
Abstract
Polyvinyl alcohol (PVA)/beryllium sulfate (BeSO4) precursor nanofibers are fabricated by electrospinning technique, mixing PVA aqueous solution with BeSO4 salt. The productivity is increased by adding polyethyleneimine (PEI) with PVA/BeSO4 spinning solution. The beryllium oxide (BeO) nanofibers are obtained by calcinating the PVA/BeSO4/PEI precursor nanofiber heated at 1000 °C or above. The crystallographic structure of BeO nanofibers is examined by X-ray diffraction. The thermal behaviors of the pure PVA nanofibers, BeSO4 salt, and PVA/BeSO4/PEI precursor nanofibers are studied by thermogravimetry analysis. The BeO nanofiber diameters are reduced with the increase in calcination temperatures. The specific surface area of the PVA/BeSO4/PEI precursor nanofibers is around 36.9 m2 g−1, and that of the BeO nanofibers calcined at 1200 °C is about 11.9 m2 g−1. The pore properties deteriorate due to sintering and blockage as the calcination temperature increases. This work introduces mesoporous BeO nanofibers for the very first time.
Article highlights
-
This work introduces mesoporous BeO nanofibers by electrospinning for the very first time.
-
Polyethylenimine was used to improve the productivity of PVA/BeSO4/PEI precursor nanofibers.
-
The BeO nanofibers prepared under this optimum condition will be valuable for further development and are expected to have the potential for industrial applications such as thermal conductive heat dissipate sheets.
Similar content being viewed by others


1 Introduction
Beryllium oxide (BeO) is a ceramic material that has a unique combination of physicochemical properties, including excellent thermal conductivity, high chemical, and thermal stability, high insulation nature, a high specific volumetric resistance, high intensity, a high melting point, transparency for vacuum, infrared, ultraviolet, visible, radiation resistance, low dielectric constant, and ultrahigh-frequency (UHF) radiation [1, 2]. Because of these outstanding characteristics, BeO-ceramic is a potential material for new technological domains, such as current electronics, vacuum electronics, nuclear technology, and microelectronics. BeO-ceramics can conduct heat from electronic technology while temperatures range from 300 to 630 K of all-ceramic materials. BeO-ceramics are generally the only option, particularly in thermal applications [3, 4].
There are few reports on BeO nanoparticle synthesis, and extensive structural characterization of produced BeO nanoparticles is limited and not reported. Wang et al. prepared BeO nanoparticles by polyacrylamide gel route. They created xerogel using acrylamide and N,N′-methylene bis acrylamide monomers and mixed with beryllium sulfate tetrahydrate (BeSO4·4H2O) aqueous solution followed by ammonium persulfate aqueous solution. Finally, they prepared BeO nanoparticles powder by calcinating at 700–1000 °C temperatures [5]. Selvaraj et al. also synthesized BeO nanoparticles. They prepared acrylamide and N,N′-methylene bisacrylamide monomer solutions, then added them drop by drop to an aqueous BeSO4·4H2O solution with continuous stirring until a translucent solution was formed. Finally, they added ammonium persulfate solution as an initiator to the complete solutions and obtained BeO nanoparticles powder after drying, crushing, and heating [6].
Nanofiber research, development, and industrial applications have received much attention in recent years. Many investigations on various nanofibers have been undertaken [7, 8]. Ohgoshi et al. fabricated magnesia nanofiber by calcinating polyvinyl alcohol (PVA)/magnesium ethoxide (C4H10MgO2) precursor nanofibers at different temperatures. They produced PVA/C4H10MgO2 precursor nanofibers mat by electrospinning technique using PVA and C4H10MgO2 mixtures [9]. Nakane et al. generated α-alumina nanofibers by calcinating PVA/boehmite precursor nanofibers. They mixed PVA solutions with boehmite nanoparticles to produce a precursor nanofibers mat with the help of an electrospinning machine [10]. Nakane et al. fabricated PVA/titanium lactate (TL) precursor nanofibers by electrospinning machine. After calcinating PVA/TL precursor nanofibers, they produced anatase-type TiO2 nanofibers [11]. Using an electrospinning process, Hwang et al. synthesized PVA/boron nitride (BN) precursor nanofibers, adding PVA solutions with BN nanoparticles. They produced BN nanofibers by calcinating PVA/BN precursor nanofibers at 1000 °C temperatures [12].
Electrospinning is becoming a popular technology for polymer nanofibers because various organic, inorganic, and organic–inorganic hybrid nanofibers are quickly produced. Electrospinning creates continuous polymer strands with nanoscale diameters by applying an external electric field to a polymer solution [13]. Electrospun nanofibers have a high specific surface area and a small pore size compared to commercial textiles. So, nanofibers can be rationally constructed to have innovative and immensely better physical, chemical, and biological properties [14]. An electric force is applied between a suspended droplet solution at a capillary tip and a collector during electrospinning. A charged jet is launched when the electrical field's intensity exceeds the surface tension of the polymer solution. It travels to the grounded target, forming fibers in the form of nonwoven mats [15].
Only a few studies on BeO nanoparticles have been published. However, the authors did not find any literature on BeO nanofiber. As a result, there have been no reports on BeO nanofiber synthesis or extensive structural study of generated BeO nanofiber. In this study, the authors created PVA/BeSO4/polyethyleneimine (PEI) mixed solution using granular PVA, BeSO4·4H2O salt, and PEI. They produced PVA/BeSO4/PEI precursor nanofibers using the electrospinning technique from the previously made mixed solution. After calcinating at various temperatures, BeO nanofibers were created. The authors also described the improved productivity of precursor nanofibers and analyzed the size, shape, morphology, structural properties, adsorption isotherms, and thermal decomposition of precursor and BeO nanofibers.
2 Materials and methods
2.1 Raw materials
Polyvinyl alcohol (PVA) (DP 1500) was purchased from Wako Pure Chemical Co., Japan. Beryllium sulfate tetrahydrate salt (BeSO4·4H2O—purity > 98%) was purchased from Kanto Chemical Co., Inc., Japan. Nacalai Tesque, Inc., Japan provided polyethyleneimine (PEI) (CH2CH2NH)n 30% in water with a molecular weight of 60,000–80,000. All materials were purchased and used without further processing.
2.2 Preparation of PVA/BeSO4/PEI precursor nanofibers
An appropriate number of PVA powders were dissolved into the deionized water and stirred for 5 h at 90 °C to make a 10% PVA solution. BeSO4·4H2O salt (Beryllium source) was added to the above-indicated 10% PVA solution and stirred slowly for 8 h at room temperature. Finally, PEI was added drop by drop to make the spinning solution. The spinning solution’s PVA, BeSO4, and PEI were 8.92, 4.23, and 1.1037 wt% respectivity. The calculated ratio of (PVA/PEI) and BeSO4 is about 90/10 wt%. The spinning solution was kept in a 3 mL plastic syringe on an electrospinning machine with an operating voltage of 20 kV and a current of 0.004 mA. The distance between the needle tip and the collector was 15 cm. An injection rate of 0.008 ml min−1 was applied for secure production. The electrospun PVA/BeSO4/PEI precursor nanofibers were collected on parchment paper.
2.3 Preparation of BeO nanofibers
An electric furnace was used for calcinating the electrospun PVA/BeSO4/PEI precursor nanofibers at a heating rate of 10 °C min−1 for 5 h from 700 to 1200 °C temperatures. Finally, white BeO nanofibers were obtained. The entire process is schematically depicted in Fig. 1.

Experimental scheme of preparation of BeO nanofiber mats
2.4 Characterization
The morphology of both PVA/BeSO4/PEI precursor and BeO nanofibers was examined using a scanning electron microscope (SEM) from Keyence VE9800, Japan. The standard deviation (σ) and mean fiber diameter (D) of the precursor and BeO nanofibers (n = 100) were calculated using Adobe Photoshop CS3 extension software measures. An X-ray diffractometer (XRD, Rigaku Mini Flex II, Japan) was used to measure X-ray diffraction using Ni-filtered CuK radiation (30 kV, 15 mA) as X-ray sources to investigate the samples’ crystallographic structure. The specific surface area is determined using a specific surface area/pore distribution measuring equipment (Belsorp-mini II, Microtrack BEL Corp., Japan). The thermal behavior and amount of BeO (wt%) in the dissipation sheets were determined using a differential thermogravimetric analyzer (Shimadzu DTG-60, Japan). An Infrared (IR) spectrometer (IR Affinity-1, Shimadzu, Japan) arrayed with an attenuated total reflection (ATR) ancillary (MIRacle 10, Shimadzu, Japan) containing a diamond/ZnSe crystal was used to perform ATR-FTIR experiments at ambient temperature.
3 Results and discussion
3.1 Reson behind improved productivity of PVA/BeSO4/PEI precursor nanofibers
PVA/BeSO4 precursor nanofibers were produced when less BeSO4·4H2O aqueous solutions were mixed with PVA aqueous solutions (BeO dry ratio is less than 1%). However, PVA/BeSO4 precursor nanofibers could not produce at a higher BeO dry ratio (> 1%). The reason is that more aqueous BeSO4·4H2O solutions reduce the concentration and decrease viscosity, as shown in Fig. 2.

Effect of BeO contents on the viscosity of PVA/BeSO4 aqueous solution
The viscosity and pH of pure PVA are 484 mPa·S and 5.12, respectively. If BeSO4·4H2O salt were integrated directly with PVA aqueous solutions, the viscosity would increase to 870 mPa·S, and pH would be reduced to 2.11. Significantly lower pH (2.11) makes the PVA/BeSO4 precursor solution acidic and becomes unbalanced. Additionally, a high concentration sometimes makes the capillary block up and reduces production [16]. As a result, an insignificant amount (0.02 g) of PVA/BeSO4 precursor nanofibers would be produced for 10 h, as shown in Fig. 3a. Productivity would increase significantly (0.12 g) by mixing a small PEI (10% of the total solution) with PVA/BeSO4 precursor nanofibers shown in Fig. 3b. The possible reason is that a small amount (10% of the whole solution) of PEI on the spinning solutions increased positive charges. At the same time, increased pH (3.39) of PVA/BeSO4/PEI precursor nanofibers solution kept the solution stable. Positively charged precursor solutions extruded from the electrospinning needle to the negatively charged collector. So a significant amount of precursor nanofibers were produced. However, the productivity of PVA/BeSO4/PEI precursor nanofibers decrease for the increasing of PEI. When PEI increases up to 18% or above total solutions, the electrospinning machine can not produce nanofiber. There are two possible reasons. The first reason is a very low viscosity (198 mPa·S), and the other is excessively positive changes that make the solution out of balance. Although, the fiber shape did not change after adding PEI.

The productivity of precursor nanofibers after 10 hours (a) without PEI and (b) with PEI
Surprisingly, the viscosity of PVA/BeSO4 precursor nanofibers drops rapidly during the first 6 h before slowing down. On the other hand, the viscosity of PVA/BeSO4/PEI precursor nanofibers decreases quickly for the first 6 h but remains constant with time. The possible reason is that PVA/BeSO4/PEI precursor nanofibers reached the equilibrium stage while PVA/BeSO4 precursor solution did not. So, the viscosity of the PVA/BeSO4/PEI precursor solution is higher than PVA/BeSO4 precursor solution. The effect of time on the viscosity of PVA/BeSO4 precursor and PVA/BeSO4/PEI precursor nanofibers is shown in Fig. 4.

Relationship between time and viscosity of PVA/BeSO4 precursor and PVA/BeSO4/PEI precursor nanofibers
3.2 Effect of calcination temperatures on the crystalline structure of BeO nanofibers
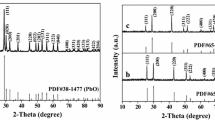
The XRD curves of BeO nanofibers obtained by calcination of the PVA/BeSO4/PEI precursor nanofibers heated at 700–1200 °C are shown in Fig. 5. Some BeO peaks with some Be compound peaks other than BeO peaks were discovered after heating to 700 °C. While heated at 800 °C and 900 °C, significant BeO peaks were found. However, a few Be compound peaks also remained at the same temperatures. The most pronounced BeO peaks were detected at 1000 °C or higher. Due to low calcination temperatures (700–900 °C), the probable mechanism is that some organic compounds will diminish but remain small. All organic compounds remove while heated at 1000 °C or above. As a result, no amorphous region while heated at 1000 °C or above; the only crystalline structure was found. The intensity of peaks also increases as the temperature grows [17].

XRD curves of BeO nanofibers ( : BeO,
: BeO,  : other than BeO compound)
: other than BeO compound)
3.3 The morphology of PVA/BeSO4/PEI precursor nanofibers and BeO nanofibers
Figure 6 shows the SEM images of PVA/BeSO4/PEI precursor and BeO nanofibers. The electrospinning method fabricated the PVA/BeSO4/PEI precursor nanofibers. On the other hand, BeO nanofibers were formed by calcination of the precursor nanofibers at 1000 °C or above temperatures. The dry ratio of (PVA/PEI) and BeO is 90/10 wt%. The productivity of PVA/BeSO4/PEI precursor nanofibers decreased due to the increased viscosity of the spinning solution. Heat treatment did not affect the precursors but diminished the average fiber diameter [17].

SEM images of nanofibers: (a) PVA/BeSO4/PEI precursor nanofibers, heated at (b) 700 °C, (c) 800 °C, (d) 900 °C, (e) 1000 °C, (f) 1100 °C, and (g) 1200 °C
Figure 7 shows fiber diameter distributions and the relationship between average fiber diameters and calcination temperatures of BeO nanofibers. The average fiber diameter of PVA/BeSO4/PEI precursor nanofibers was 396 nm. On the other hand, calcinated BeO nanofiber’s average diameters were 240 nm, 225 nm, 217 nm, 213 nm, 210 nm, and 207 nm at 700 °C, 800 °C, 900 °C, 1000 °C, 1100 °C, and 1200 °C temperatures, respectively. Thus, the average diameters are inversely proportional to the calcination temperatures, with 200–250 nm values. The following equation determines the shrinkage ratio:
where B is the apparent fabric area after calcinating at 1200 °C for 5 h and A is the evident fabric area before calcination). Before calcination, the PVA/BeSO4/PEI precursor nanofibers were square (3 cm in length and width), where the thickness of the fabric was unconsidered. The shrinkage ratio is about 60%. The possible reason is that the space between the PVA/BeSO4/PEI precursor composite became denser and the fiber length of the nanofibers became shorter due to thermal degradation of the PVA matrix.

Histograms of average fiber diameter (a) PVA/BeSO4/PEI precursor nanofibers, heated at (b) 700 °C, (c) 800 °C, (d) 900 °C, (e) 1000 °C, (f) 1100 °C, (g) 1200 °C and (h) effect of calcination temperatures on average fiber diameter of BeO nanofibers
3.4 Analysis of pore-size distribution curves and adsorption isotherms
The calcination temperature can influence the surface area and morphologies of fibers, tuning the crystalline structure. The nitrogen adsorption isotherms and pore-size distributions of PVA/BeSO4/PEI precursor nanofibers and calcined BeO nanofibers at 700–1200 °C are shown in Fig. 8. The micropores and mesopores gradually decreased with the increase in heating temperatures.

Nitrogen adsorption isotherms and pore-size distribution curves of BeO nanofibers at each temperatures
The relationship between the pore characteristics (the specific surface area and the pore volume) of the BeO nanofibers is shown in Fig. 9. The PVA/BeSO4/PEI precursor nanofibers’ specific surface area is 36.3 m2 g−1 and increases significantly to 122 m2 g−1 for BeO nanofibers while calcinated at 700 °C. However, the specific surface area gradually reduced with rising heating temperatures, and at 1200 °C, the specific surface area decreased to 11.9 m2 g−1. The results show that removing organics improves specific surface areas at appropriate calcination temperatures. A high calcination temperature (above 1000 °C) inhibits the increase in specific surface area, resulting in a low specific surface area. This could be due to sintering or metal particle blockage. As a result, heating PVA/BeSO4/PEI precursor nanofibers to 1200 °C resulted in mesoporous BeO nanofibers. However, BeO nanofibers samples became brittle when heat-treated at 1100 °C and 1200 °C temperatures [18].

Effect of calcination temperatures upon pore characteristics of BeO nanofibers
Figure 10 shows the relationship between calcination temperatures and the average pore diameter of the BeO nanofibers. Heated at 700–1200 °C temperatures, all average pore diameters of BeO nanofibers range from 2 to 50 nm, indicating a mesoporous structure.

Effect of calcination temperatures upon average pore diameter of BeO nanofibers
3.5 Thermal decomposition of the pure PVA, PVA/BeSO4/PEI precursor nanofibers, and the BeSO4·4H2O salt
Thermogravimetric (TG) curves of pure PVA, PVA/BeSO4/PEI precursor nanofibers, and BeSO4·4H2O salt are shown in Fig. 11. As can be seen from curve (a) in Fig. 11, the decomposition of pure PVA was a multi-step process. At about 290 °C, no significant weight loss occurred, but a consequential weight loss (about 83%) at 290–500 °C. However, a weight loss of nearly 100% has happened after 500–600 °C temperature, and it occurred due to organic PVA decomposing at shallow temperatures. Curve (b) in Fig. 11 shows PVA/BeSO4/PEI precursor nanofibers decomposition curve is simpler than pure PVA curve (a). Roughly 10% weight residues (BeO contents in the precursor nanofibers) at 600 °C reflect the BeSO4 contents in the spinning solutions. The BeSO4·4H2O salt decomposition was a multi-step process, as seen in curve (c) in Fig. 11. At temperatures below 304 °C, planar and crystal water departed, resulting in a mass loss of around 40.3%. From 300 to 600 °C, the mass did not change, indicating that the salt is stable in the BeSO4 form. Finally, from 700 to 840 °C, [5] the curve dropped sharply, which was decomposed entirely at about 840 °C, and the total weight loss was 85.7% at the same temperatures.

Thermogravimetric curves under airflow at a heating rate of 10 °C/min for (a) pure PVA, (b) PVA/BeSO4/PEI precursor nanofibers, and (c) BeSO4·4H2O salt
3.6 FTIR of the pure PVA, PVA/BeSO4/PEI precursor nanofibers, BeSO4·4H2O salt, and the BeO nanofibers
FTIR was used to investigate the chemical structures of BeO nanofibers. In contrast, pure PVA, BeSO4.4H2O salt, and PVA/BeSO4/PEI precursor nanofibers were used as controls (Fig. 12). C–H rocking of pure PVA has been assigned to the absorption peak at 853 cm−1 (Fig. 12a) which is shifted to 823 cm−1 for PVA/BeSO4/PEI precursor nanofibers (Fig. 12c) [19]. The C–O stretching of acetyl groups on the PVA backbone corresponds to the band at about 1098 cm−1. This peak is shifted to 1068 cm−1 and 1078 cm−1 for BeSO4·4H2O salt and PVA/BeSO4/PEI precursor nanofibers. In the complexed system, CH2 bending exhibited at 1452 cm−1 in pure PVA is shifted to higher wavenumbers (1430 cm−1) for precursor nanofibers. Broadband at around 3200–3500 cm−1 is attributed to the stretching vibration of a strong O–H bond of pure PVA BeSO4·4H2O salt and PVA/BeSO4/PEI precursor is displaced. The main reason is the hydrogen bonds of the water contained in the sample. In the complexed system, the C=O stretching of pure PVA [20] is observed to be steered to higher wavenumbers. For instance, in BeSO4·4H2O salt and PVA/BeSO4/PEI precursor, the C=O peak is shifted to 1651 cm−1 and 1734 cm−1 from 1736 cm−1. This clearly shows that the interaction between the salt and PVA arises from O–H groups and C–O groups of pure PVA. In addition, it is considered that, like other inorganic metal oxide compounds, the BeO bending vibration was found below 1000 cm−1 [21,22,23,24,25,26,27]. It is assumed that BeO’s sharp peak was found at around 889 cm−1. The possible reason is that water molecules are eliminated at higher calcinating temperatures (1100 °C) [28]. On the other hand, the low peak is ʋ (Be–O) vibrations at 1500 cm−1 [29].

FTIR spectra of (a) pure PVA, (b) BeSO4·4H2O salt, (c) PVA/BeSO4/PEI precursor nanofibers, and (d) BeO nanofibers
4 Conclusion
The authors have successfully synthesized mesoporous BeO nanofibers for the first time in the two steps. Firstly, PVA/BeSO4/PEI precursor nanofibers were formed by electrospinning using a PVA aqueous solution mixing with BeSO4·4H2O salt and PEI. The productivity was increased by adding PEI with PVA/BeSO4 spinning solution. Secondly, high calcination temperatures (1000–1200 °C) were applied to PVA/BeSO4/PEI precursor nanofibers to make BeO nanofibers. The calcination temperatures reduce the diameter of the BeO nanofibers. The calcination temperatures also impacted the specific surface area and average pore area. The specific surface area of PVA/BeSO4/PEI precursor nanofibers (36.3 m2 g−1) increased to 122 m2 g−1 for BeO nanofibers while calcined at 700 °C. This indicates that removing organics from PVA/BeSO4/PEI precursor nanofibers is beneficial to improving the low specific surface area of BeO nanofibers calcined at 700 °C. On the contrary, a very high calcination temperature (1200 °C) will decrease specific surface area (11.9 m2 g−1) due to sintering or blocking by metal particles. Due to the sintering, the pore volume of the BeO nanofibers decreased with increased calcination temperatures. Mesoporous BeO nanofibers were obtained by calcination of the PVA/BeSO4/PEI precursor nanofibers at 1200 °C. However, BeO nanofibers samples became brittle when heat-treated at 1100 °C and 1200 °C. Thus, the BeO nanofibers prepared under this optimum condition will be valuable for further development and are expected to have industrial applications such as thermal conductive heat dissipate sheets.
References
Nabihah MF, Shanmugan S (2016) Structural parameter and tem analysis of beryllium oxide nanoparticles synthesized by polyacrylamide gel route. Dig J Nanomater Biostruct (DJNB) 11:349–356
Wang L, Liu L, Chen J et al (2020) Synthesis of honeycomb-structured beryllium oxide via graphene liquid cells. Angew Chem Int Ed 59:15734–15740. https://doi.org/10.1002/anie.202007244
Kiiko VS, Vaispapir VYa (2015) Thermal conductivity and prospects for application of BeO ceramic in electronics. Glass Ceram 71:387–391. https://doi.org/10.1007/s10717-015-9694-6
Akishin GP, Turnaev SK, Vaispapir VYa et al (2009) Thermal conductivity of beryllium oxide ceramic. Refract Ind Ceram 50:465–468. https://doi.org/10.1007/s11148-010-9239-z
Wang X, Wang R, Peng C et al (2010) Synthesis and sintering of beryllium oxide nanoparticles. Prog Nat Sci Mater Int 20:81–86
Selvaraj V, Morri B, Nair LM, Krishnan H (2019) Experimental investigation on the thermophysical properties of beryllium oxide-based nanofluid and nano-enhanced phase change material. J Therm Anal Calorim 137:1527–1536. https://doi.org/10.1007/s10973-019-08042-w
Buchko CJ, Chen LC, Shen Y, Martin DC (1999) Processing and microstructural characterization of porous biocompatible protein polymer thin films. Polymer 40:7397–7407
Doshi J, Reneker DH (1995) Electrospinning process and applications of electrospun fibers. J Electrostat 35:151–160
Ohgoshi A, Takahashi K, Nakane K (2019) Polymer/magnesia nanofiber composite sheets with anisotropic high thermal conductivity. J Mater Sci Mater Electron 30:20566–20573
Nakane K, Seto M, Irie S et al (2011) Alumina nanofibers obtained from poly(vinyl alcohol)/boehmite nanocomposites. J Appl Polym Sci 121:1774–1779. https://doi.org/10.1002/app.33319
Nakane K, Shimada N, Ogihara T et al (2007) Formation of TiO2 nanotubes by thermal decomposition of poly(vinyl alcohol)-titanium alkoxide hybrid nanofibers. J Mater Sci 42:4031–4035. https://doi.org/10.1007/s10853-006-0241-2
Hwang HJ, Barakat NA, Kanjwal MA et al (2010) Boron nitride nanofibers by the electrospinning technique. Macromol Res 18:551–557
Deitzel JM, Kleinmeyer J, Harris DEA, Tan NB (2001) The effect of processing variables on the morphology of electrospun nanofibers and textiles. Polymer 42:261–272
Lu JY, Norman C, Abboud KA, Ison A (2001) Crystal engineering of an inclusion coordination polymer with cationic pocket-like structure and its property to form metal–organic nanofibers. Inorg Chem Commun 4:459–461
Lee KH, Kim HY, Ryu YJ et al (2003) Mechanical behavior of electrospun fiber mats of poly(vinyl chloride)/polyurethane polyblends. J Polym Sci B 41:1256–1262
Rwei S-P, Huang C-C (2012) Electrospinning PVA solution-rheology and morphology analyses. Fibers Polym 13:44–50
Hossain MS, Nakane K (2022) Development of a high-performance heat dissipation sheet with three-dimensional alumina fibrous structure fabricated by freeze-drying. Results Mater 13:100241
Yang H, Sugita N, Nakane K (2019) Factors influencing the PVA polymer-assisted freeze-drying synthesis of Al2O3 nanofibers. Ceram Int 45:16731–16739. https://doi.org/10.1016/j.ceramint.2019.05.190
Liebenow C (1998) A novel type of magnesium ion conducting polymer electrolyte. Electrochim Acta 43:1253–1256. https://doi.org/10.1016/S0013-4686(97)10026-3
Karthikeyan A, Vinatier P, Levasseur A (2000) Study of lithium glassy solid electrolyte/electrode interface by impedance analysis. Bull Mater Sci 23:179–183. https://doi.org/10.1007/BF02719906
Alexander S, Morrow L, Lord AM et al (2015) pH-responsive octylamine coupling modification of carboxylated aluminium oxide surfaces. J Mater Chem A 3:10052–10059. https://doi.org/10.1039/C5TA01294E
Habte L, Shiferaw N, Mulatu D et al (2019) Synthesis of nano-calcium oxide from waste eggshell by sol–gel method. Sustainability 11:3196. https://doi.org/10.3390/su11113196
Department of Chemistry, Faculty of Mathematics and Natural Sciences, Sriwijaya University, Mohadi R, Sueb A et al (2018) Calcium oxide catalyst based on quail eggshell for biodiesel synthesis from waste palm oil. J Pure Appl Chem Res 7:129–138. https://doi.org/10.21776/ub.jpacr.2018.007.02.390
Shatan AB, Venclíková K, Zasońska BA et al (2019) Antibacterial silver-conjugated magnetic nanoparticles: design, synthesis and bactericidal effect. Pharm Res 36:147. https://doi.org/10.1007/s11095-019-2680-x
Kandiban M, Vigneshwaran P. Potheher IV synthesis and characterization of MgO nanoparticles for photocatalytic applications. 5
Liang Y, Ouyang J, Wang H et al (2012) Synthesis and characterization of core–shell structured SiO2@YVO4:Yb3+,Er3+ microspheres. Appl Surf Sci 258:3689–3694. https://doi.org/10.1016/j.apsusc.2011.12.006
Saravanan S, Dubey RS. Synthesis of SiO2 nanoparticles by sol–gel method and their optical and structural properties. 9
Shahrokh Abadi MH (2015) Effects of annealing temperature on infrared spectra of SiO2 extracted from rice husk. J Ceram Sci Technol. https://doi.org/10.4416/JCST2014-00028
Altunal V (2016) BeO OSL dosimeter: its synthesis and luminescence mechanisms. https://doi.org/10.13140/RG.2.2.12449.84329
Acknowledgements
Md. Shakhawat Hossain wishes to express his gratitude to the Japanese government for awarding the MONBUKAGAKUSHO scholarship, which has enabled him to pursue his Ph.D.
Funding
The authors state that no financial incentives were received for this study, and all expenditures were self-funded.
Author information
Authors and Affiliations
Contributions
MSH: Data curation, Writing—original draft, Investigation, Software, Methodology, Visualization. KN: Design the research, Conceptualization, and Supervision Resources, Formal analysis, Writing-review & editing.
Corresponding author
Ethics declarations
Competing interests
The authors state that they have no known competing financial interests or personal ties that would have influenced the work presented in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hossain, M.S., Nakane, K. Formation of beryllium oxide nanofibers by polyvinyl alcohol/beryllium sulfate/polyethyleneimine composite precursors. SN Appl. Sci. 4, 325 (2022). https://doi.org/10.1007/s42452-022-05135-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-022-05135-4