoV-2
Molecular dynamic simulation of Tinosponone with PLpro
The molecular dynamic simulation was carried out for the docked complex of Tinosponone with papain-like protease using GROMACS (version 2022.1) through Google Colab pro. The conformation of the molecule binding of the compound was found to be stable with a mean RMSD value deviation in the range of 0.2 nm when simulation was done for the period of 50 ns using Google colab pro. RMSD value was plotted as the function of time frame was plotted as shown in Fig. 2. RMSF fluctuation of binding of Tinosponone with PLprowas shown in Fig. 3 (Silva et al. 2020). The number of hydrogen bonds formed during 50 ns Molecular dynamic simulation was shown in Fig. 4

Time dependence RMSD plot of binding of Tinosponone with PLpro using MD simulation

Time dependence RMS fluctuation plot of binding of Tinosponone with PLpro using MD simulation

Number of Hydrogen Bonds of Tinosponone binding with PLpro in 50 ns MD simulation
Discussion
In the current study, Tinosponone derived from the plant Tinospora cordifolia showed better binding activity with the binding energy of − 9.6 kcal/mol using Autodock Vina, Similarly in another study by Krupanidhi et al. (2020), Tinosponone showed a better binding affinity with 3CL pro also with binding activity of − 7.7 kcal/mol (Krupanidhi et al. 2020). Results of molecular dynamic simulation with Papain protease of SARS CoV-2 with Tinosponone also indicates the stable binding when the simulation was performed in the 50 ns range (Table 4).
Conclusion
Virtual screening of SARS CoV-2 papain-like protease (PDB ID: 6W9C) with various phytoligands demonstrated the five phytoligands, Tinosponone, Rhoifolin, Rosmanol, Berberin, and Nimbin with the best inhibitory potential in terms of higher binding affinities. Tinosponone had a binding affinity of − 9.3 kcal/mol and obeyed all Lipinski rules, making it a potential inhibitor for SARS Cov-2 PLpro. Tinosponone's binding site with the target PLpro was identified as P246 on chain C of SARS CoV-2's papain-like protease (PLpro). Therefore, Tinosponone could be used as a potential inhibitor of papain like protease of SRS CoV-2 based on further in-vitro and in-vivo investigations.
References
Abdellatiif MH, Ali A, Ali A, Hussien MA (2021) Computational studies by molecular docking of some antiviral drugs with COVID-19 receptors are an approach to medication for COVID-19. Open Chem 19(1):245–264. https://doi.org/10.1515/chem-2021-0024
Alfaroa M, Alfaro I, Ange C (2020) Identification of potential inhibitors of SARS-CoV-2 papain-like protease from tropane alkaloids from Schizanthusporrigens: a molecular docking study. Chem Phys Lett 761:138068. https://doi.org/10.1016/j.cplett.2020.138068
Bharadwaj S, Lee KE, Dwivedi VD, Kanga SG (2020) Computational insights into tetracyclines as inhibitors against SARS-CoV-2 Mpro via combinatorial molecular simulation calculations. Life Sci 257:118080. https://doi.org/10.1016/j.lfs.2020.118080
Chakraborty K, ERS Member (2020) ACE2 receptor blockers: a novel therapeutic approach for COVID-19
Dutt P, Roy P (2020) Molecular docking unmasks potent phyto ligands against SARS-CoV-2 Spike glycoprotein, main protease, papain-like protease, and RNA-dependent RNA polymerase. J Biomol Struct Dyn 39:236–244. https://doi.org/10.1080/07391102.2020.1796808
Dwarka D, Agonib C, Mellem JJ, Soliman ME, Baijnath H (2020) Identification of potential SARS-CoV-2 inhibitors from South African medicinal plant extracts using molecular modelling approaches. S Afr J Bot 133:273–284. https://doi.org/10.1016/j.sajb.2020.07.035
El-hoshoudy AN (2020) Investigating the potential antiviral activity drugs against SARS-CoV-2 by molecular docking simulation. J Mol Liquids 318:113968. https://doi.org/10.1016/j.molliq.2020.113968
Ferreira LG, Ricardo N, Oliva G, Andricopulo AD (2015) Molecular docking and structure-based drug design strategies. Molecules 20(7):13384–13421. https://doi.org/10.3390/molecules200713384
Garg S, Anand A, Roy A (2020) Molecular docking analysis of selected phytochemicals against SARS-CoV-2 Mpro receptor. Vegetos. https://doi.org/10.1007/s42535-020-00162-1
Kodchakorn K, Poovorawan Y, Suwannakarn K, Kongtawelert P (2020) Molecular modelling investigation for drugs and nutraceuticals against protease of SARS-CoV-2. J Mol Graph Model 101:107717. https://doi.org/10.1016/j.mgm.2020.107717
Krupanidhi S, Abraham Peele KTC, Venkateswarulu TC, Ayyagari VS, Nazneen-Bobby M, Babu DJ, Venkata-Narayana A, Aishwarya G (2020) Screening of phytochemical compounds of Tinospora cordifolia for their inhibitory activity on SARS-CoV-2: an in silico study. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1787226
Li D, Luan J, Zhang L (2020) Molecular docking of potential SARS-CoV-2 papain-like protease inhibitors. Biochem Biophys Res Commun 538:72–79. https://doi.org/10.1016/j.bbrc.2020.11.083
Meng X-Y, Zhang H-X, Mezei M, Cui M (2020) Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 7:146–157. https://doi.org/10.2174/157340911795677602
Meyer-Almes F-J (2020) Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: virtual screening and structure based drug design. Comput Biol Chem 88:107351. https://doi.org/10.1016/j.combiolchem.2020.10735
Mohanraj K, Karthikeyan BS, Vivek-Ananth RP, Bharath-Chand RP, Aparna SR, Mangalapandi P, Samal A (2018) IMPPAT: a curated database of Indian Medicinal Plants. Phytochem Therap Sci Rep 8:4329
Ni W, Yang X, Yang D et al (2020) Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit Care 24(1):422. https://doi.org/10.1186/s13054-020-03120-0
Pall S, Zhmurov A, Baur P et al (2020) Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J Chem Phys 153:134110. https://doi.org/10.1063/5.0018516
Salsbury FR (2010) Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr Opin Pharmacol 10:738–744. https://doi.org/10.1016/j.coph.2010.09.016
Shah B, Modi P, Sagar SR, LJ Institute of Pharmacy (2020) In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci 252:117652–117652. https://doi.org/10.1016/j.ifs.2020.117652
Sharma K, Morla S, Goyal A, Kumar S (2020) Computational guided drug repurposing for targeting 2′-O-ribose methyltransferase of SARS-CoV-2. Life Sci 259:118169. https://doi.org/10.1016/j.ifs.2020.118169
Shawk E, Nada AA, Ibrahim RS (2020) Potential role of medicinal plants and their constituents in the mitigation of SARS-CoV-2: identifying related therapeutic targets using network pharmacology and molecular docking analyses. RSC Adv 47:27961–27983. https://doi.org/10.1039/dora05126h
Shin D, Mukherjee R, Grewe D, Bojkov D, Baek K, Bhattacharya A, Schulz L, Widera M, Mehdipour AR, Tascher G, Geurink PP, Wilhelm A, van der Heden-van-Noort GJ, Schulman B, Cinat J, Hummer G, Ciesek S, Dikic I (2020) Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587:657–662. https://doi.org/10.1038/s41586-020-2601-5
Silva TL, Toffano L, Fernandes JB, das Graças-Fernandes-da-Silva MF, de Sousa LRF, Vieira PC (2020) Mycotoxins from Fusariumproliferatum: new inhibitors of papain-like cysteine proteases. Braz J Microbiol. https://doi.org/10.1007/s42770-020-00256-7
Swain SS, Panda SK, Luyten W (2020) Phytochemicals against SARS-CoV as potential drug leads. Biomed J 44:74–85. https://doi.org/10.1016/j.bj.2020.12.002
Tripathi L, Kumar P, Haneef J, Singh R (2011) Molecular docking softwares: an overview. Curr Bioact Compd 5:160–168. https://doi.org/10.20174/157340709788452019
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31(2):455–461. https://doi.org/10.1002/jcc.21334
Vardhan S, Sahoo SK (2020) In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoid. Comput Biol Med 124:103936. https://doi.org/10.1016/j.compbiomed.2020.103936
Venkateshana M, Suresha J, Muthu M, Ranjithkumar R (2020) Azaphenantherene derivatives as inhibitor of SARS-CoV-2 Mpro: synthesis, physiochemical, quantum chemical and molecular docking analysis. Chem Dtat Collect. https://doi.org/10.1016/j.moistruc.2020.128741
Yuan SH, Chan S, Hu Z (2017) Using PyMOL as a platform for computational drug design. Comput Mol Sci. 7(2):e1298. https://doi.org/10.1002/wcms.1298
Zhu W, Xu M, Chen CZ, Guo H, Shen M, Hu X, Shinn P, Klumpp-Thomas C, Michael SG, Zheng W (2020) Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol Transl Sci 5:1008–1016. https://doi.org/10.1021/acsptsci.0c00108
Acknowledgements
The author hasn't received any funds from the funding agencies. The author would like to acknowledge the permission given to access the Bioinformatics facilities of the Department of Industrial Biotechnology, Government College of Technology, Coimbatore.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Saranya, P., Karunya, R., Keerthi Varshini, G. et al. In-silico docking studies of selected phytochemicals against papain like protease of SARS-Cov-2. Vegetos 36, 188–194 (2023). https://doi.org/10.1007/s42535-022-00525-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42535-022-00525-w