
Abstract
Nanostructured bilayer thin films with superhydrophobic and superhydrophilic surfaces were prepared using Ti6Al4V alloy substrates which allowed for the comparative analysis of polyvinyl acetate (PVAc) particle adsorption as a function of the interface structure. The PVAc particles were obtained from emulsion polymerization of vinyl acetate. A superhydrophilic TiO2 nanofiber-based 3D network was created on the Ti6Al4V alloy substrate by application of a hydrothermal method. Subsequent UV-grafting of ultra-thin polydimethylsiloxane (PDMS) layers resulted in a superhydrophobic surface. The modification steps were followed via Diffuse Reflectance Infrared Fourier Transform Spectroscopy, X-ray Photoelectron Spectroscopy, Field Emission-Scanning Electron Microscopy, contact angle and Electrochemical Impedance Spectroscopy. A mechanism for the adsorption of PVAc at the two electrolyte/substrate interfaces could be revealed.
Article Highlights
-
Superhydrophobic and superhydrophilic surfaces were created based on a Ti-alloy by combining topography and chemistry on a nanometer scale.
-
Polymeric polyvinyl acetate particle fouling was significantly reduced by generation and employment of a superhydrophobic surface.
-
Particle/surface interactions were explained based on the performed experiments and the results were summarized in an interface model.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Alongside inorganic, organic and biological fouling, polymer fouling represents an additional research field that is becoming increasingly important due to its high industrial relevance. The initial adsorption of polymers onto oxide surfaces is a main factor when discussing polymer deposit formation and the related unwanted accumulation of solid material at the solid/liquid interface [1,2,3,4,5]. However, even though in times of resource conservation and energy saving this kind of fouling is a well discussed topic in the polymer-producing industry, the in-depth polymer/solid surface interactions behind are only slightly researched, moreover, largely unknown. This is particularly the case when it comes to specifically modified surfaces.
While the formation of polymer gels during the radical polymerization of acrylic acid in aqueous solution is reported to be the result of crosslinking reactions in combination with low intermixing [6], side reactions forming terminal double bonds and a subsequent branching and crosslinking are main processes leading to deposit formation during the radical polymerization of N-Vinyl-2-pyrrolidone [7, 8]. On the other side, conversion and monomer composition is reported to play a key role in the fouling processes observed during co-polymerization of vinyl acetate (VAc) [1].
Such interfacial polymer deposits significantly reduce heat transfer properties [9], which, under the need for constant product quality, inevitably leads to larger heat transfer surfaces and correspondingly increased energy and resource consumption e.g. due to cleaning procedures. Constant heat transfer properties are of particular importance during emulsion polymerization since complex latex dispersions exhibit numerous possibilities for destabilization, which is often considered as major source of polymeric deposit formation [10, 11]. This particle instability is well studied [12], for instance, in terms of ionic strength [13], implementation of organic solvents [14] or using surfactants [15]. A system of high industrial relevance which tends to form severe polymeric deposits during the process even after short contact times with oxidic solid surfaces, is the aqueous emulsion polymerization of VAc. The issue could be shown in relation to studies carried out for fouling prediction [1] or fouling monitoring [16]. Further, fouling quantification on heat transfer surfaces was performed in [5].
To overcome the general issue of fouling, several coating technologies were applied [17,18,19,20]. The adsorption of polymers at the solid/liquid interface strongly depends on the surface chemistry and topography of the substrate material. Surfaces with low surface free energy and reduced roughness are predicted to have promising anti-fouling characteristics according to Malayeri et al. [21]. In contrast, Horseman et al. [4] found a reduction in both the adhesion force and the distance of the attractive interactions of polystyrene and polyethylene particles when a hydrophilic surface with increased roughness was used. Moreover, hydrophilic surfaces showed overall reduced fouling tendencies in the same study. Such examples illustrate the complexity of the relation of fouling with the microstructure and surface chemistry of the interacting material [22,23,24,25].
Ultra-thin low surface energy films based on perfluorinated organosilane films showed anti-fouling properties during polymerization of polyvinyl pyrrolidone [26]. Zhou et al. [27] created superhydrophobic surfaces by combining a perfluorinated organosilane with a nanostructured iron surface and showed anti-corrosive and poor adhesive properties against bacteria, water and blood. Further, Keshmiri et al. [28] increased surface hydrophobicity by combining polydimethylsiloxane (PDMS) with a perfluorinated organosilane. The application of slippery liquid-infused porous surface (SLIPS) as anti-adhesive surface against various organic and inorganic substances was presented by Wong et al. [29]. A combination of PTFE surface structuring and perfluorinated fluids as wetting fluid was used. However, the underlying mechanisms responsible for the anti-adhesive tendencies is usually not analyzed in detail. Further, the application of perfluorinated polymer chemistry should be avoided in the future as the transfer of such materials into the environment is precarious [30]. Therefore, there is a strong need to improve the understanding of polymer/substrate interaction in polymerization reactors and to find alternative sustainable surface film chemistries.
The aim of this study is to generate surfaces based on biocompatible chemicals and sustainable process routes that exhibit anti-adhesive properties against polymeric fouling during the polymerization of VAc, which is prone to significant deposit formation. The results can be used to reduce fouling in real reactors for polymer production, which is important to save energy and resources. Further, it is intended to enhance the understanding of polyvinyl acetate (PVAc) adsorption on complex model surfaces that exhibit different surface topography as well as wettability characteristics. It enables the transfer to other systems.
For this purpose, bilayer surface films formed on common corrosion resistant Ti6Al4V alloy were prepared based on sustainable process routes. Superhydrophilic TiO2 3D nanostructured films were created based on a hydrothermal process. In a second step, PDMS could be grafted onto the surface via UV-light, which completely reversed the surface wetting properties from superhydrophilic to superhydrophobic, as determined by contact angle measurements. The modified surfaces were sequentially analyzed by Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS), X-ray Photoelectron Spectroscopy (XPS), Field Emission-Scanning Electron Microscopy (FE-SEM) as well as Electrochemical Impedance Spectroscopy (EIS). The interface adsorption of polymer particles was then comparatively investigated by immersing both surfaces into a dilute PVAc-based dispersion at room temperature. Subsequent surface analysis was carried out by EIS and FE-SEM. Finally, a mechanism is presented for the adsorption of PVAc onto both the superhydrophilic and superhydrophobic surface.
2 Material and methods
2.1 Materials and chemicals
Ti90/Al6/V4 alloy plates (20 mm × 20 mm, Grade 5, ASTM B265, annealed) (referred to as Ti6Al4V alloy below) were supplied by Goodfellow. Chemicals for solvent cleaning, namely tetrahydrofuran (THF), acetone, propan-2-ol and ethanol was all in p.a. grade, purchased from Sigma Aldrich. Potassium hydroxide (KOH) from Sigma Aldrich was used in p.a. grade and polydimethylsiloxane (PDMS, OH-terminated, liquid) with a molecular weight of 4200 g/mol was used as supplied from Alfa Aesar. The polyvinyl acetate (PVAc) dispersion VINNAPAS® LL6999 was supplied by Wacker Chemie AG, which is a copolymer of vinyl ester, ethylene and acrylic acid ester as dispersion in water (solid content 54.5 m%) with a mass content of 6.5 m% partly hydrolyzed polyvinyl alcohol (4/88). The dispersion pH is 4.6 and the average particle size equals 1.271 µm. Sodium perchlorate hydrate was purchased from Sigma Aldrich in p.a. quality. All water utilized in this study was obtained from Carl Roth GmbH + Co. KG in HPLC quality.
2.2 Variation of Ti6Al4V surface topography
Ti6Al4V alloy plates served as base substrates for surface modification and were subjected to solvent cleaning before usage. The latter consisted of a consecutive treatment in THF, acetone, propan-2-ol and ethanol in an ultrasonic bath for 15 min each. The cleaning procedure has been completed by drying the samples in a nitrogen stream. Afterwards, the Ti6Al4V alloy surface was nanostructured by a 2-step process, consistent of (I) a hydrothermal treatment and (II) air annealing, as proposed by Anitha et al. [31]. Step (I) was performed by immersing the Ti6Al4V alloy substrates into a freshly prepared 10 M KOH solution in water, followed by a 30 min treatment in an autoclave at 121 °C. The treatment should lead to the formation of a dense nanostructure that is time adapted to the PVAc particle size obtained during emulsion polymerization of vinyl acetate (VAc). Step I is completed by thoroughly rinsing the surfaces with water.
For step (II), the as-described etched samples were placed in an oven and heated to 400 °C for 3 h. The cooling took place gradually at 10 °C/min to room temperature. During the process a TiO2-based nanostructure forms, since at the chosen conditions, which is high aqueous alkali molarity and high temperature, metal Ti dissolution to TiIV(OH3)+ is supposed to dominate the oxide growth based on the reaction kinetics [31, 32]. Further, polytitanate anions develop which probably precipitate as potassium-titanate (K-titanate) due to the presence of KOH [32]. This has been even observed in [31]. In this study, the chemical surface composition is analyzed by XPS and DRIFTS. FE-SEM images were recorded for surface topography and water contact angle measurements for surface wetting property analysis.
2.3 Surface modification by PDMS grafting
For subsequent surface chemistry modification, PDMS was grafted onto the nanostructured surface via mild UV-light as described by Tesler et al. [33]. For this, 200 µl of as-delivered PDMS was dropped onto the freshly prepared surface until it is completely and homogeneously wetted. Afterwards, the surface was UV-lighted under employment of an OmniCure S1000 UV curing system (EXFO Life Science & Industrial Division) equipped with a 250–450 nm filter. The measured power density at the working distance equals 50 mW/cm2. After 30 min of illumination, the samples were rinsed with toluene, followed by a consecutive cleaning step in an ultrasonic bath (performance 120 W) in toluene and ethanol for 15 min each. The modification is completed by rinsing the sample with water and drying in a nitrogen stream.
Several processes induced by UV-illumination which promote the grafting process of PDMS have been discussed in the literature [33,34,35].
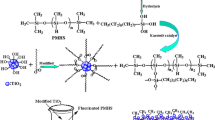
The formation of electron hole pairs in the TiO2 network, which migrate to the surface and react with the adsorbed species such as oxygen, water and hydroxyl groups, form hydroxyl radicals which can bind to Si-atoms with a dangling bond (Fig. 1a). [34, 35] Photo dissociation of Si–CH3 bonds within the PDMS, which are expected to be activated in the selected UV wavenumber range based on the bond dissociation energy leads to Si-atoms with dangling bonds (Fig. 1b). Both processes lead to a direct grafting of PDMS to the nanostructured TiO2 surface via Si–O–Ti bonds (Fig. 1c). [33] However, multiple Si-CH3 bond dissociations are possible within one PDMS chain, which could lead, for instance, to a bridging of two silicon atoms onto two surface oxygen groups. Finally, a binding mechanism via hydrolysis and interfacial condensation was proposed by Teisala et al. in the absence of UV-light [36].

Schematic of the nanostructured TiO2 surface with additional PDMS modification including the proposed radical formation by UV illumination for stable PDMS-surface bond formation
All prepared samples were analyzed comparatively to the nanostructured substrates by DRIFTS, XPS and FE-SEM. The water contact angle was determined to show water repellant surface characteristics.
2.4 Surface analysis
2.4.1 X-ray photoelectron spectroscopy (XPS)
X-ray photoelectron spectroscopy (XPS) was performed using an Omicron ESCA + system (Omicron NanoTechnology GmbH) equipped with a hemispherical energy analyzer at a base pressure of < 1·10–9 mbar. Spectra were recorded at pass energies of 100 eV for survey spectra and 20 eV for element spectra. A monochromatic Al Kα (1486.3 eV) X-ray source with a spot diameter of 0.6 mm was used. The take-off angle of the detected photoelectrons was set at 30° in relation to the surface plane. Spectra were internally calibrated to the C1s peak (binding energy 285.0 eV). Peak fitting and data analysis were performed with CasaXPS software (V 2.3.23, Casa Software Ltd.). Quantifications were performed through integration of the peaks with respect to the corresponding relative sensitivity factor values. A Shirley type background correction and a Gaussian Lorentzian peak shape (70%/30%) were used to fit the measured peaks.
2.4.2 Field emission-scanning electron microscopy (FE-SEM)
Field Emission-Scanning Electron Microscopy (FE-SEM) images were obtained by means of a NEON 40 FE-SEM microscope (Carl Zeiss SMT AG), which was equipped with an InLens detector and a SE2. Images were recorded with different magnifications as indicated under the respective figures at an accelerating voltage of 0.50 to 2.00 kV.
2.4.3 Contact angle measurement
The static contact angle was determined under employment of an OCA 15 plus goniometer (Dataphysics) at 25 °C using the sessile drop method. For each measurement a water drop with a volume of 6 µl was dispensed onto the surfaces using a motor-driven syringe. The contact angle formed at the solid/air/liquid interface was evaluated with the Laurentian method. The standard error was calculated from measuring 3 drops on three distinct, but identical prepared surfaces each. For surface free energy calculation, the OWRK method was employed since it is generally valid for planar surfaces based on Young’s equation. For this, three liquids with variations in the polar (ɣp) and dispersive component (ɣd), which is ultrapure water (ɣd: 21.8 mN/m, ɣp: 51.0 mN/m [37]), Dimethyl sulfoxide (DMSO, > 99.9%, Sigma-Aldrich, ɣd: 34.9 mN/m, ɣp: 8.7 mN/m [38]) and Diiodomethane (for synthesis, Merck, ɣd: 50.8 mN/m, ɣp: 0 mN/m [37]) were used. The contact angles were measured both on a PDMS-modified silicon wafer (water contact angle 102°) and a smooth TiO2-surface as prepared by atomic layer deposition, modified with PVAc forming a homogeneous layer. Each value was determined from measuring at least 3 drops on three distinct, but identical prepared surfaces.
2.4.4 Diffuse reflectance infrared fourier transform spectroscopy (DRIFTS)
DRIFTS analysis was performed using a Bruker Vertex 70 spectrometer (Bruker Optics) equipped with a LN-MCT detector and a Harrick Praying Mantis measurement cell, which is continuously floated with dry air. All spectra were recorded in a wavenumber range from 750 to 4000 cm−1 with a resolution of 4 cm−1. 256 individual spectra were recorded and averaged for each spectrum. The OPUS Software (V6.5 Bruker Optics GmbH) was used for data treatment. Appropriate surface states with similar topography served as reference in all cases.
2.4.5 Electrochemical impedance spectroscopy
Electrochemical analysis was carried out by using a Reference 1000 potentiostat (Gamry Instruments) in a 2 mM sodium perchlorate electrolyte solution or dilute PVAc dispersion (1:10 in 2 mM NaClO4, v:v), while the temperature was kept constant at 20 °C by a thermostat. A three-electrode setup, equipped with a Pt-pseudo reference wire, graphene as counter electrode and the Ti6Al4V alloy, nanostructured TiO2 or additional PDMS modified surface serving as working electrode, was employed for the analysis of the electric properties in Sect. 3.4. An Ag/AgCl reference electrode was used instead of Pt for the experiments in the diluted polymer dispersion (Sect. 3.6). Data were obtained in a frequency range from 100 kHz to 1 Hz with 10 points per decade. The amplitude of the sine-wave voltage signal was set to 20 mV (rms). The measurements were performed with the OCP being measured for 2 min in each cycle, followed by the record of the impedance and phase angle up to a time of 24 h.
3 Results and discussion
3.1 Microstructure of the prepared surfaces
The change in the surface topography was analyzed by recording FE-SEM images of the solvent cleaned Ti6Al4V alloy (referred to as Ti6Al4V below), the latter after etching and calcination (referred to as ns-TiO2|Ti6Al4V or nanostructured TiO2 below) and subsequent PDMS deposition (referred to as PDMS|ns-TiO2|Ti6Al4V below) (Fig. 2).

FE-SEM images of a the solvent cleaned Ti6Al4V surface, b the ns-TiO2|Ti6Al4V and c the latter with additional PDMS deposition, all obtained with a magnification of 10000x
The applied time-adapted etching and calcination process results in nanostructure forming strands, which are cross-linked to build a fine 3D network (Fig. 2b). Due to the etching process in the autoclave reduced to only 30 min, the single strands are slightly thickened compared to the results by Anitha et al. [31] and exhibit a total height of 300 to 600 nm as obvious from FIB-SEM image (Supplementary Information S1). Thus, the time-adapted treatment leads to a denser nanostructure of the surface oxide, which is intended to prevent the irreversible embedding of the particles into the oxide cavities. Further, smaller cracks are visible, where the nanostructure is only partly connected. After PDMS modification (Fig. 2c), the nanostructure is slightly finer, but overall, only minor changes in the surface topography appear.
Thus, a surface was created whose fine network has been adapted to the particle size found in the used PVAc polymer dispersion and which further allows PDMS modification without major surface topography changes.
3.2 Chemical composition
XPS high-resolution (HR) spectra were recorded for the Ti6Al4V surface as well as for the respective surface after etching and calcination, which leads to the TiO2 nanostructure, and the nanostructured surface after PDMS modification. The analysis is meant to analyze the surface chemistry in the different surface transformation steps in detail, mainly based on the C1s and O1s peak regions (Fig. 3).

XPS HR spectra of the a, d Ti6Al4V surface, the b, e ns-TiO2|Ti6Al4V surface and c, f the latter after subsequent PDMS deposition in the a–c C1s and d–f O1s peak region
The C1s peak region of the Ti6Al4V surface (Fig. 3a) can be fitted into contributions of C–C/C–H (285.0 eV), C–O (286.5 eV) and O–C=O bonds (288.7 eV). These can be attributed to a superimposition of the surface itself and adventitious carbon. After etching and calcination (Fig. 3b) these contributions are still present with peaks centred at 285.0 eV, 285.8 eV and 288.7 eV, however, with significantly decreased intensity. A shoulder appearing at lower binding energies is present, however, was not fitted to any component. C–Ti bonds are reported at similar binding energies [39] but the species is not expected to form during the process. No Ti6Al4V alloy characteristic bands can be observed in the survey spectrum of the nanostructured TiO2 surface anymore (Supplementary Information S2). In combination with the higher intensity of the peaks for titanium (cf. especially Ti2s at 565.3 eV and Ti2p at 458.5 eV) and oxygen in the survey spectrum after the etching process compared to the reference surface state, these observations clearly confirm complete surface conversion and successful TiO2 modification. The results are reflected by the determination of the molecular surface composition in at.−% (Table 1).
It is obvious that the carbon content on the surface is reduced from 37 at.−% on the Ti6Al4V surface to 3 at.−% for the respective surface after the etching and calcination process (Table 1). Concurrently, the Ti:O ratio is slightly increased to about 1:2.5 compared to the theoretical value of 1:2. Since peaks for potassium (K2s and K2p) were detected in the survey spectrum (Supplementary Information S2), the increased oxygen to metal ratio could be due to a slight precipitation of potassium titanates on the surface. The slight formation of this species is discussed in literature [31, 32] and Sect. 2.2.
After PDMS modification, both a reduction of the TiO2 characteristic peaks and an increase of the carbon amount can be observed (Table 1). Together with the clear presence of the signal for silicon in the Si2p peak region of the XPS HR spectrum (Supplementary Information S2), this unambiguously illustrates the successful PDMS deposition on the nanostructured TiO2. Due to the low signals from the underlying substrate, a film thickness in the low nanometer range, as suggested by Tesler et al. [33], is assumed. A decomposition of the C1s peak region (Fig. 3c) further shows that the spectrum is composed exclusively of C–H (285.0 eV) and minor C–O (286.0 eV) bonds, which originate from PDMS [40] and atmospheric contributions, respectively. However, the C–H region is affected by C–Si bonds [41].
For the Ti6Al4V surface, the O1s peak region is mainly composed of metal–oxygen bonds (530.2 eV), most of which can be attributed to Ti4+-oxides (Fig. 3d) [42]. Further, minor amounts of non-lattice oxygen and surface hydroxyl groups (531.7 eV) [43] as well as contributions from the adsorbed water species and/or CO bonds (533.3 eV) [44] could be detected. The O1s peak region of the nanostructured TiO2 surface exhibits similar peaks for Ti4+-oxides (530.2 eV), non-lattice oxygen/surface hydroxyl groups (531.3 eV) [43, 45] and adsorbed water/CO-groups (Fig. 3e) [44]. Especially the oxide peak is increased in intensity, which is due to complete surface transformation to TiO2. Finally, the O1s peak region after PDMS modification can be deconvoluted into two main peaks, which are for Ti–O (530.1 eV) and Si–O–Si bonds from PDMS (Fig. 3f) [46]. Based on the XPS probing depth [47] and the used take-off angle of 30°, this indicates the presence of a PDMS layer thickness in the low nanometer range. However, contributions from the underlying nanostructured TiO2 substrate or PDMS film defects are likely. The latter is of importance for the discussion of the in-situ hydroxylation of the PDMS-modified substrate based on DRIFT spectroscopy (Sect. 3.5).
3.3 Wetting properties
The surface wettability was analyzed sequentially on the Ti6Al4V surface as well as on the nanostructured TiO2 and PDMS terminated substrate by water contact angle measurements (Fig. 4).

Water contact angles as measured on a the Ti6Al4V and b the ns-TiO2|Ti6Al4V surface, as well as c the latter after subsequent PDMS modification
The water contact angle was measured to be 84 ± 2° on the solvent cleaned Ti6Al4V surface (Fig. 4a), which is in excellent agreement with the results from Lo and Chang et al. [48]. After etching and calcination, the surface became wettable instantly (Fig. 4b). The water drop spread on the surface directly after contact with the solid, which makes the determination of a specific contact angle unfeasible. Accordingly, the surface exhibits superhydrophilic properties. It is assumed that these surface characteristics result from a combination of the generally hydrophilic properties based on the TiO2 surface chemistry and the rough morphology in form of the nanostructure, in which the water can sink and drain. After additional PDMS modification, the hydrophilic surface properties are entirely reversed to superhydrophobicity with complete water repellence, which is generally correlated with a water contact angle above 170° (Fig. 4c). The surface hydrophobicity is slightly increased compared to the water contact angles of 140 ± 1° and 148 ± 3° as obtained by Tesler et al. [33] for PDMS-modified TiO2 nanotubular arrays and sponge-like nanoporous stainless steel, respectively. The presented improved non-wetting characteristics are attributable to the fine TiO2 nanostructure chosen in this work, since generally lower water contact angles were determined on various PDMS-modified metal oxides with low roughness [33] or on cured, flat PDMS [49], respectively. The same trend could be shown by Wooh et al. [34]. The surface free energy of the PDMS layer deposited onto a conventional, mirror-polished silicon wafer was determined to be 21 mN/m, which is composed of mainly dispersive (ɣd: 19 mN/m) and to a minor extend of polar interactions (ɣp: 2 mN/m).
3.4 Electrochemical analysis
The electrochemical barrier properties were analyzed by applying EIS to the Ti6Al4V as well as the nanostructured TiO2 surface and the latter after subsequent PDMS modification (Fig. 5). To obtain detailed information about interface processes, the EIS data were fitted with an equivalent electric circuit (EC) by using the Gamry Echem Analyst Software (Gamry Instruments).

i Bode Plots as obtained from electrochemical impedance spectroscopy performed on a the Ti6Al4V as well as on b the ns-TiO2|Ti6Al4V surface and c additionally PDMS modified surface in a 2 mM NaClO4 electrolyte at room temperature with ii the corresponding EIS equivalent circuits used for data fitting a–c
While the impedance (|Z|) of the Ti6Al4V surface increases slightly with time at low frequencies under otherwise unchanged frequency-dependent values and the phase angle during the electrolyte exposure time of 24 h (Fig. 5i(a)), |Z| at 1 Hz decreased by around one half compared to the initial value for the nanostructured TiO2 surface in the same time frame (Fig. 5i(b)). Even for the PDMS coated substrate, the corresponding value in the low-frequency range decreases over 24 h in about the same order of magnitude, however, it is still increased compared to the non-coated surface (Fig. 5i(c)).
For quantification and further interpretation, the data were fitted with different adapted ECs (Fig. 5ii). All equivalent circuits consist of the solution resistance (Rs) and the capacity and resistance of the porous layer (Cpo and Rpo) as well as of the respective components for the barrier layer (Cbar, Rbar), which are meant to describe the oxide layer of the Ti6Al4V being in contact with the electrolyte (Fig. 5ii(a)) [50]. After etching and calcination, a TiO2-nanostructure is formed, which is considered as a further capacity and resistance (Rns and Cns) (Fig. 5ii(b)). Due to heterogeneous surface characteristics of the thin films a constant phase element (CPE) was used to describe the coated nanostructures (CPEns) instead of the capacity (Fig. 5ii(c)). From the values presented in Table 2 several observations can be made.
The minor increase of |Z| at 1 Hz on the Ti6Al4V alloy surface can be explained by the evaluated data (Table 2). Ti and its alloys are generally accepted to be composed of a two layer oxide film forming the outermost alloy surface part (consistent of an inner compact layer and an outer porous film) and the inner metal part [50, 51]. Since according to the capacitor model, the capacity is inverse proportional to the thickness (\(C=\varepsilon \bullet {\varepsilon }_{0}/d\)), with \(\varepsilon\) equaling the dielectric constant of the film, \({\varepsilon }_{0}\) describing the dielectric constant in vacuum and \(d\) is the thickness of the layer of interest [52]), a slight decrease of Cbar in combination with an increase of Rbar, as observed in this study, indicates a minor thickening of the barrier part of the oxide layer of the alloy. However, this trend, as well as the expected water incorporation into the pores of the oxide layer [50] is negligible, so that overall, the surface is assumed to remain stable within the analyzed time scale.
For the nanostructured TiO2 surface, the impedance decreases over the time of immersion (Fig. 5i(b)). Correspondingly, the phase shift decreases. This observation is mainly dominated by the significant decrease of Rbar and Rpo from 156.1 to 80.7 kΩ cm2 and 65.2 to 3.6 kΩ cm2 during 24 h (Table 2), respectively, and implies the presence of water contacting and penetrating the oxide film during the exposition time.
In contrast, the impedance in the low and mid-frequency range is increased significantly after PDMS modification (Fig. 5i(c)). This implies barrier properties of the film even after 24 h of electrolyte exposure. However, a change in the course of the impedance is observed over time in the same frequency region, which seems to be dominated by a substantial time-dependent decrease in the resistance of the nanostructure (Rns) (Table 2). This indicates the progressive incorporation of the electrolyte in the PDMS layer. Similar observation were reported by Le Floch et al. [53] for elastomers like PDMS.
The shape of the curve (Fig. 5i(c)) fits very well with the impedance data presented in the same publication by the authors [53]. In their study it could be shown that ions gradually diffuse from the electrolyte into the dielectric elastomer. The conductivity is thus significantly increased in contrast to the pristine film. The interpretation is in further agreement with the observed time course of the value for the electrolyte resistance. From fitting our data, it is obvious that Rs slightly increases with time and equals 9.4 kΩ cm2 after 24 h (compared to 8.2 kΩ cm2 and 8.1 kΩ cm2 on the Ti6Al4V alloy and nanostructured TiO2- surfaces, respectively). This, again, mirrors the increased ion incorporation into the PDMS network compared to the other analyzed surfaces, since the electrolyte resistance is inversely proportional to the conductivity [54].
Therefore, it can tentatively be assumed that, in contrast to the nanostructured TiO2, ions are primarily incorporated into the PDMS film with the nanostructure being filled with electrolyte in both cases.
3.5 Surface layer stability under simulated process conditions
DRIFTS was applied to obtain further information about PDMS film stability and water conformation characteristics in dilute electrolytes under neutral pH (after 24 h of EIS) as well as in slightly acidic (pH4.5) conditions (Fig. 6a). The latter corresponds to the pH value of the used polymer dispersion and the time scale of 1 h is related to the immersion time.

DRIFT spectra of a the PDMS|ns-TiO2|Ti6Al4V surface (green), the corresponding surface after 1 h at pH4.5 (red) as well as after 24 h EIS in 2 mM NaClO4 (blue) and b zoom of the OH-peak region from 3050 to 3800 cm−1 for the different surface states. All spectra were referenced against a pristine nanostructured TiO2 surface
For the PDMS modified surfaces, the DRIFT spectrum (Fig. 6a, green spectrum) shows a distinct absorption band in the range from 3650 to 3120 cm−1 as well as at 1627 cm−1, which is attributable to the OH-stretching and HOH-bending modes, respectively [55, 56]. The two peaks at 2964 cm−1 and 2905 cm−1 can be assigned to CH-stretching vibrations of CH3, which are part of the PDMS structure. The peak located at 1260 cm−1 shows CH3 symmetric bending, while absorption bands at 1098 cm−1 and 1028 cm−1 are distinctive of Si–O–Si in PDMS [57, 58]. The peak at 804 cm−1 is assumed to be an overlap of mainly rocking vibrations in Si–CH3 due to PDMS [57, 58] and minor TiO2 lattice vibrations from the underlying TiO2 substrate [59]. However, the presence of PDMS on the TiO2 nanostructured surface can clearly be concluded, which is in complete accordance with XPS analysis shown in Sect. 3.2.
The characteristic peaks could be detected on the surfaces even after immersion into electrolytes exhibiting varying pHs (Fig. 6a, red and blue spectra). This illustrates PDMS film stability over the analyzed time scale.
However, a significant increase in the region of the OH-stretching (> 3000 cm−1) and HOH-bending mode (1627 cm−1) appears together with an additional shoulder for interfacial water absorptions (876 cm−1) [59] after substrate immersion at neutral as well as at slight acidic pH. This indicates water uptake, a slight change in the chemical structure of PDMS or in that of adsorbed water molecules.
To clarify this, a further detailed analysis was performed in the wavenumber range from 3050 to 3800 cm−1 (Fig. 6b). In this regard, the bending vibrations at 1640 cm−1 are not suitable for such an investigation due to their lack of sensitivity to changes that might appear in the configuration of water molecules [56].
According to literature [60, 61] the maxima in the low wavenumber range correspond to hydrogen bonded OH stretching vibrations (Fig. 6b). In detail, the local maxima in the 3146 cm−1 and 3237 cm−1 region indicate the presence of water in the “ice-like” configuration, which is meant to describe the hydrogen-bonds appearing between the first and second adsorbed water monolayer on the surface [61]. However, while the elevation at 3400 cm−1 clearly describes bulk liquid water, also the maximum at 3237 cm−1 could rather be attributed to “liquid” H2O, regardless of the name. The latter is due to symmetric and asymmetric stretching vibrations of liquid water exhibiting asymmetric hydrogen-bond strength [62]. The underlying component at 3146 cm−1 is only slightly pronounced or not present at all in the spectra, since it describes the interactions between adsorbed water monolayers with orientations similar to the water/air interface [63]. These are only expected to occur at high relative humidity [60] and appear on the PDMS layer only after extensive contact with the electrolyte solution (Fig. 6b, blue spectrum).
Other than water band analysis performed on conventional, i.e. SiO2 [60], substrates, where the peak above 3600 cm−1 corresponds to non-hydrogen bonded OH stretching vibrations (“free OH groups”) [60, 61, 64], on TiO2-based materials the 3500 cm−1 to 3800 cm−1 region is dominated by a broad absorption band accompanied by a bunch of narrow peaks [65,66,67]. While the one at the highest wavenumber of 3730 cm−1 can clearly be ascribed to Ti–OH groups [66,67,68], the underlying signals in the lower wavenumber range cannot be accurately assigned, as their peak positions vary significantly dependent on the literature. However, an overlap of Ti–OH2 groups bound to the TiO2 surface [67] with Ti–OH–Ti groups, which are also reported as acidic OH-groups [66, 68], is assumed in this region. Si–OH groups would result in a peak at even higher wavenumbers (around 1745 cm−1 according to literature [66, 68]). Since no maximum appears in this region of the recorded DRIFT spectra, a hydroxylation of the PDMS layer is tentatively excluded in this study.
Finally, the following conclusions can be made: (I) Since “liquid” water makes up the largest percentage of the whole OH-band area, it can be concluded that bulk liquid water is mainly present on the pristine PDMS-coated surface and the incorporated water molecules exhibit the same configuration after electrolyte contact, even though in a higher magnitude. (II) The detected maxima in the high wavenumber range after electrolyte contact in contrast to the pristine PDMS surface indicates a slight in-situ hydroxylation of the PDMS-modified substrate. This is most probably related to the existence of non-bound surface TiO2 (compare XPS analysis, Fig. 3f), which is not occupied by PDMS and undergoes hydroxylation in the working pH range [69]. The latter is discussed in Sect. 3.6 with respect to particle adsorption analysis.
However, surface hydrophobicity completely remains after all. Therefore, it is assumed that the observed chemical changes of the TiO2 nanostructured and PDMS surface via DRIFTS and EIS as well as the change of water molecule configuration quantity do not deteriorate the performance of the outer part of the low free surface energy coating. Rather, electrolyte penetrates the pores of the coated nanostructure, while water being in contact with the outer part of the surface mostly rolls and dries off immediately in air.
3.6 Adsorption of PVAc particles
In order to study the adsorption and interaction characteristics of the modified surfaces with respect to PVAc deposit formation, the analyzed surfaces presented in this chapter were analogously treated as followed: (I) 1 h exposure to a diluted PVAc dispersion solution (1:10 in water, w:w) (pH 4.5–5.0) and (II) 1 h immersion into water, followed by rinsing the surface carefully with water. The FE-SEM images were taken after drying the surfaces under atmospheric conditions (Fig. 7).

FE-SEM images of a the Ti6Al4V surface, b the ns-TiO2|Ti6Al4V and c the additional PDMS modified surface as well as d the Ti6Al4V surface after PDMS modification, each obtained with a magnification of 5000× by a SE2 detector after 1 h immersion into an 1:10 diluted PVAc dispersion, followed by a 1 h water immersion and subsequent water rinsing. Particles are coloured for better visualization
As obvious from the FE-SEM image of the Ti6Al4V surface after contact with the polymer dispersion, the polymeric particles are homogeneously distributed over the entire surface and accumulate both in surface rifts and on the raises based on the surface roughness (Fig. 7a).
Adsorbed particles were also detected on the TiO2 nanostructured surface (Fig. 7b). However, it becomes apparent that the particles appear to spread on the superhydrophilic surface. They are arranged flat and seem to maximize the contact area with the high energetic nanostructure. As a result, a precise particle structure is often no longer recognizable. This demonstrates a high affinity of the PVAc particles to the nanostructured TiO2 surface in comparison to the reference surface state, which exhibits significantly less high energetic surface properties than the nanostructured TiO2 surface. Therefore, hydrophilic repulsion is no mechanism that inhibits polymeric particle adsorption. The observations can further tentatively be attributed to the fact that at the pH range from 4.5 to 5.0 TiO2 is reported to carry mainly TiOH and slightly TiOH2+ surface groups [69] and acetate groups adsorb preferably to the hydroxyl moiety [70]. Therefore, the observed particle spreading could be a result of spontaneous adsorption to this high amount of hydrophilic functional groups, which are slightly present on the initial surface state and are further formed in-situ by electrolyte exposition at pH 4.5–5.0.
After PDMS surface modification of the nanostructured TiO2 surface, the polymeric particle adsorption is reduced (Fig. 7c). This mirrors that most particles do not even adsorb or only weakly physisorb on the surface since they can be rinsed off by a gentle flow of water.
Interestingly, particle adsorption is not favoured in surface cracks. It is further evident that particle–particle interactions seem to be favoured compared to particle–surface interactions, since at a few points particles accumulate on the surface. Therefore, in this study, the low surface interactions can be attributed to a combination of water repellent surface properties, the role of which was discussed before, and the non-polarizability of the PDMS based on the high symmetry of the macromolecule.
Nevertheless, initial particle attachment at apparently thermodynamically favoured adsorption sites is possible at a few points. This is related to a partial in-situ hydroxylation of the TiO2 moiety present on the PDMS surface upon contact with the particle-containing electrolyte solution at pH 4.5–5.0, as shown by DRIFT spectroscopy (Fig. 6). Therefore, it might be possible that PVAc particles adsorb onto these in-situ formed OH groups. Due to the significant lower surface OH-group concentration on PDMS, which is also obvious from constant superhydrophobic properties even after electrolyte immersion, no particle spreading, and less particle adsorption is observed compared to the pure nanostructured TiO2 surface. These adsorbates then allow and facilitate further polymer particle attachment, probably by interacting with surface OH groups as present on the particle due to polyvinyl alcohol.
To illustrate the influence of the microstructure on the particle adsorption, PDMS was deposited directly onto the Ti6Al4V surface for comparison (Fig. 7d). The successful surface modification as well as the hydrophobic surface properties were shown by contact angle measurement and DRIFTS (Supplementary Information S3).
Similar to the results shown in Fig. 7c, the image taken on the corresponding smooth surface (Fig. 7d) also shows reduced particle adsorption and an increased aggregation of particles compared to the non-modified Ti6Al4V surface (Fig. 7a). This is interpreted as possibility to reduce surface interactions with the PDMS and is in full agreement with the results obtained from the PDMS|ns-TiO2|Ti6Al4V surface.
While images obtained by the SE2 detector can best be used to analyze topographic surface characteristics, an InLens detector can give extending information on existing material contrasts. Respective images were taken from the PDMS|Ti6Al4V surface in comparison to the pristine Ti6Al4V surface after particle contact as described previously (Fig. 8).

FE-SEM images of a the Ti6Al4V surface and b the PDMS|Ti6Al4V surface, each obtained with a magnification of 5000× by an InLens detector after 1 h immersion into an 1:10 diluted PVAc dispersion, followed by a 1 h water immersion and subsequent water rinsing
When looking closely at the images acquired by the InLens detector, dark and round imprints are obvious on the PDMS-modified surface (Fig. 8b, marked in yellow), which are absent on the non-coated substrate (Fig. 8a). Due to the similarity in terms of size and distribution of the particles, it is concluded that many polymeric particles could be removed by the water rinsing process and were thus only weakly adsorbed.
In order to further underpin the qualitative results, a quantitative analysis was carried out for all discussed surface states. For this, the surface area occupied by the particles is related to the total available surface area and percentage data was generated (Table 3).
The determined percentage data again reflects the high affinity of PVAc particles towards the superhydrophilic ns-TiO2 surface. Further, the employment of the superhydrophobic PDMS|ns-TiO2|Ti6Al4V surface halves the amount of the surface area, which is covered by the polymeric particles compared to the pristine Ti6Al4V alloy. As expected from FE-SEM images, the amount could also be reduced by modification of the Ti6Al4V surface with PDMS. However, the effect is less pronounced.
Therefore, qualitative and quantitative evaluation consistently show that fewest particles are adsorbed on the nanostructured surface with PDMS modification in comparison, which mirrors well the necessity for highly water repellent surface characteristics.
The previously discussed quantity of adsorbed polymer particles correlates well with the intensity of the PVAc characteristic peaks obtained from DRIFT spectroscopy (Supplementary Information S4).
Finally, it can be concluded that the PVAc dispersion exhibiting a high surface free energy of 34 mN/m with a low polar component of 4 mN/m and a high dispersive component of 30 mN/m interacts significantly less with the low surface energy PDMS coating compared to the high-energetic substrates.
To further follow the particle adsorption on the surfaces of interest during 24 h, EIS was applied using a diluted polymer dispersion (1:10 in 2 mM NaClO4, w/w) as electrolyte (Fig. 9).

Bode Plots as obtained from electrochemical impedance spectroscopy performed on a the Ti6Al4V as well as on b the ns-TiO2|Ti6Al4V and c additionally PDMS modified surface, obtained in an 1:10 diluted PVAc dispersion as electrolyte at room temperature
On the Ti6Al4V alloy surface (Fig. 9a), the same trend, which is a minor time-dependent increase of |Z| at 1 Hz, is observed as for EIS performed in NaClO4 as electrolyte (Sect. 3.4). This increase could be attributed to particle adsorption, which is expected to appear according to FE-SEM and DRIFTS analysis, or the oxide layer growth of the barrier part of the alloy surface, as previously observed for EIS performed in NaClO4 as electrolyte (Sect. 3.4). It could also be a superposition of both effects, however, this cannot be clarified in this study.
On the nanostructured TiO2 (Fig. 9b) and additionally PDMS-modified surface (Fig. 9c), similar trends as detected on the respective surfaces under employment of NaClO4 as electrolyte could be observed, which is decrease of |Z| at 1 Hz over time. This was expected since electrolyte from the diluted dispersion is supposed to penetrate into the nanostructure leading to the same trends as for the pure electrolyte. However, the analysis confirms and extends the observations made from FE-SEM analysis, that the particles do not penetrate into the TiO2 fibrous network or into surface cracks but interact only with the upper part of the nanostructure.
4 Proposed interaction model based on experimental results
Based on the results a model for the interaction of PVAc particles with the nanostructured TiO2-surface and additionally PDMS-modified surface is presented (Fig. 10).

Schematic of the proposed interactions of polyvinyl acetate dispersion with a the ns-TiO2|Ti6Al4V and b the PDMS|ns-TiO2|Ti6Al4V upon contact with polymer solution
The nanostructured TiO2 surface exhibits superhydrophilic surface properties with strong polar characteristics, as shown by complete water wetting under employment of contact angle analysis. During contact with the diluted PVAc dispersion at pH 4.5–5.0, TiOH2+ and TiOH-groups form, which allow binding between the carbonyl groups of the PVAc and the surface hydroxyl moiety according to literature [69, 70]. Due to the high affinity of the polymer for the surface groups as well as the high concentration of possible adsorption sites, a spreading of the particles was observed by FE-SEM (Fig. 10a).
In comparison, the PDMS-modified nanostructured TiO2 surface results in superhydrophobic and complete water repellent properties with low polar characteristics (Fig. 10b). Based on DRIFTS and detailed OH-absorption band analysis, it is assumed that the particle adsorption is, as analogous to the nanostructured TiO2 surface, possible via in-situ hydroxylation of the surface. In this context, it is assumed that the underlying TiO2-nanostructure, which might not be completely covered by the hydrophobic film, is hydroxylated. Basically, a reduced particle adsorption is observed complementary from FE-SEM and DRIFTS analysis after PDMS modification, which can be attributed to a significantly less amount of favoured adsorption sites.
During EIS analysis it could further be shown that water penetrates into the nanostructure of the TiO2 over time for both the PDMS- and non-modified rough substrates. However, this is not relevant for the analyzed particle adsorption in this study since the nanostructure was adapted to the particle size. Therefore, the PVAc particles only interact with the outer part of the nanostructure, which makes hydroxylation in the inner part of the substrate irrelevant and the groups not available for interactions. Analysis showed that the outer part of the PDMS surface is not affected in performance.
Less contact points are most likely also the reason for minor particles being chemisorbed on the nanostructured compared to the smooth surfaces.
5 Conclusions
In this study, nanostructured surfaces exhibiting superhydrophobic and superhydrophilic surface properties were generated from a Ti6Al4V alloy by a simple etching and calcination process and subsequent polydimethylsiloxane (PDMS) modification by grafting via UV-light. Thereby the modification process was adapted to the particle size of the exemplary used polymer dispersion as obtained from polymerization of vinyl acetate with the aim of analyzing the interactions between polyvinyl acetate (PVAc) particles with surfaces of different wetting and structural characteristics.
On the one side, less adhesion of PVAc is observed on the PDMS-modified nanostructured TiO2 surface compared to the non-coated substrate. This is attributable to a strongly minimized OH-group concentration that forms in-situ from the underlying TiO2 upon contact with the polymer solution. Further, the superhydrophobic properties remain after extended contact with electrolytes in the pH range from 4.5 to 7.0. Irreversible aqueous electrolyte diffusion into the PDMS coating was observed with predominant ion incorporation being assumed, however, it does not seem to influence the performance of the film when deposited onto the nanostructured surface.
Even though the same adsorption via carbonyl groups from the polymer with the OH-moiety of the surface is considered, it can be predicted that the observed quantitative and structural changes during adsorption of PVAc particles onto the TiO2-nanostructured surface appear due to the higher amount of in-situ formed OH-groups. The less favored interactions with the nanostructured surfaces compared to smooth substrates can be explained by the reduced surface points where interactions can take place.
The results can be used to reduce and prevent polymeric particle fouling during polymer synthesis. Due to the high adaptability of the nanostructure towards a particular problem, the approach is highly transferrable to other systems.
Data availability
FIB-SEM data supporting the findings of this study can be found in the Supplementary Information S1. XPS survey spectra of the analyzed surfaces as well as the HR spectrum in the Si2p peak region of the PDMS-modified surface are available from Supplementary Information S2. DRIFTS and contact angle analysis of the PDMS-modified Ti6Al4V surface is available from the Supplementary Information S3 and complementary DRIFTS studies of the surfaces after immersion into the polymer solution can be found in the Supplementary Information S4. Raw and original data of this study are available from the corresponding author upon reasonable request.
References
Rust S, Pauer W. Formulation and process determined fouling prediction for the continuous emulsion co polymerisation of vinyl acetate. J Polym Res. 2023;30(6):242. https://doi.org/10.1007/s10965-023-03588-8.
Neßlinger V, Orive AG, Meinderink D, Grundmeier G. Combined in-situ attenuated total reflection-fourier transform infrared spectroscopy and single molecule force studies of poly(acrylic acid) at electrolyte/oxide interfaces at acidic pH. J Colloid Interface Sci. 2022;615:563–76. https://doi.org/10.1016/j.jcis.2022.01.175.
Srebnik S. Polymer adsorption on multicomponent surfaces with relevance to membrane fouling. Chem Eng Sci. 2003;58(23–24):5291–8. https://doi.org/10.1016/j.ces.2003.05.008.
Horseman T, Wang Z, Lin S. Colloidal interactions between model foulants and engineered surfaces: interplay between roughness and surface energy. Chem Eng J Adv. 2021;8:100138. https://doi.org/10.1016/j.ceja.2021.100138.
Hohlen A, Augustin W, Scholl S. Quantification of polymer fouling on heat transfer surfaces during synthesis. Macro React Eng. 2020;14(1):1900035. https://doi.org/10.1002/mren.201900035.
Kohlmann D, Chevrel M-C, Hoppe S, Meimaroglou D, Chapron D, Bourson P, Schwede C, Loth W, Stammer A, Wilson J, Ferlin P, Falk L, Engell S, Durand A. Modular, flexible, and continuous plant for radical polymerization in aqueous solution. Macro React Eng. 2016;10(4):339–53. https://doi.org/10.1002/mren.201500079.
Deglmann P, Hellmund M, Hungenberg K-D, Nieken U, Schwede C, Zander C. Side reactions in aqueous phase polymerization of n -vinyl-pyrrolidone as possible source for fouling. Macro React Eng. 2019;13(5):1900021. https://doi.org/10.1002/mren.201900021.
Zander C, Hungenberg K-D, Schall T, Schwede C, Nieken U. Modeling strategies for the propagation of terminal double bonds during the polymerization of n-vinylpyrrolidone and experimental validation. Macro React Eng. 2020;14(3):2000009. https://doi.org/10.1002/mren.202000009.
Fries S, Castañeda-Zúñiga DM, Duchateau J, Neuteboom P, Porras CT, Busch M. Fouling in the high pressure LDPE process: experimental and computational investigation approach. Macromol Symp. 2016;360(1):78–86. https://doi.org/10.1002/masy.201500112.
Urrutia J, Peña A, Asua JM. Reactor fouling by preformed latexes. Macro React Eng. 2017;11(1):1600043. https://doi.org/10.1002/mren.201600043.
Urrutia J, Asua JM. Reactor fouling in emulsion polymerization. Ind Eng Chem Res. 2021;60(29):10502–10. https://doi.org/10.1021/acs.iecr.1c00097.
Kiparissides C, Macgregor JF, Hamielec AE. Continuous emulsion polymerization of vinyl acetate. Part I: experimental studies. Can J Chem Eng. 1980;58(1):48–55. https://doi.org/10.1002/cjce.5450580108.
Chern CS, Chen YC. Stability of the polymerizable surfactant stabilized latex particles during semibatch emulsion polymerization. Colloid Polym Sci. 1997;275(2):124–30. https://doi.org/10.1007/s003960050061.
Bianco H, Cohen Y, Narkis M. Coagulum formation and elimination in emulsion copolymerization of tribromostyrene with styrene. Polym Adv Technol. 1996;7(10):809–12. https://doi.org/10.1002/(SICI)1099-1581(199610)7:10%3c809::AID-PAT582%3e3.0.CO;2-0.
Lü T, Shan G, Shang S. Stability of two-phase polymerization of acrylamide in aqueous poly(ethylene glycol) solution. J Appl Polym Sci. 2011;122(2):1121–33. https://doi.org/10.1002/app.34243.
Neßlinger V, Rust S, Atlanov J, Pauer W, Grundmeier G. Monitoring polymeric fouling in a continuous reactor by electrochemical impedance spectroscopy. Chem Ing Tec. 2024;96(3):291–9. https://doi.org/10.1002/cite.202300032.
Idumah CI, Obele CM, Emmanuel EO, Hassan A. Recently emerging nanotechnological advancements in polymer nanocomposite coatings for anti-corrosion, anti-fouling and self-healing. Surf Interfaces. 2020;21:100734. https://doi.org/10.1016/j.surfin.2020.100734.
Dong W, Li B, Wei J, Tian N, Liang W, Zhang J. Environmentally friendly, durable and transparent anti-fouling coatings applicable onto various substrates. J Colloid Interface Sci. 2021;591:429–39. https://doi.org/10.1016/j.jcis.2021.02.014.
Del Sabaté RJ, Henry OYF, Jolly P, Ingber DE. An antifouling coating that enables affinity-based electrochemical biosensing in complex biological fluids. Nat Nanotechnol. 2019;14(12):1143–9. https://doi.org/10.1038/s41565-019-0566-z.
Tian S, Jiang D, Pu J, Sun X, Li Z, Wu B, Zheng W, Liu W, Liu Z. A new hybrid silicone-based antifouling coating with nanocomposite hydrogel for durable antifouling properties. Chem Eng J. 2019;370:1–9. https://doi.org/10.1016/j.cej.2019.03.185.
Malayeri MR, Al-Janabi A, Müller-Steinhagen H. Application of nano-modified surfaces for fouling mitigation. Int J Energy Res. 2009;33(13):1101–13. https://doi.org/10.1002/er.1599.
ElSherbiny IMA, Khalil ASG, Ulbricht M. Influence of surface micro-patterning and hydrogel coating on colloidal silica fouling of polyamide thin-film composite membranes. Membranes. 2019;9(6):67. https://doi.org/10.3390/membranes9060067.
Hoek EMV, Bhattacharjee S, Elimelech M. Effect of membrane surface roughness on colloid−membrane DLVO interactions. Langmuir. 2003;19(11):4836–47. https://doi.org/10.1021/la027083c.
Albert F, Augustin W, Scholl S. Roughness and constriction effects on heat transfer in crystallization fouling. Chem Eng Sci. 2011;66(3):499–509. https://doi.org/10.1016/j.ces.2010.11.021.
Wang M, Wang J, Jiang J. Membrane fouling: microscopic insights into the effects of surface chemistry and roughness. Advcd Theory Sims. 2022;5(1):2100395. https://doi.org/10.1002/adts.202100395.
Neßlinger V, Welzel S, Rieker F, Meinderink D, Nieken U, Grundmeier G. Thin organic-inorganic anti-fouling hybrid-films for microreactor components. Macro React Eng. 2023;17(1):2200043. https://doi.org/10.1002/mren.202200043.
Zhou W, Yang F, Yuan L, Diao Y, Jiang O, Pu Y, Zhang Y, Zhao Y, Wang D. Construction of superhydrophobic coating on iron surface with enhanced anti-corrosion, anti-adhesive and anti-bacterial properties. Materials. 2022;15(23):8634. https://doi.org/10.3390/ma15238634.
Keshmiri K, Huang H, Nazemifard N. Compatibility of poly(dimethylsiloxane) microfluidic systems with high viscosity hydrocarbons. SN Appl Sci. 2019;1(7):711. https://doi.org/10.1007/s42452-019-0666-2.
Wong T-S, Kang SH, Tang SKY, Smythe EJ, Hatton BD, Grinthal A, Aizenberg J. Bioinspired self-repairing slippery surfaces with pressure-stable omniphobicity. Nature. 2011;477(7365):443–7. https://doi.org/10.1038/nature10447.
Stahl T, Mattern D, Brunn H. Toxicology of perfluorinated compounds. Environ Sci Eur. 2011;23(1):38. https://doi.org/10.1186/2190-4715-23-38.
Anitha VC, Banerjee AN, Joo SW, Min BK. Morphology-dependent low macroscopic field emission properties of titania/titanate nanorods synthesized by alkali-controlled hydrothermal treatment of a metallic Ti surface. Nanotechnology. 2015;26(35):355705. https://doi.org/10.1088/0957-4484/26/35/355705.
Divya Rani VV, Manzoor K, Menon D, Selvamurugan N, Nair SV. The design of novel nanostructures on titanium by solution chemistry for an improved osteoblast response. Nanotechnology. 2009;20(19):195101. https://doi.org/10.1088/0957-4484/20/19/195101.
Tesler AB, Prado LH, Khusniyarov MM, Thievessen I, Mazare A, Fischer L, Virtanen S, Goldmann WH, Schmuki P. A one-pot universal approach to fabricate lubricant-infused slippery surfaces on solid substrates. Adv Funct Mater. 2021;31(27):2101090. https://doi.org/10.1002/adfm.202101090.
Wooh S, Encinas N, Vollmer D, Butt H-J. Stable hydrophobic metal-oxide photocatalysts via grafting polydimethylsiloxane brush. Adv Mater. 2017. https://doi.org/10.1002/adma.201604637.
Vormoor M (2009) Untersuchungen zur superhydrophilie von titandioxid-beschichtungen. Cuvillier, Göttingen
Teisala H, Baumli P, Weber SAL, Vollmer D, Butt H-J. Grafting silicone at room temperature-a transparent, scratch-resistant nonstick molecular coating. Langmuir. 2020;36(16):4416–31. https://doi.org/10.1021/acs.langmuir.9b03223.
Ström G, Fredriksson M, Stenius P. Contact angles, work of adhesion, and interfacial tensions at a dissolving hydrocarbon surface. J Colloid Interface Sci. 1987;119(2):352–61. https://doi.org/10.1016/0021-9797(87)90280-3.
Schuster JM, Schvezov CE, Rosenberger MR. Analysis of the results of surface free energy measurement of Ti6Al4V by different methods. Proc Mater Sci. 2015;8:732–41. https://doi.org/10.1016/J.MSPRO.2015.04.130.
Mu M, Zhou X, Xiao Q, Liang J, Huo X. Preparation and tribological properties of self-lubricating TiO2/graphite composite coating on Ti6Al4V alloy. Appl Surf Sci. 2012;258(22):8570–6. https://doi.org/10.1016/j.apsusc.2012.05.051.
He Z, Zhuo Y, Wang F, He J, Zhang Z. Design and preparation of icephobic PDMS-based coatings by introducing an aqueous lubricating layer and macro-crack initiators at the ice-substrate interface. Prog Org Coat. 2020;147:105737. https://doi.org/10.1016/j.porgcoat.2020.105737.
Lv X, Wang L, Fu J, Li Y, Yu L. A one-step tannic acid coating to improve cell adhesion and proliferation on polydimethylsiloxane. New J Chem. 2020;44(35):15140–7. https://doi.org/10.1039/d0nj02663h.
Reggio C, Barberi J, Ferraris S, Spriano S. Functionalization of Ti6Al4V alloy with polyphenols: the role of the titanium surface features and the addition of calcium ions on the adsorption mechanism. Metals. 2023;13(8):1347. https://doi.org/10.3390/met13081347.
Bharti B, Kumar S, Lee H-N, Kumar R. Formation of oxygen vacancies and Ti(3+) state in TiO2 thin film and enhanced optical properties by air plasma treatment. Sci Rep. 2016;6:32355. https://doi.org/10.1038/srep32355.
Müller G, Benkhai H, Matthes R, Finke B, Friedrichs W, Geist N, Langel W, Kramer A. Poly (hexamethylene biguanide) adsorption on hydrogen peroxide treated Ti–Al–V alloys and effects on wettability, antimicrobial efficacy, and cytotoxicity. Biomaterials. 2014;35(20):5261–77. https://doi.org/10.1016/j.biomaterials.2014.03.033.
Fan C, Chen C, Wang J, Fu X, Ren Z, Qian G, Wang Z. Black hydroxylated titanium dioxide prepared via ultrasonication with enhanced photocatalytic activity. Sci Rep. 2015;5:11712. https://doi.org/10.1038/srep11712.
Cha B, Woo T, Han S, Saqlain S, Seo H, Cho H, Kim J, Kim Y. Surface modification of TiO2 for obtaining high resistance against poisoning during photocatalytic decomposition of toluene. Catalysts. 2018;8(11):500. https://doi.org/10.3390/catal8110500.
Fredriksson W, Malmgren S, Gustafsson T, Gorgoi M, Edström K. Full depth profile of passive films on 316L stainless steel based on high resolution HAXPES in combination with ARXPS. Appl Surf Sci. 2012;258(15):5790–7. https://doi.org/10.1016/j.apsusc.2012.02.099.
Lo Y-S, Chang C-C, Lin P-C, Lin S-P, Wang C-L. Direct growth of structurally controllable hydroxyapatite coating on Ti–6Al–4V through a rapid hydrothermal synthesis. Appl Surf Sci. 2021;556:149672. https://doi.org/10.1016/j.apsusc.2021.149672.
Gayani B, Senarathna D, Weerasekera MM, Kottegoda N, Ratnaweera DR. Improving superhydrophobicity of polydimethylsiloxanes using embedding fluorinated polyhedral oligomeric silsesquioxanes cages. SN Appl Sci. 2020;2(12):1. https://doi.org/10.1007/s42452-020-03721-y.
Gudić S, Vrsalović L, Kvrgić D, Nagode A. Electrochemical behaviour of Ti and Ti–6Al–4V alloy in phosphate buffered saline solution. Materials. 2021;14(24):7495. https://doi.org/10.3390/ma14247495.
Tamilselvi S, Raman V, Rajendran N. Corrosion behaviour of Ti–6Al–7Nb and Ti–6Al–4V ELI alloys in the simulated body fluid solution by electrochemical impedance spectroscopy. Electrochim Acta. 2006;52(3):839–46. https://doi.org/10.1016/j.electacta.2006.06.018.
Wypych A, Bobowska I, Tracz M, Opasinska A, Kadlubowski S, Krzywania-Kaliszewska A, Grobelny J, Wojciechowski P. Dielectric properties and characterisation of titanium dioxide obtained by different chemistry methods. J Nanomater. 2014;2014:1–9. https://doi.org/10.1155/2014/124814.
Le Floch P, Molinari N, Nan K, Zhang S, Kozinsky B, Suo Z, Liu J. Fundamental limits to the electrochemical impedance stability of dielectric elastomers in bioelectronics. Nano Lett. 2020;20(1):224–33. https://doi.org/10.1021/acs.nanolett.9b03705.
Ochs H (2003) Physical-chemical testing of boundary-layers on metallic surface. Dissertation, University of Stuttgart
Berthomieu C, Hienerwadel R. Fourier transform infrared (FTIR) spectroscopy. Photosynth Res. 2009;101(2–3):157–70. https://doi.org/10.1007/s11120-009-9439-x.
Asay DB, Kim SH. Evolution of the adsorbed water layer structure on silicon oxide at room temperature. J Phys Chem B. 2005;109(35):16760–3. https://doi.org/10.1021/jp053042o.
Lee J, Kim J, Kim H, Bae YM, Lee K-H, Cho HJ. Effect of thermal treatment on the chemical resistance of polydimethylsiloxane for microfluidic devices. J Micromech Microeng. 2013;23(3):35007. https://doi.org/10.1088/0960-1317/23/3/035007.
Shi D, Li P. Preparation of PDMS/PVDF composite pervaporation membrane modified with hydrophobic TiO2 nanoparticles for separating formaldehyde solution. Polym Sci. 2016. https://doi.org/10.4172/2471-9935.100012.
Connor PA, Dobson KD, McQuillan AJ. Infrared spectroscopy of the TiO2/aqueous solution interface. Langmuir. 1999;15(7):2402–8. https://doi.org/10.1021/la980855d.
Torun B, Kunze C, Zhang C, Kühne TD, Grundmeier G. Study of water adsorption and capillary bridge formation for SiO(2) nanoparticle layers by means of a combined in situ FT-IR reflection spectroscopy and QCM-D set-up. Phys Chem Chem Phys. 2014;16(16):7377–84. https://doi.org/10.1039/C3CP54912G.
Fan Y, Chen X, Yang L, Cremer PS, Gao YQ. On the structure of water at the aqueous/air interface. J Phys Chem B. 2009;113(34):11672–9. https://doi.org/10.1021/jp900117t.
Zhang C, Khaliullin RZ, Bovi D, Guidoni L, Kühne TD. Vibrational signature of water molecules in asymmetric hydrogen bonding environments. J Phys Chem Lett. 2013;4(19):3245–50. https://doi.org/10.1021/jz401321x.
Kühne TD, Pascal TA, Kaxiras E, Jung Y. New insights into the structure of the vapor/water interface from large-scale first-principles simulations. J Phys Chem Lett. 2011;2(2):105–13. https://doi.org/10.1021/jz101391r.
Chamerski K, Korzekwa W, Filipecki J, Shpotyuk O, Stopa M, Jeleń P, Sitarz M. Nanoscale observation of dehydration process in PHEMA hydrogel structure. Nanoscale Res Lett. 2017;12(1):303. https://doi.org/10.1186/s11671-017-2055-3.
Deiana C, Fois E, Coluccia S, Martra G. Surface structure of TiO2 P25 Nanoparticles: infrared study of hydroxy groups on coordinative defect sites. J Phys Chem C. 2010;114(49):21531–8. https://doi.org/10.1021/jp107671k.
Bonne M, Pronier S, Batonneau Y, Can F, Courtois X, Royer S, Marécot P, Duprez D. Surface properties and thermal stability of SiO2-crystalline TiO2 nano-composites. J Mater Chem. 2010;20(41):9205. https://doi.org/10.1039/c0jm01184c.
Arrouvel C, Digne M, Breysse M, Toulhoat H, Raybaud P. Effects of morphology on surface hydroxyl concentration: a DFT comparison of anatase–TiO2 and γ-alumina catalytic supports. J Catal. 2004;222(1):152–66. https://doi.org/10.1016/j.jcat.2003.10.016.
Zaki MI, Hasan MA, Al-Sagheer FA, Pasupulety L. In situ FTIR spectra of pyridine adsorbed on SiO2–Al2O3, TiO2, ZrO2 and CeO2: general considerations for the identification of acid sites on surfaces of finely divided metal oxides. Colloids Surf A. 2001;190(3):261–74. https://doi.org/10.1016/S0927-7757(01)00690-2.
Chibowski, Paszkiewicz M. Studies of the influence of acetate groups from polyvinyl alcohol on adsorption and electrochemical properties of the TiO2 polymer solution interface. J Dispers Sci Technol. 2001;22(2–3):281–9. https://doi.org/10.1081/DIS-100105215.
Koopal LK, Lyklema J. Characterization of adsorbed polymers from double layer experiments. J Electroanal Chem Interfacial Electrochem. 1979;100(1–2):895–912. https://doi.org/10.1016/S0022-0728(79)80208-9.
Acknowledgements
We thank Nadine Buitkamp from Paderborn University for FE-SEM measurements and assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL. The financial support of the German Federal Ministry for Economic Affairs and Climate Action (BMWK) under grant number 03EN2004J (KoPPonA 2.0) is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Conceptualization: Vanessa Neßlinger, Guido Grundmeier; Methodology: Vanessa Neßlinger, Jan Atlanov; Formal analysis and investigation: Vanessa Neßlinger, Jan Atlanov; Writing – original draft: Vanessa Neßlinger; Writing – review and editing: Guido Grundmeier; Funding Acquisition: Guido Grundmeier; Resources: Guido Grundmeier; Supervision: Guido Grundmeier
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no known competing financial and non-financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Neßlinger, V., Atlanov, J. & Grundmeier, G. Interactions of polyvinyl acetate dispersions with nanostructured superhydrophilic and superhydrophobic Ti6Al4V alloy surfaces. Discov Appl Sci 6, 294 (2024). https://doi.org/10.1007/s42452-024-05916-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-024-05916-z