
Abstract
Compared to fullerene based electron acceptors, n-type organic semiconductors, so-called non-fullerene acceptors (NFAs), possess some distinct advantages, such as readily tuning of optical absorption and electronic energy levels, strong absorption in the visible region and good morphological stability for flexible electronic devices. The design and synthesis of new NFAs have enabled the power conversion efficiencies (PCEs) of organic photovoltaic (OPV) devices to increase to around 19%. This review summarises the important breakthroughs that have contributed to this progress, focusing on three classes of NFAs, i.e. perylene diimide (PDI), diketopyrrolopyrrole (DPP) and acceptor–donor–acceptor (A-D-A) based NFAs. Specifically, the PCEs of PDI, DPP, and A-D-A series based non-fullerene OPVs have been reported up to 11%, 13% and 19%, respectively. Structure–property relationships of representative NFAs and their impact on OPV performances are discussed. Finally, we consider the remaining challenges and promising directions for achieving high-performing NFAs.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Organic photovoltaic cells (OPVs) use blends of carbon-based materials to produce electricity from sunlight by the photovoltaic effect and are a prospectively scalable approach to harvest solar energy. Compared to traditional silicon based cells, OPVs have the potential advantages of lower cost, lighter weight, flexibility and large area fabrication [1, 2]. Research into OPVs has raised intensive academic and industrial interest over the past two decades and has resulted in cell power conversion efficiency (PCEs) improving to over 19% [3].
The photoactive layer of an OPV cell generally consists of a p-type organic semiconductor as the electron donor and an n-type counterpart as the electron acceptor. Fullerenes, especially PC61BM and PC71BM, due to their excellent electron affinity and charge transporting ability, have served as the dominate acceptor materials for a long period [4]. The PCEs of fullerene based OPVs have exceeded 10% based on blends of narrow bandgap polymer donors and fullerene acceptors for single-junction OPV devices [5,6,7,8,9,10]. However, fullerenes have some inherent shortcomings, such as high synthesis costs, narrow and weak absorption in the visible region, limited electronic variability in their energy levels and spectral absorption, and heating induced morphological instability. Therefore, there was a practical need to develop alternative, so-called non-fullerene acceptors (NFAs) [11].
To date, the PCEs of non-fullerene OPVs have improved rapidly and new efficiency records are continually reported due to the structural innovations of NFAs [12, 13]. Without doubt, the photovoltaic performances of NFAs have outperformed fullerene acceptors in the OPV field. Perylene diimide (PDI), diketopyrrolopyrrole (DPP), and acceptor–donor–acceptor (A-D-A) series have been the most successful and well-studied NFAs (Fig. 1). The PCEs of PDI, DPP, and A-D-A based non-fullerene OPVs have been reported over 11% [14, 15], 13% [16] and 19% [3], respectively. Here, we focus our discussion on representative molecules with high PCEs from these three classes and finish with a brief discussion of the remaining challenges and future prospects of NFAs.

Chemical structures of fullerenes (PC61BM and PC71BM) and representative non-fullerene acceptors from the PDI, DPP and A-D-A series (ITIC and Y6)
2 Perylene diimide (PDI) based NFAs
PDI possesses relatively high electron affinity and mobility, tunable energy levels, and excellent chemical, thermal and photochemical stabilities, making it amongst the earliest and most widely investigated NFA in OPVs [17,18,19]. The first PDI based acceptor was reported by Tang and coworkers in 1986, based on a bilayer heterojunction OPVs [17]. Until now the best PCE of OPV devices based on a PDI monomer is only 3.7% [20,21,22], lower than that of fullerene based OPVs. This is because PDI tends to readily form unfavorable micrometer scale crystallinity, due to its intrinsic molecular planarity and strong intermolecular forces. The undesirable large crystalline domains reduce exciton splitting in the polymer blend, decrease the photocurrent, and finally result in poor device performance. Hence studies have focused on functionalizing the backbone modifiable positions (a1) to reduce molecule crystallinity, but the results are unsatisfying. Disrupting the molecular crystallinity without adversely compromising its electron transporting abilities is the main challenge for developing new PDI NFAs. Twisted and star-shaped PDIs have been successfully proposed, which have demonstrated their potential for high-performance NFAs [23].
2.1 Twisted PDI dimers
Narayan and coworkers designed a PDI dimer acceptor a2a by introducing a twisted central N–N single bond as a linker to connect two PDI monomers (Fig. 2, Table 1) [24]. As the four imide oxygen atoms carry partial negative charges, the perylene units tended to achieve a perpendicular orientation to each other to minimize their electronic repulsion, which significantly disrupted the planarity of the perylene backbones. When blended with a polymer donor PBDTTT-C-T, a promising PCE of 3.2% was obtained, attributed to the weakened aggregation and increased short circuit current (JSC) density. Larger alkyl groups on the imide nitrogen atoms can increase the solubility of NFAs and orient molecular stacking spatially, however they can adversely affect the intermolecular charge mobility and to some extent decrease device efficiencies. To investigate the side chain effects, a2b was synthesized with slightly shorter alkyl chain [25]. OPVs based on PBDTTT-C-T:a2b delivered a PCE of 3.78%, higher than that of PBDTTT-C-T:a2a based OPVs. When blending a polymer donor PBDT-TS1 with a2b, a high PCE of 5.58% was realized, benefiting from a more balanced hole and electron mobility.

Chemical structures of twisted and star-shaped PDI based NFAs
Except from modifying alkyl substitutions and using hydrazine as a linker on imide nitrogen atoms to decrease molecule planarity and restrict the tendency of crystallization, one effective way of twisting PDI is through dimerization on its bay position. Using a simple thiophene as the bridge, a PDI dimer a3 was designed and synthesized [26]. When blended with PBDTTT-C-T, the efficiency of single-junction OPV devices improved to 6.1% after solvent annealing, significantly enhanced with respect to a value of 1.44% without such treatment [27, 28]. Wang and coworkers reported a bay-linked perylene bisimide dimer a4a, in which each perylene was bridged with a selenium atom [29]. The molecular structure of a4a was largely twisted owing to the selenium bridge, due to the reduced flexibility and increased steric repulsion between the two PDI units. A high PCE of 8.42% was achieved, higher than its sulfur counterpart based OPVs (7.16%) [30]. Compared to sulfur, selenium had a larger and more polarisable electron cloud and an extra empty orbital, which was proposed to improve orbital overlap increasing the charge carrier mobility and electron-accepting ability.
Jen and coworkers developed three twisted PDI dimer derivatives through the oxidative cyclization of oxygen/thiophene/selenium-bridged PDI dimers [31]. By density functional theory (DFT) calculations, these fused structures significantly enhanced the overall planarity, compared with the previously reported semi-flexible thiophene bridged PDI acceptors [26, 32]. With the increased chalcogen size, the twist angles between two PDI subunits enlarged gradually. Compared with furan (a5a) or selenophene (a5c), thiophene incorporated fused PDI dimer a5b had a modestly twisted structure to modulate the paradox between enhanced π-π stacking for improving charge mobility and nonplanar structure for suppressing large aggregation formation. A highest PCE of 6.72% was obtained for PTB7-Th:a5b based OPVs, higher than those of a5a and a5c based devices.
Cyano substituents can increase the electron affinity and help to promote efficient electron transport and have therefore been utilized in NFAs. Cyano moieties can also induce molecular torsion and therefore realizing balanced aggregation with efficient charge transport in cyano substituted structures remains challenging. Molecule a6 was designed and synthesized through cracking the PDI backbone and incorporating cyano units [33]. OPVs based on P3HT:a6 showed an initial PCE of 2.55%. By blending a6 with a semi-crystalline low-bandgap donor PPDT2FBT, a remarkably high VOC of 1.03 V and a maximum efficiency of 7.6% were achieved, due to their complementary absorption and well-intermixed crystalline blend film morphology [34].
To compare the effectiveness of ortho-position and bay-position functionalization, Yu and coworkers synthesized a series of PDI dimers using benzo[1,2-b:4,5-b’]dithiophene (BDT) as a bridge [35]. Compared with the molecule with a torsion linker on the bay-position (β-isomers), ortho-position functionalized PDIs (α-isomers) showed blue-shifted absorption and better planarity. The α-isomer a7 based OPVs obtained the highest PCE of 4.92% due to the higher charge transport and more efficient charge separation when blended with polymer donor PTB7-Th.
A helical PDI NFA a8 was reported by Nuckolls and coworkers, by fusing two PDI subunits with a two-carbon bridge [36]. The best efficiencies based on PTB7:a8 and PBDTT-TT:a8 were 5.21% and 6.05% respectively, due to the blend films of PBDTT-TT:a8 having broader absorption, better matched energy levels and higher electron mobility. Subsequently, two helical conjugated PDI oligomers with mesh-like network were synthesized and the efficiencies were further improved up to 8.3% [37].
Meanwhile, other research groups have prepared a variety of twisted PDI analogues. For example, Jenekhe and coworkers reported a imide dimer derivative a9 by rationally twisting the tetraazabenzodifluoranthene diimide [38] with a 3,4-ethylenedioxythiophene bridge [39]. Compared to their planar subunits, such a twisted bulky structure improved morphological compatibility with the polymer donor and hence enhanced charge separation. A high PCE of 8.1% and a VOC of 0.93 V were obtained in the optimized inverted PSEHTT:a9 devices, suggesting that fine tuning the twisted framework was a rational design for PDI NFAs to obtain high performance in OPVs.
2.2 Star-shaped PDI oligomers
In order to further suppress the self-aggregation of planar PDI monomers, star-shaped molecules with pseudo-3D/3D structures have been introduced to design PDI based NFAs. This strategy can not only facilitate nanoscale phase domain formation with donor materials, but also enhance isotropic charge transport, due to the similar geometry with fullerenes [40]. In recent years, many studies about star-shaped PDI derivatives have been reported and exciting record efficiencies have been achieved.
In 2012, Zhan and coworkers reported the nonplanar star-shaped PDI trimer b1 with triphenylamine as the core and three PDI monomers as the propellers (Fig. 2, Table 1) [41]. This quasi-3D nonplanar structure facilitated weak intermolecular interactions and molecular aggregation, resulting in isotropic optical and charge-transporting properties. Solution-processed OPVs based on the PBDTTT-C-T:b1 blend exhibited maximum PCEs as high as 3.32%, which demonstrated a star-shaped molecular structure could be applied to develop PDI NFAs for OPVs.
Similar to a4a and a5c, a highly fused star-shaped NFA b2 was developed, based on three-bladed PDI propellers [42]. Molecule b2 displayed an extremely distorted configuration whilst the three PDI subunits showed strong π-π intermolecular interactions with other PDI subunits from neighboring molecules. The integration of selenophene endowed b2 with a more distorted spatial configuration and a more compact intermolecular network assembly due to the Se···O interactions. When using PDBT-T1 as the donor, a high efficiency of 9.28% was yielded, higher than that of the contrasting acceptor without selenophene (8.28%). In recent years, integration of nitrogen on the PDI bay region, so-called N-annulation, has become another effective strategy to develop new PDI based NFAs [43]. Similar to Se-annulated PDI acceptors, N-annulation can modulate the molecular optoelectronic properties and enable a higher LUMO level for a larger VOC. More importantly, N-annulated PDI based NFAs provide an additional grafting site for functionalization. Therefore, solvent solubility and thin-film morphology can be finely tuned to obtain high performance OPVs.
Yan and coworkers systematically studied the influence of twisting angles of star-shaped molecules on the photovoltaic performance by developing three star-shaped PDI NFAs using tetraphenylmethane (b3a), tetraphenylethylene (b3b) and tetraphenylpyrazine (b3c) as the core, flanked with four PDI monomers [44,45,46]. The calculated twisting angle between the phenyl rings and double bond in compound b3b was 55° and the twisting extent between the pyrazine unit and the phenyl groups in compound b3c was reduced to 40°, due to the larger size of pyrazine. Moreover, the dihedral angle between the pyrazine ring and PDI was only 25°, indicating the four PDI subunits laid nearly parallel. The lower twisting angles of b3c were concluded to contribute to its higher aggregation tendency and electron mobility. Importantly, the domain size of the active layer was more optimal when blended with a polymer donor PffBT-T3(1,2)-2, yielding an impressive PCE of 7.1%, higher than the non-fullerene acceptors based on b3a (4.7%) and b3b (6.0%).
Yu and coworkers have demonstrated that the α-substituted PDI dimer (a7) could show higher planarity and superior photovoltaic performance over its β-substituted counterpart. Subsequently, they developed a cross-like oligomer b4, based on coplanar π-conjugated BDT-Th as the core and four α-functionalized PDI subunits as the arms [47]. A high JSC of 18.3 mA cm−2 and maximum PCE of 8.47% were obtained when blended with PTB7-Th, comparable with those of PTB7-Th:PC71BM based solar cells. This work demonstrated that, compared to PDI dimers, twisted star-shaped PDI NFA could improve the morphological compatibility with the donor polymers, leading to enhanced photovoltaic performance.
Compound b5 was designed and synthesized based on a spiro-bifluorene core connected with four PDI building blocks, which effectively suppressed molecular aggregation and facilitated excitation energy transfer among PDI subunits [48]. A 3D interlocking geometry was exhibited in b5 between the central spiro-bifluorene core and four PDI subunits and this dependence was suggested to afford conformational uniformity and structural rigidity of compound b5. Choosing polymer PV4T2FBT as the donor, the PCEs of OPVs based on PV4T2FBT:b5 were up to 5.98%, as a result of the combination of matched electronic and optical properties and well-mixed interpenetrating morphology.
He and coworkers designed and synthesized two novel star-shaped PDI tetramers (b6) based on a tetrathienylethene core, with non-fused and fused PDI structures [49]. These two NFAs possessed similar energy levels but very different UV–Vis absorptions, with the fused b6 showing strong broad-band absorption with multiple sharp peaks in the 300–600 nm region, which contributed more to the charge generation and JSC and led to higher OPV efficiency (6.6%). The broad absorption spectrum in the fused b6 was explained by time-dependent DFT calculations, and arose from the combination of multiple bright transitions in the visible region with a strong vibronic progression. PDI based NFAs are wide bandgap materials, so donor materials with narrow optical bandgap and proper energetics are essential to match these acceptors.
3 Diketopyrrolopyrrole (DPP) based NFAs
DPPs are a classic and chemically modifiable dye with strong visible region absorption and photochemical stability. Due to high backbone planarity and strong intermolecular π-π stacking, DPPs have been used to build a variety of intermolecular charge transfer systems. DPP molecules possess strong electron affinity, good electron mobility and low-lying LUMO energy levels [50,51,52]. Therefore, DPP derivatives have been designed and investigated as NFAs for efficient OPVs.

Chemical structures of DPP based NFAs
3.1 Twisted DPP dimers
Compound c1 was developed as an early example of a NFA based on a dibenzosilole core encapped with two DPPs (Fig. 3, Table 1) [53]. The LUMO energy level (− 3.28 eV) and optical absorption range (broad absorption of 300–700 nm) of c1 matched well with the donor P3HT. A best PCE of 2.05% based on P3HT:c1 blended films was achieved, due to improved charge transport after solvent annealing, which was useful to enhance JSC in OPVs (4.91 mA cm−2 > 3.05 mA cm−2). This PCE value was among the highest reported values at that time, indicating DPP was a promising unit to construct NFAs.
Compound c2 was designed by connecting a DPP core with two electron-deficient dicyano substituted electron-rich triphenylamine [54]. With this push–pull structure, c2 possessed high electron-accepting ability and proper energy levels as a NFA. This push–pull structure also extended the absorption spectra toward the near-infrared region. A maximum PCE of 4.95% was achieved along with an improved JSC of 10.28 mA cm−2 after a solvent annealing treatment.
Bhosale and coworkers developed a DPP dimer acceptor c3 based on carbazole as the central core [55]. The calculated torsional angle between the thiophene planes of DPP units and the carbazole core was 25.8°, indicating c3 was spatially a nonplanar structure. When blending c3 with P3HT to form the active layers, a very high VOC of 1.17 V and a maximum PCE of 2.30% were afforded. Chen and coworkers developed a bipolar twisted DPP dimer c4, in which a fluorene ring functioned as the core and two DPP units encapped with benzene ring as the arms [56]. It showed intense absorption in the visible light region of 550–700 nm, complementing well with that of P3HT (400–600 nm), which was favorable to achieve a high JSC. The offset between the LUMO (− 3.39 eV) of c4 and the HOMO (− 4.76 eV) of P3HT, as well as that between the HOMO (− 5.21 eV) of c4 and the LUMO (− 4.0 eV) of PC71BM were both large, resulting in a high VOC. The blended films based on P3HT:c4 and c4:PC71BM gave champion PCEs of 3.17% and 3.26% respectively.
Two benzothiadiazole flanked DPP dimers c5a and c5b, with or without fluorine substitutions, were reported as non-fullerene acceptors [57]. The coplanar backbone structure with extended conjugation length provided good electron transport. Both c5a and c5b showed a low bandgap of 1.50 eV and energy levels well matched with polymer donor PTB7. By incorporating fluorine on the central benzothiadiazole core, both the HOMO and LUMO energy levels were lowered, resulting in a lower VOC of PTB7:a5b than that of PTB7:a5b. OPVs based on PTB7:a5a and PTB7:a5b exhibited the highest PCEs of 3.03% and 5.00% after optimization employing chloronaphthalene (CN) as an additive. The JSC of PTB7:a5b (12.10 mA cm−2) was much higher than that of PTB7:a5a (7.77 mA cm−2), due to the larger photoresponse of the a5b based devices.
Cabanetos and coworkers designed compound c6 by grafting acetylene-substituted phthalimides onto a central DPP core [58]. By DFT calculations, c6 exhibited a quasi-planar structure with a torsion angle of just 0.04° between the phthalimide block and the central core. Besides, c6 possessed broad absorption in the range of 500–750 nm and energy levels well matched with polymer donor P3HT. The maximum PCE of inverted BHJ OPVs based on P3HT:c6 was achieved 3.28%.
3.2 Star-shaped DPP oligomers
Zhan and coworkers reported a star-shaped acceptor d1 based on TPA as the central core and DPP as the flanking arms (Fig. 3, Table 1), with a strong absorption in the 500–700 nm range [59]. Single-junction OPVs based on P3HT:d1 showed a high VOC up to 1.18 V, due to the large difference between the LUMO of d1 (− 3.26 eV) and the HOMO of P3HT (− 4.76 eV). Along with this high VOC, a PCE of 1.2% was reached, demonstrating that star-shaped DPP oligomers might be promising alternatives to fullerene.
Through installing a spirobifluorene core with four benzene endcapped DPP arms, d2 was designed and synthesized as a potential electron acceptor [60]. Due to its cruciform molecular configuration, strong intermolecular aggregation was suppressed and active layers based on P3HT:d2 exhibited a well intermixed microstructure. A maximum PCE of 5.16% along with a high VOC of 1.14 V was achieved owing to the well-matched energy levels of the donor and acceptors and fine phase separation of the blended films. These values were higher than those of the P3HT: PC61BM blended films (3.18%, 0.62 eV) and the P3HT:d2 devices showed much better thermal stability. Using a central phenyl moiety flanked with three DPP moieties (d3) has also been investigated [61]. The dihedral angle between the core and the DPP subunits was calculated to be about 25° and the triangular non-planar structure reduced the crystallinity compared to linear DPP derivatives. However the PCE of the P3HT:d3 OPVs fabricated on the ITO-coated glass substrates with spin-coating process was only 0.31% while a higher PCE of 0.54% was obtained when fabricating roll coated, large area, ITO- and vacuum-free flexible OPVs. Replacing the phenyl core with TPE and four DPP units afforded d4 whose star-shaped molecular arrangement was crucial to prevent strong intermolecular aggregation whilst sustaining an elongated conjugation length [62]. It showed a small domain size of 2.5 nm when blended with P3HT, which was favorable for exciton dissociation. Owing to the matched energy levels and the well interlaced blend film morphology, a maximum PCE of 3.86% with a high VOC of 1.18 V was obtained.
Zhang and coworkers designed a conjugated oligomer d5 based on a [2,2]paracyclophane core with four DPP moieties as flanks [63]. This molecular scaffold possessed suitable lower energy levels and good electron accepting abilities. A PCE of 2.69% was obtained from P3HT:d5 blended films after thermal annealing treatment, which afforded deceased domain size and increased thin film smoothness compared to films without any treatments.
A star-shaped DPP oligomer d6, containing a benzodithiophene core and four diketopyrrolopyrrole arms was designed and synthesized by He and coworkers [64]. This molecule showed good thermal stability, a high-lying LUMO level and strong aggregation behaviour from thin film absorption measurements complemented by computational studies. The utility of the molecule was demonstrated in both solar cell and field-effect transistor devices. In the former, d6 displayed low efficiency when used as an acceptor in blends with P3HT but exhibited promising performance as a donor, in blends with either a fullerene or a non-fullerene acceptor. In field-effect transistors d6 exhibited typical p-type transistor behavior, which was in accordance with its better donor performance in solar cell devices. Therefore, core selection is important to regulate the energy levels and charger transport properties of the DPP oligomers. For DPP based NFAs, donor materials with high-lying LOMO levels (e.g. P3HT) are needed to get ideal device performance.
4 A-D-A-type NFAs
One approach to NFA’s that has proven very successful is that of A-D-A type acceptors. A-D-A-type NFAs consist of an electron-rich core (D) flanked with two electron-deficient terminals (A). The D and A parts can be modified separately to fine-tune the energy levels, bandgap, molecular packing, and other properties. ITIC (e1) [65], as reported by the Zhan group, was one of the first examples, and more recently high performing Y6 (f1) [66] are typical A-D-A type NFAs, which consist of a fused ladder-type arene as the backbone and electron-withdrawing units as the flanking arm. These molecules exhibit high charge mobility due to their planar skeletal structures and ordered π-π stacking and enhanced and extended optical absorption to NIR region induced by push–pull effect. As shown in Fig. 4, the strategies for developing new A-D-A type NFAs include the design of electron-donating cores, electron-withdrawing end groups, soluble sidechains and π-conjugated spacers.

Structural modification strategies for A-D-A-type NFAs
Similar to PDIs and DPPs, each part of A-D-A type NFAs can be easily and independently modified, which affords numerous structural possibilities. As shown in Fig. 4, these can be characterized according to their principal changes:
-
1.
Backbone modification. Extension of the electron-rich backbones can red-shift and broaden the absorption of A-D-A type NFAs for better light-absorbing capability [67]. Replacing the sp3-hybridized carbon bridge in the fused heterocyclic or introducing different chalcogen atoms, commonly selenium, can influence the fundamental electronic properties and molecular packing on the resultant NFAs [68]. Symmetric and asymmetric extension can also influence the electronic properties as asymmetric functionalisation modulates the molecular properties in a stepwise manner and increases the dipole moment [69]. Introducing hydrogen bonding can restrain the backbone into a relatively rigid and planar structure and, at the same time, reduce the synthetic complexity [70]. Introducing banana shaped cores can help to facilitate multi-dimensional charge transport which is beneficial for higher JSC and FF [71, 72].
-
2.
End group modification. End groups will affect the optical and electrochemical properties of NFAs dependent on their electron-withdrawing ability. The selection of different end groups is important for designing new NFAs with different optical bandgap for specific applications. End groups with weak electron-withdrawing capability include 3-ethylrhodanin (BR) and N,N'-diethyl-2-thiobarbituric acid (TA), and those with strong electron-withdrawing capability are 3-(1,1-dicyanomethylene)-1-indanone (IC) and its derivatives.
-
3.
Side chain modification. Although side chains are usually considered to provide solubility and have minor influence on the electronic properties, they are crucial to regulate molecule packing and intermolecular interactions. Modification of the side chains from bulky phenylalkyl to simple linear alkyl chains could potentially improve the packing ability and the charge transport mobility [73].
-
4.
Spacer modification. Inserting π-conjugated spacers between electron-rich backbone and end groups can further alter the electronic properties of A-D-A type NFAs. These spacers, with either electron-rich or electron-deficient capabilities, can enlarge effective conjugation length, leading to more red-shifted absorption and smaller bandgap.
In this review, we discuss several representative A-D-A-type NFAs (Figs. 5 and 6, Table 1). ITIC (e1) [65] was a seminal material, one of the first to outperform the established fullerene acceptors and it aroused wide interest from the scientific community. Similarly the high efficiency reported for the first Y6 (f1) derivatives [66] prompted much interest. Y6 is a similar A-D-A type material in which the donor core contains an additional electron-deficient benzothiadiazole unit resulting in a banana shaped material. The high reported PCE of 15.7% was unprecedented at that time, and induced another wave in the OPV research and can be classified as A-DA’D-A type NFAs for clarification.

Chemical structures of A-D-A-type NFAs

Chemical structures of A-DA’D-A-type NFAs
Since the molecule e1, also well known as ITIC [65], was reported by Zhan and coworkers, indacenodithiophene (IDT) derivatives have attracted great attention in non-fullerene OPVs. Tremendous endeavors have been dedicated to manipulating the aromatic core, the bridge, the end-capped electron deficient groups, and the sidechains in order to fine tune the optical and electrochemical properties. e1 exhibited a strong and broad absorption in the visible and near- infrared regions and appropriate energy levels matched with low bandgap donor polymers. Since the original publication, the PCE of e1 based OPVs has been improved to 11.4% utilizing polymer donor J71 [74,75,76,77,78]. Changing the sidechains of e1 from bulky alkylphenyl to linear octyl sidechains resulted in further efficiency improvements, to a best of 13.2% PCE when used with a fluorinated PBDB-T type polymer. This improvement was related to a combination of reduction in the optical bandgap, higher absorptivity, and an increased propensity to crystallize for the alkylated ITIC versus the phenylalkylated e1 [79].
Zhan et al. also reported the planar five-fused-ring IDT electron acceptor using linear alkyl sidechians (e2, known as IDIC) [80]. The as-cast OPVs based on PDBT-T1:e2 without any treatments exhibited a maximum PCEs as high as 8.71%. The performance of e2 based non-fullerene OPVs have been further improved to over 11% through blending with different polymer donors [81].
The NFA e3 was synthesized in the McCulloch group and its energy levels and optical bandgap (1.63 eV) matched well with P3HT [82]. Consequently, e3 based OPVs achieved a high JSC of 14.1 mA cm−2 and a high PCE of 6.4%, one of the highest with the readily scalable P3HT donor polymer. More importantly, the P3HT:e3 blend system was more air stable compared with the benchmark P3HT:PC61BM devices. Subsequently the PCE of e3 based OPVs was further improved to 9.95% by blending with the another donor PffBT4T-2DT, due to the reduced non-radiative recombination loss [83]. Zhu and coworkers synthesized the related e4 by combining a thieno[3,4-b]thiophene-rhodanine functional end group with the IDT core [84]. e4 possessed a planar conjugated framework, intensive absorption in the visible region and suitable energy levels well matched with PTB7-Th. The PTB7-Th:e4 blended films exhibited increased crystal sizes and preferential face-on orientation after adding 1,8-diiodooctane (DIO) as a processing additive. A best PCE of 10.07% was obtained based on the as-cast DIO-processed PTB7-Th:e4 OPVs.
Hou and coworkers reported a small molecule acceptor e5 based on the IDT core, by introducing alkoxy groups into an earlier reported NFA IEIC [85, 86]. Compared to IEIC, the resulting e5 (also known as IEICO) exhibited a similar LUMO level but a narrower optical bandgap of 1.34 eV with a high-lying HOMO of − 5.32 eV. A maximum PCE of 8.4% with a high JSC of 17.7 mA cm−2 were obtained for the PBDTTT-E-T:e5 based single-junction OPV devices. More impressively, tandem devices using PBDTTT-E-T:e5 as the rear subcell and PBDD4T-2F:PC71BM as the front subcell, afforded a maximum PCE of 10.7%, along with a VOC of 1.70 V, a JSC of 10.3 mA cm−2, and an FF of 0.61, indicating e5 is a promising candidate for high efficient single-junction and tandem OPVs.
To further elevate the LUMO level of molecule e1, e6 was designed by rationally incorporating methyl units onto the end groups [87]. Due to the weak electron-donating property of methyl, the LUMO level of e6 was slightly elevated by 0.04 eV and the whole absorption spectra was blue-shifted by 6 nm with a slightly larger bandgap. The morphology of the PBDB-T:e6 blend films was highly ordered with high purity domains. The optimal PCE of 12.05% was obtained for the PBDB-T:e6 OPVs. Through sidechain isomerization, Li and coworkers developed a low bandgap electron acceptor e7 to further improve the device performance of e1 based OPVs [88]. e7 showed a higher film absorption coefficient of 1.06 × 105 cm−1, a larger crystalline coherence, and higher electron mobility of 2.45 × 10−4 cm2 V−1 s−1 than e1 (1.00 × 105 cm−1 and 1.60 × 10−4 cm2 V−1 s−1) and these inherent advantages resulted in a higher efficiency of 11.77% based on the J61:e7 blended films. Notably, the J61:e7 based devices showed less thickness-dependent photovoltaic behavior, with a minimum PCE over 8.50% retained as the film thickness increased from 60 to 360 nm.
Liao et al. [89] developed a non-fullerene acceptor e8, employing a ten-fused-heterocyclic-ring as its donor core. e8 possessed a highly planar structure with extremely small torsion angles of 0.14°, facilitating π-electron delocalization and enhancing charge mobility. The introduction of Se atoms into the multi fused-ring compound resulted in a narrower bandgap compared to the S analogues [90]. In practice, this compound exhibited a low bandgap of 1.52 eV. Due to complementary absorption of the large bandgap polymer J51 and e8, broad absorption coverage from 300 to 800 nm was obtained in the blended films, which contributed to a high JSC of 15.16 mA cm−2 and finally a high PCE of 8.02%.
Two fused carbazole-based small molecules e9, based on an electron-donating fully alkylated dithienocyclopentacarbazole core flanked by electron withdrawing nonfluorinated or fluorinated 1,1-dicyanomethylene-3-indanone (IC or IC-4F), were prepared and utilized in OPVs [91]. The two molecules revealed planar structures and strong aggregation behavior, and fluorination was shown to red-shift the optical bandgap and downshift energy levels. OPVs based on fluorinated e9 exhibited a power conversion efficiency of 12.6%, much higher than that of nonfluorinated-based devices (6.2%). Microstructural studies revealed that while both acceptors were highly crystalline, bulk heterojunction blends based on the nonfluorinated e9 resulted in overly coarse domains, while blends based on the fluorinated e9 exhibited a more optimal nanoscale morphology. These results highlighted the importance of end group fluorination in controlling molecular aggregation and miscibility.
Acceptor e10 was designed based on a curved, electron-rich cyclopentadithienothiophene core, which was synthesized via a facile aromatic extension strategy [71]. As-cast, single-junction OPVs based on PFBDB-T:e10 blends exhibited very high short-circuit currents up to 26.2 mA cm−2 in combination with power conversion efficiencies over 11% without any additional processing treatments. The high photocurrent resulted from the near-infrared absorption of the e10 acceptor and the well-intermixed blend morphology of polymer donor PFBDB-T and e10. This work demonstrated a useful fused ring extension strategy and promising solar cell results, indicating the great potential of the e10 derivatives as electron-rich building blocks for constructing high-performance small molecule acceptors in organic solar cells.
With the assistance of non-covalent interactions, non-fused A-D-A acceptor e11 was designed to reduce the synthetic complexity associated with fused-ring cores [70]. Such synthetic complexity may be an impediment to the scale-up and ultimate commercialization of NFA based OPVs. The compound e11 has the structural feature of a freely rotatable conformation in solution, which helps with solubility, while being restrained into a relatively rigid and planar conformation upon stacking in the film via the assistance of intramolecular non-covalent interactions. Non-fused e11 showed an efficient performance of 10.27% PCE in single-junction OPVs and 13.97% PCE in tandem OPVs. In addition, e11-based OPVs exhibited good stability under continuous illumination.
Bo and coworkers developed a fully non-fused ring acceptor e12, which exhibited a planar central backbone, possibly induced by the encapsulation effect of four ortho substituted side chains [92]. This promoted thin-film crystallinity and red-shifted absorption upon thermal annealing. This planar structure and ordered intermolecular stacking was favorable for higher charge carrier mobility in its devices. A PCE of 7.34% was achieved for OPV devices based on as-cast PBDB-T:e12 blends and then further improved to 10.26% after thermal annealing, which is among the highest reported PCEs employing fully non-fused A-D-A type acceptors.
Although, many new acceptors based on the A-D-A type design have been developed and applied in various device structures, the PCE improvement in the OPV field slowed and approached a plateau. A significant breakthrough came with molecule f1, also known as Y6, which is a A-DA’D-A-type NFA employing a ladder-type central fused core containing an electron deficient benzothiadiazole group [72]. Ladder-type multi-fused rings with an electron-deficient core had previously demonstrated a narrow bandgap [93] and the electron affinity could be fine-tuned by the introduction of an electron-withdrawing moiety in the middle of the central core to create a charge-deficient region [94]. OPVs made from f1 in conventional and inverted architectures both exhibited a very high efficiency of 15.7%. In addition, f1-based devices maintained an efficiency of 13.6% even with an active layer thickness of 300 nm. With the emergence of f1, many modifications have been made to develop related NFAs, and the efficiency has sharply risen to over 19% [3], which indicates that A-D-A-type NFAs incorporating an electron-deficient core are an effective strategy to prepare high-performance NFAs.
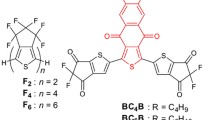
Similar to the chemical modifications of ITIC, new A-DA’D-A type NFAs can be developed by changing the backbones, end groups, and soluble sidechains. Typical OPV acceptors exhibited higher Urbach energies (ca. 25–50 meV) than their inorganic counterparts, resulting in higher non-radiative recombination and reduced device efficiency. Lin and coworkers[95] developed a new A-DA’D-A NFA f2, a selenium-containing analog to f1, which showed lower Urbach energy (20.4 meV), broader and enhanced absorption, and higher electron mobility. As-cast single junction D18:f2 based OPVs showed a highest efficiency of 17.7%, better than that of the D18:f1-based control device.
The compound f3 [96] was designed by combining benzotriazole and selenophene together, compared to the model molecule f1. Benzotriazole possesses a weaker electron-withdrawing ability than benzothiadiazole and selenophene has a stronger electron-donating strength than thiophene. As a result, this novel A-DA’D-A type NFA showed an absorption edge approaching 1000 nm due to the efficient stronger intra- and intermolecular interactions. The PM6:f3 based binary OPV devices showed an optimal PCE of 17.02% with an remarkable JSC of 27.72 mA cm−2 and a low energy loss of 0.446 eV.
Molecule f4 was designed and synthesized to explore the asymmetric isomer effects [97]. By changing from symmetric f1 to asymmetric f4, the dielectric constant in f4 was significantly enhanced and the exciton binding energy was also reduced, facilitating a more efficient charge dissociation process. The weaker crystallization behavior of molecule f4 also enables an optimized micromorphology due to its good miscibility with the donor materials. Excessive molecular aggregation and unfavorable edge-on orientation were inhibited and PM6:f4 based OPV devices achieved a PCE of 15.8%.
Eco-compatible processability with less toxic solvents is important for scalable large-area production of OPV devices. By modifying the flexible alkyl chains on the nitrogen atoms of compound f1, NFA f5 was synthesized [98]. With longer alkyl chains, f5 can be processed in various non-halogenated solvents, such as o-xylene, 1,2,4-TMB, and THF. The THF-processed T1:f5 based OPV cells exhibited a best PCE of 16.1%, comparable to the devices fabricated with halogenated solvents. when using the blade coating method to fabricate the OPV cells with a 1.07 cm2 active area, a high PCE of 14.4% was still maintained, demonstrating fine-tuning of the side chains can facilitate large-scale printing with greener solvents.
The side chains on the β-position of the outermost thieno[3,2-b]thiophene units, can also be substituted to adjust the molecular properties. By modifying the linear alkyl chains of f1, f6 with branched alkoxy side chains was synthesized [99]. The PM6:f6 based blend films exhibited a more ordered molecular packing and enhanced crystallinity, compared to the f1 based blend film. The f6 based OPV devices exhibited a higher VOC (0.963 eV) but a smaller JSC (21.5 mA cm–2) than f1 based devices (0.839 eV and 25.6 mA cm–2). Through varying the ratio of these two acceptors in the ternary devices, the VOC could be linearly tuned, and a maximum PCE of 17.5% was achieved after the acceptor ratio optimization.
Modifying the end groups of the A-DA’D-A type NFAs has been regarded as a relatively simple but effective strategy to improve the OPV device performance. Wang and coworkers facilely synthesized a series of NFAs with different hetero-dihalogenated IC terminals (FCl-IC, FBr-IC and ClBr-IC), which contain a fluorine/ chlorine, fluorine/ bromine or bromine/chlorine pair on the same IC skeleton [100]. Among them, molecule f7 with FCl-IC as the end groups exhibited the most planar molecular geometry and ordered intermolecular packing, indicated by the crystallographic and theoretical analysis. A optimal PCE of 17.52% was obtained for PM6:f7 based OPV devices, higher than the other two NFAs.
Non-fused A-DA’D-A type NFA molecules have also been explored to reduce the synthetic complexity and maintain the high device efficiency. f8 was a representative molecule that showed a record PCE of 14.82% [101]. Combining the effects of noncovalent intramolecular interaction and steric terminal side chains, f8 possesses a rigid molecular structure with lower reorganization free energy. With this conformational lock strategy, f8 based device exhibited a high PCE of 14.82%, which provided an approachto effectively design non-fused NFAs and demonstrate their high potential application in the OPV research.
Molecule f9 was design with the all-fused-ring NFA concept proposed by Zhu and coworkers [102]. State-of-the-art high performance NFAs can be unstable due to the vulnerable exocyclic double bond, which limits the device lifetime of their OPV devices. With the elimination of C = C double bond linkage between the D and A parts, the innovative f9 showed better chemical, photochemical, and thermal stability. Moe importantly, this molecule could also be synthesized on a large scale with a high yield in a short time, unlike many other reported acceptors. Benefitted by its near-infrared responsiveness and 3D honeycomb-type stacking, a high PCE of 13% was yielded for D18:f9 based OPV devices.
5 Summary and outlook
Developing effective technology for the utilization of solar energy is a research hotspot in the field of new energy and new materials. To improve the performance of OPV devices, one of the most direct and effective approaches has been the design and development of novel donor and acceptor materials [103]. Over the past few years, there has been a tremendous effort to develop NFAs for high PCEs of OPVs. In this review, we discussedthe basic structure–property relationship and clarified the rational design of PDI, DPP and A-D-A derivatives, which is crucial for further improving the performance of non-fullerene OPVs. PDI based electron acceptors have been investigated for over 30 years, due to their high electron mobility, good thermal ability, and broad absorption in the visible range. Excessive self-aggregation of PDI molecules is detrimental to effective blend morphology and the efficiencies of OPV devices. To reduce the intermolecular packing, structural modifications have been made on the nitrogen, bay and ortho position of PDI monomer. Constructing twisted or star-shaped PDI based NFAs with two or more PDI monomers is another effective approach to overcome the over-aggregation of its monomeric derivatives in blend films. However, there is a trade-off relationship between nanometer-sized phase-separated domains and electron transport in PDI based NFAs, which is important when designing new twisted and star-shaped PDI derivatives. To maintain the favorable properties of subunits in a star-shaped molecule fine structural tuning like ring fusion (e.g. b6) has been proposed. Constructing twisted PDI dimers is one of the most synthetically accessible approaches to suppress the over-aggregation, and constructing multidimensional PDI based NFAs has enabled multidimensional charge transport, similar to fullerenes. As the optical, electronic and morphological properties of PDI based NFAs are sensitive to their intrinsic molecular geometry, conjugation length and electron distribution, judicious consideration should be taken when designing new PDI based NFAs and more effort is required to understand the relationship between the molecular structure, optoelectronic properties, and photoelectric performances [104].
DPP based NFAs possess NIR light-absorbing characteristics and high electron mobility, and are therefore attractive for non-fullerene OPVs. DPP based small molecule can be further classified into DPP-cored acceptors and DPP-terminated acceptors, according to their different molecular design strategies. However, the reported NFAs cannot provide satisfactory OPV performance so far due to their high-lying energy levels and strong aggregation behavior. Only a few donor materials can be used to match these acceptors and the P3HT donor is among the most representative ones. Therefore, it is essential to propose an efficient way to lower the energy levels of DPP based NFAs, in order to match well with existing high-performance donors. To reduce the severe phase separation issues in DPP based blend films, the twisted or star-shaped design is an effective solution. By improving the miscibility of these DPP NFAs, smaller domain sizes can be obtained, which is beneficial for exciton dissociation and diffusion to the D/A interface. DPP based polymers display high hole and electron mobilities in organic transistors [105] and their light absorption in the NIR region is comparable to naphthalene diimide based polymers, such as N2200. Therefore, DPP-polymers also have the potential to be applied in non-fullerene OPVs [52]. Most of the DPP based polymer acceptors have similar issues as their small molecule acceptors. The situation is also more complex when using polymer donors to fabricate OPV devices as the microphase separation between the polymer donors and polymer acceptors is difficult to optimize. Thus, developing new donor materials, especially small molecule donors, to match the optoelectronic properties of DPP based acceptors and form favorable morphology in their blends is critical to realize high efficiency OPVs.
The achievement of the highest efficiencies has thus far mainly resulted from A-D-A-type acceptors. Synergistic adjustment of the conjugated donor core, end group, side chain and spacer will enable further improvements in efficiency and stability in the OPV field. Firstly, by extending the fused backbone, the light-harvesting ability and charge transport properties can be improved in the resultant NFAs. By incorporating electron-rich heteroatoms such as nitrogen and selenium into the backbone, the donor strength is increased and intermolecular interactions can be enhanced, which leds to red-shifted and enhanced absorption. By changing the symmetric backbone into an asymmetric design, a higher dipole moment can be induced, together with enhanced π–π stacking in face-on orientation. To reduce the synthetic complexity and cost, introducing noncovalent interactions such as hydrogen bonds to promote molecular planarity has been an effective design idea. The reported f1 (well known as Y6) was another innovative backbone modification example for efficient OPV devices. With an electron-deficient benzothiadiazole unit introduced, this A-DA’D-A type structure contributes to a large number of high-performance NFAs so far. By employing different side chains, end groups and π-spacers, the solubility, crystallinity, and miscibility of the A-D-A type NFAs can be further modulated.
There remain some challenges existing in the current development of A-D-A type NFAs. Simple synthetic routes should be developed to promote scalable and low-cost materials. As most of the A-D-A acceptors are designed with narrow bandgap and NIR absorption, it is also important to develop wide bandgap donors that have well matched energy levels, complementary absorption and form ideal blend morphology. As all polymer OPVs offer improved morphological stability and mechanical durability than polymer donor/small molecule NFA system, developing novel high-performing polymer acceptors is a prerequisite to meet this need. With regards to the device optimization, photovoltaic materials with relatively less ratio sensitivity need be developed to improve the reproducibility of ternary OPVs. For tandem solar cells, novel wide bandgap acceptors are in demand for the front-cell and weak electron-deficient end groups are scarce. Photovoltaic materials with less thickness sensitivity are also coveted as thick-film devices can harvest more incident photons and are more suitable for roll-to-roll process [103].
The device stability is another major barrier for NFAs based OPVs towards commercialization and should be given more focus in the development of non-fullerene acceptors [106, 107]. NFAs exhibit diverse chemical, thermal, photo-oxidative stabilities because of various reactive sites in their distinct chemical structures and differing aggregation behavior in blend films. Chemical reactions between limiting factors (such as oxygen and moisture) and photoactive materials can gradually deteriorate the photoactive materials and result in the irreversible degradation of OPV devices. For example, IC and its derivatives are the most popular end groups employed in A-D-A type NFAs. The NFAs with these electron-deficient terminals are prepared by kinetically reversible Knoevenagel condensation, which has been recognized as a major factor for chemical instability upon reaction photo-oxidation. The exocyclic double bonds between the backbone and end groups are highly reactive and easily broken upon ZnO-catalyzed photodegradation and base-induced decomposition, which can limit the lifetime of OPV devices. Therefore, new terminal groups need be explored to tackle this molecular stability issue, such as the benzothiadiazole-rhodanine terminal group in molecule e3 (o-IDTBR). e3 and its derivatives exhibited high photo-oxidative resistance in neat films and these type of NFAs based OPVs possessed good photo-stability. Secondly, new molecular structures need to be proposed to improve the molecule and device stability. It is promising to see that several strategies like non-fused NFAs with intramolecular noncovalent bonds, all-fused NFAs with elimination of exocyclic double bond, and asymmetric structures with larger dipole moments, have demonstrated their high photo-oxidation stability in OPV devices, presumably due to the improved crystallinity and restrained morphology variation. Compared to the photo-oxidative stability of NFAs, the metastable morphology of blend films is another factor that can accelerate the degradation of OPVs.
In addition to chemical structure, molecular packing has a great influence in the miscibility and aggregation of blend films. Favorable miscibility between polymer donors and NFAs with high crystalline behavior is vital to accomplish reasonable exciton separation and transport. Recently ternary OPVs have shown promising performance in their thermal-stability devices, which is due to the stabilized morphological phase in blend films by introducing a third component. However, it is worthy to note that procedures for formation of blend films and assembly of OPV devices should be simplified to low the whole production cost. Finally, interlayer materials (such as PEDOT:PSS in conventional devices and ZnO in inverted devices) have nonnegligible effects on the instability of NFA based OPVs. Conjugated structures of NFAs can be damaged by interacting with these interlayers and charge traps will be formed in the blend films. Modification of current interlayers and development of new stable interlayers are possible methods to avoid potential chemical reactions at the interface and thus improve the stability of OPV devices.
References
Cheng Y-J, Yang S-H, Hsu C-S (2009) Synthesis of conjugated polymers for organic solar cell applications. Chem Rev 109:5868–5923. https://doi.org/10.1021/cr900182s
Søndergaard R, Hösel M, Angmo D, Larsen-Olsen TT, Krebs FC (2012) Roll-to-roll fabrication of polymer solar cells. Mater Today 15:36–49. https://doi.org/10.1016/S1369-7021(12)70019-6
Chong K, Xu X, Meng H, Xue J, Yu L, Ma W et al (2022) Realizing 19.05% efficiency polymer solar cells by progressively improving charge extraction and suppressing charge recombination. Adv Mater 34:2109516. https://doi.org/10.1002/adma.202109516
He Y, Li Y (2011) Fullerene derivative acceptors for high performance polymer solar cells. Phys Chem Chem Phys 13:1970–1983. https://doi.org/10.1039/C0CP01178A
Chen JD, Cui C, Li YQ, Zhou L, Ou QD, Li C et al (2015) Single-junction polymer solar cells exceeding 10% power conversion efficiency. Adv Mater 27:1035–1041. https://doi.org/10.1002/adma.201404535
Wang Z, Li Z, Xu X, Li Y, Li K, Peng Q (2016) Polymer solar cells exceeding 10% efficiency enabled via a facile star-shaped molecular cathode interlayer with variable counterions. Adv Funct Mater 26:4643–4652. https://doi.org/10.1002/adfm.201504734
Jin Y, Chen Z, Dong S, Zheng N, Ying L, Jiang XF et al (2016) A novel aphtha[1,2-c:5,6-c’]bis([1,2,5]thiadiazole)-based narrow-bandgap pi-conjugated polymer with power conversion efficiency over 10%. Adv Mater 28:9811–9818. https://doi.org/10.1002/adma.201603178
Liu X, Li X, Li Y, Song C, Zhu L, Zhang W et al (2016) High-performance polymer solar cells with pce of 10.42% via al-doped zno cathode interlayer. Adv Mater 28:7405–7412. https://doi.org/10.1002/adma.201601814
Wan Q, Guo X, Wang Z, Li W, Guo B, Ma W et al (2016) 10.8% efficiency polymer solar cells based on ptb7-th and pc71bm via binary solvent additives treatment. Adv Funct Mater 26:6635–6640. https://doi.org/10.1002/adfm.201602181
Lee J, Sin DH, Moon B, Shin J, Kim HG, Kim M et al (2017) Highly crystalline low-bandgap polymer nanowires towards high-performance thick-film organic solar cells exceeding 10% power conversion efficiency. Energy Environ Sci 10:247–257. https://doi.org/10.1039/C6EE02466A
Anthony JE (2011) Small-molecule, nonfullerene acceptors for polymer bulk heterojunction organic photovoltaics. Chem Mater 23:583–590. https://doi.org/10.1021/cm1023019
Liu T, Pan X, Meng X, Liu Y, Wei D, Ma W et al (2016) Alkyl side-chain engineering in wide-bandgap copolymers leading to power conversion efficiencies over 10%. Adv Mater 29:1604251. https://doi.org/10.1002/adma.201604251
Li Z, Jiang K, Yang G, Lai JY, Ma T, Zhao J et al (2016) Donor polymer design enables efficient non-fullerene organic solar cells. Nat Commun 7:13094. https://doi.org/10.1038/ncomms13094
Chen S, Meng D, Huang J, Liang N, Li Y, Liu F et al (2021) Symmetry-induced orderly assembly achieving high-performance perylene diimide-based nonfullerene organic solar cells. CCS Chem 3:78–84. https://doi.org/10.31635/ccschem.021.202000538
Zhang G, Feng J, Xu X, Ma W, Li Y, Peng Q (2019) Perylene diimide-based nonfullerene polymer solar cells with over 11% efficiency fabricated by smart molecular design and supramolecular morphology optimization. Adv Funct Mater 29:1906587. https://doi.org/10.1002/adfm.201906587
Privado M, Dahiya H, de la Cruz P, Keshtov ML, Langa F, Sharma GD (2021) A ternary organic solar cell with 15.6% efficiency containing a new dpp-based acceptor. J Mater Chem C 9:16272–16281. https://doi.org/10.1039/D1TC02241E
Tang CW (1986) Two-layer organic photovoltaic cell. Appl Phys Lett 48:183–185. https://doi.org/10.1063/1.96937
Li C, Wonneberger H (2012) Perylene imides for organic photovoltaics: Yesterday, today, and tomorrow. Adv Mater 24:613–636. https://doi.org/10.1002/adma.201104447
Zhan X, Facchetti A, Barlow S, Marks TJ, Ratner MA, Wasielewski MR et al (2011) Rylene and related diimides for organic electronics. Adv Mater 23:268–284. https://doi.org/10.1002/adma.201001402
Sharenko A, Proctor CM, van der Poll TS, Henson ZB, Nguyen TQ, Bazan GC (2013) A high-performing solution-processed small molecule: perylene diimide bulk heterojunction solar cell. Adv Mater 25:4403–4406. https://doi.org/10.1002/adma.201301167
Hartnett PE, Timalsina A, Matte HS, Zhou N, Guo X, Zhao W et al (2014) Slip-stacked perylenediimides as an alternative strategy for high efficiency nonfullerene acceptors in organic photovoltaics. J Am Chem Soc 136:16345–16356. https://doi.org/10.1021/ja508814z
Singh R, Aluicio-Sarduy E, Kan Z, Ye T, MacKenzie RCI, Keivanidis PE (2014) Fullerene-free organic solar cells with an efficiency of 3.7% based on a low-cost geometrically planar perylene diimide monomer. J Mater Chem A 2:14348–14353. https://doi.org/10.1039/c4ta02851a
Zhan C, Yao J (2016) More than conformational “twisting” or “coplanarity”: Molecular strategies for designing high-efficiency nonfullerene organic solar cells. Chem Mater 28:1948–1964. https://doi.org/10.1021/acs.chemmater.5b04339
Shivanna R, Shoaee S, Dimitrov S, Kandappa SK, Rajaram S, Durrant JR et al (2014) Charge generation and transport in efficient organic bulk heterojunction solar cells with a perylene acceptor. Energy Environ Sci 7:435–441. https://doi.org/10.1039/c3ee42484g
Ye L, Sun K, Jiang W, Zhang S, Zhao W, Yao H et al (2015) Enhanced efficiency in fullerene-free polymer solar cell by incorporating fine-designed donor and acceptor materials. ACS Appl Mater Interfaces 7:9274–9280. https://doi.org/10.1021/acsami.5b02012
Lu Z, Zhang X, Zhan C, Jiang B, Zhang X, Chen L et al (2013) Impact of molecular solvophobicity vs. solvophilicity on device performances of dimeric perylene diimide based solution-processed non-fullerene organic solar cells. Phys Chem Chem Phys 15:11375–11385. https://doi.org/10.1039/c3cp51475g
Zhang X, Lu Z, Ye L, Zhan C, Hou J, Zhang S et al (2013) A potential perylene diimide dimer-based acceptor material for highly efficient solution-processed non-fullerene organic solar cells with 4.03% efficiency. Adv Mater 25:5791–5797. https://doi.org/10.1002/adma.201300897
Zhang X, Zhan C, Yao J (2015) Non-fullerene organic solar cells with 6.1% efficiency through fine-tuning parameters of the film-forming process. Chem Mater 27:166–173. https://doi.org/10.1021/cm504140c
Meng D, Sun D, Zhong C, Liu T, Fan B, Huo L et al (2016) High-performance solution-processed non-fullerene organic solar cells based on selenophene-containing perylene bisimide acceptor. J Am Chem Soc 138:375–380. https://doi.org/10.1021/jacs.5b11149
Sun D, Meng D, Cai Y, Fan B, Li Y, Jiang W et al (2015) Non-fullerene-acceptor-based bulk-heterojunction organic solar cells with efficiency over 7. J Am Chem Soc 137:11156–11162. https://doi.org/10.1021/jacs.5b06414
Zhong H, Wu CH, Li CZ, Carpenter J, Chueh CC, Chen JY et al (2016) Rigidifying nonplanar perylene diimides by ring fusion toward geometry-tunable acceptors for high-performance fullerene-free solar cells. Adv Mater 28:951–958. https://doi.org/10.1002/adma.201504120
Yan Q, Zhou Y, Zheng Y-Q, Pei J, Zhao D (2013) Towards rational design of organic electron acceptors for photovoltaics: a study based on perylenediimide derivatives. Chem Sci 4:4389–4394. https://doi.org/10.1039/c3sc51841h
Kwon OK, Park J-H, Park SK, Park SY (2015) Soluble dicyanodistyrylbenzene-based non-fullerene electron acceptors with optimized aggregation behavior for high-efficiency organic solar cells. Adv Energy Mater 5:1400929. https://doi.org/10.1002/aenm.201400929
Kwon OK, Uddin MA, Park JH, Park SK, Nguyen TL, Woo HY et al (2016) A high efficiency nonfullerene organic solar cell with optimized crystalline organizations. Adv Mater 28:910–916. https://doi.org/10.1002/adma.201504091
Zhao D, Wu Q, Cai Z, Zheng T, Chen W, Lu J et al (2016) Electron acceptors based on α-substituted perylene diimide (PDI) for organic solar cells. Chem Mater 28:1139–1146. https://doi.org/10.1021/acs.chemmater.5b04570
Zhong Y, Trinh MT, Chen R, Wang W, Khlyabich PP, Kumar B et al (2014) Efficient organic solar cells with helical perylene diimide electron acceptors. J Am Chem Soc 136:15215–15221. https://doi.org/10.1021/ja5092613
Zhong Y, Trinh MT, Chen R, Purdum GE, Khlyabich PP, Sezen M et al (2015) Molecular helices as electron acceptors in high-performance bulk heterojunction solar cells. Nat Commun 6:8242. https://doi.org/10.1038/ncomms9242
Li H, Kim FS, Ren G, Hollenbeck EC, Subramaniyan S, Jenekhe SA (2013) Tetraazabenzodifluoranthene diimides: building blocks for solution-processable n-type organic semiconductors. Angew Chem Int Ed 52:5513–5517. https://doi.org/10.1002/anie.201210085
Hwang YJ, Li H, Courtright BA, Subramaniyan S, Jenekhe SA (2016) Nonfullerene polymer solar cells with 8.5% efficiency enabled by a new highly twisted electron acceptor dimer. Adv Mater 28:124–131. https://doi.org/10.1002/adma.201503801
Roncali J (2009) Molecular bulk heterojunctions: an emerging approach to organic solar cells. Acc Chem Res 42:1719–1730. https://doi.org/10.1021/ar900041b
Lin Y, Wang Y, Wang J, Hou J, Li Y, Zhu D et al (2014) A star-shaped perylene diimide electron acceptor for high-performance organic solar cells. Adv Mater 26:5137–5142. https://doi.org/10.1002/adma.201400525
Meng D, Fu H, Xiao C, Meng X, Winands T, Ma W et al (2016) Three-bladed rylene propellers with three-dimensional network assembly for organic electronics. J Am Chem Soc 138:10184–10190. https://doi.org/10.1021/jacs.6b04368
Sharma V, Koenig JDB, Welch GC (2021) Perylene diimide based non-fullerene acceptors: top performers and an emerging class featuring n-annulation. J Mater Chem A 9:6775–6789. https://doi.org/10.1039/D0TA11197J
Liu Y, Lai JYL, Chen S, Li Y, Jiang K, Zhao J et al (2015) Efficient non-fullerene polymer solar cells enabled by tetrahedron-shaped core based 3d-structure small-molecular electron acceptors. J Mater Chem A 3:13632–13636. https://doi.org/10.1039/c5ta03093e
Liu Y, Mu C, Jiang K, Zhao J, Li Y, Zhang L et al (2015) A tetraphenylethylene core-based 3d structure small molecular acceptor enabling efficient non-fullerene organic solar cells. Adv Mater 27:1015–1020. https://doi.org/10.1002/adma.201404152
Lin H, Chen S, Hu H, Zhang L, Ma T, Lai JY et al (2016) Reduced intramolecular twisting improves the performance of 3d molecular acceptors in non-fullerene organic solar cells. Adv Mater 28:8546–8551. https://doi.org/10.1002/adma.201600997
Wu Q, Zhao D, Schneider AM, Chen W, Yu L (2016) Covalently bound clusters of alpha-substituted PDI-rival electron acceptors to fullerene for organic solar cells. J Am Chem Soc 138:7248–7251. https://doi.org/10.1021/jacs.6b03562
Lee J, Singh R, Sin DH, Kim HG, Song KC, Cho K (2016) A nonfullerene small molecule acceptor with 3D interlocking geometry enabling efficient organic solar cells. Adv Mater 28:69–76. https://doi.org/10.1002/adma.201504010
He Q, Eisner FD, Pearce D, Hodsden T, Rezasoltani E, Medranda D et al (2020) Ring fusion in tetrathienylethene cored perylene diimide tetramers affords acceptors with strong and broad absorption in the near-UV to visible region. J Mater Chem C 8:17237–17244. https://doi.org/10.1039/d0tc04110f
Nielsen CB, Turbiez M, McCulloch I (2013) Recent advances in the development of semiconducting DPP-containing polymers for transistor applications. Adv Mater 25:1859–1880. https://doi.org/10.1002/adma.201201795
Liu Q, Bottle SE, Sonar P (2020) Developments of diketopyrrolopyrrole-dye-based organic semiconductors for a wide range of applications in electronics. Adv Mater 32:1903882. https://doi.org/10.1002/adma.201903882
Zhao C, Guo Y, Zhang Y, Yan N, You S, Li W (2019) Diketopyrrolopyrrole-based conjugated materials for non-fullerene organic solar cells. J Mater Chem A 7:10174–10199. https://doi.org/10.1039/C9TA01976F
Lin Y, Li Y, Zhan X (2013) A solution-processable electron acceptor based on dibenzosilole and diketopyrrolopyrrole for organic solar cells. Adv Energy Mater 3:724–728. https://doi.org/10.1002/aenm.201200911
Patil Y, Misra R, Keshtov ML, Sharma GD (2016) 1,1,4,4-tetracyanobuta-1,3-diene substituted diketopyrrolopyrroles: An acceptor for solution processable organic bulk heterojunction solar cells. J Phys Chem 120:6324–6335. https://doi.org/10.1021/acs.jpcc.5b12307
Raynor AM, Gupta A, Patil H, Ma D, Bilic A, Rook TJ et al (2016) A non-fullerene electron acceptor based on central carbazole and terminal diketopyrrolopyrrole functionalities for efficient, reproducible and solution-processable bulk-heterojunction devices. RSC Adv 6:28103–28109. https://doi.org/10.1039/c6ra01558a
Shi H, Fu W, Shi M, Ling J, Chen H (2015) A solution-processable bipolar diketopyrrolopyrrole molecule used as both electron donor and acceptor for efficient organic solar cells. J Mater Chem A 3:1902–1905. https://doi.org/10.1039/c4ta06035k
Jung JW, Jo WH (2015) Low-bandgap small molecules as non-fullerene electron acceptors composed of benzothiadiazole and diketopyrrolopyrrole for all organic solar cells. Chem Mater 27:6038–6043. https://doi.org/10.1021/acs.chemmater.5b02480
Josse P, Dalinot C, Jiang Y, Dabos-Seignon S, Roncali J, Blanchard P et al (2016) Phthalimide end-capped thienoisoindigo and diketopyrrolopyrrole as non-fullerene molecular acceptors for organic solar cells. J Mater Chem A 4:250–256. https://doi.org/10.1039/c5ta09171c
Lin Y, Cheng P, Li Y, Zhan X (2012) A 3d star-shaped non-fullerene acceptor for solution-processed organic solar cells with a high open-circuit voltage of 1.18 v. Chem Commun 48:4773–4775. https://doi.org/10.1039/c2cc31511d
Li S, Liu W, Shi M, Mai J, Lau T-K, Wan J et al (2016) A spirobifluorene and diketopyrrolopyrrole moieties based non-fullerene acceptor for efficient and thermally stable polymer solar cells with high open-circuit voltage. Energy Environ Sci 9:604–610. https://doi.org/10.1039/c5ee03481g
Brandt RG, Zhang F, Andersen TR, Angmo D, Shi M, Gurevich L et al (2016) Roll coated large area ito- and vacuum-free all organic solar cells from diketopyrrolopyrrole based non-fullerene acceptors with molecular geometry effects. RSC Adv 6:41542–41550. https://doi.org/10.1039/c6ra06898g
Rananaware A, Gupta A, Li J, Bilic A, Jones L, Bhargava S et al (2016) A four-directional non-fullerene acceptor based on tetraphenylethylene and diketopyrrolopyrrole functionalities for efficient photovoltaic devices with a high open-circuit voltage of 1.18 v. Chem Commun 52:8522–8525. https://doi.org/10.1039/c6cc03730e
Yang Y, Zhang G, Yu C, He C, Wang J, Chen X et al (2014) New conjugated molecular scaffolds based on [2,2]paracyclophane as electron acceptors for organic photovoltaic cells. Chem Commun 50:9939–9942. https://doi.org/10.1039/c4cc04384g
He Q, Shahid M, Panidi J, Marsh AV, Huang W, Daboczi M et al (2019) A versatile star-shaped organic semiconductor based on benzodithiophene and diketopyrrolopyrrole. J Mater Chem C 7:6622–6629. https://doi.org/10.1039/c9tc00905a
Lin Y, Wang J, Zhang ZG, Bai H, Li Y, Zhu D et al (2015) An electron acceptor challenging fullerenes for efficient polymer solar cells. Adv Mater 27:1170–1174. https://doi.org/10.1002/adma.201404317
Li S, Li C-Z, Shi M, Chen H (2020) New phase for organic solar cell research: emergence of y-series electron acceptors and their perspectives. ACS Energy Lett 5:1554–1567. https://doi.org/10.1021/acsenergylett.0c00537
Dai S, Zhao F, Zhang Q, Lau T-K, Li T, Liu K et al (2017) Fused nonacyclic electron acceptors for efficient polymer solar cells. J Am Chem Soc 139:1336–1343. https://doi.org/10.1021/jacs.6b12755
Huang C, Liao X, Gao K, Zuo L, Lin F, Shi X et al (2018) Highly efficient organic solar cells based on s, n-heteroacene non-fullerene acceptors. Chem Mater 30:5429–5434. https://doi.org/10.1021/acs.chemmater.8b02276
Yang L, Song X, Yu J, Wang H, Zhang Z, Geng R et al (2019) Tuning of the conformation of asymmetric nonfullerene acceptors for efficient organic solar cells. J Mater Chem A 7:22279–22286. https://doi.org/10.1039/C9TA07634D
Yu Z-P, Liu Z-X, Chen F-X, Qin R, Lau T-K, Yin J-L et al (2019) Simple non-fused electron acceptors for efficient and stable organic solar cells. Nat Commun 10:2152. https://doi.org/10.1038/s41467-019-10098-z
He Q, Shahid M, Wu J, Jiao X, Eisner FD, Hodsden T et al (2019) Fused cyclopentadithienothiophene acceptor enables ultrahigh short-circuit current and high efficiency >11% in as-cast organic solar cells. Adv Funct Mater 29:1904956. https://doi.org/10.1002/adfm.201904956
Yuan J, Zhang Y, Zhou L, Zhang G, Yip H-L, Lau T-K et al (2019) Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core. Joule 3:1140–1151. https://doi.org/10.1016/j.joule.2019.01.004
Fei Z, Eisner FD, Jiao X, Azzouzi M, Rohr JA, Han Y et al (2018) An alkylated indacenodithieno[3,2-b]thiophene-based nonfullerene acceptor with high crystallinity exhibiting single junction solar cell efficiencies greater than 13% with low voltage losses. Adv Mater. https://doi.org/10.1002/adma.201705209
Bin H, Zhang Z-G, Gao L, Chen S, Zhong L, Xue L et al (2016) Non-fullerene polymer solar cells based on alkylthio and fluorine substituted 2d-conjugated polymers reach 9.5% efficiency. J Am Chem Soc 138:4657–4664. https://doi.org/10.1021/jacs.6b01744
Zheng Z, Awartani OM, Gautam B, Liu D, Qin Y, Li W et al (2016) Efficient charge transfer and fine-tuned energy level alignment in a THF-processed fullerene-free organic solar cell with 113% efficiency. Adv Mater 29:1604241. https://doi.org/10.1002/adma.201604241
Qin Y, Uddin MA, Chen Y, Jang B, Zhao K, Zheng Z et al (2016) Highly efficient fullerene-free polymer solar cells fabricated with polythiophene derivative. Adv Mater 28:9416–9422. https://doi.org/10.1002/adma.201601803
Gao L, Zhang Z-G, Bin H, Xue L, Yang Y, Wang C et al (2016) High-efficiency nonfullerene polymer solar cells with medium bandgap polymer donor and narrow bandgap organic semiconductor acceptor. Adv Mater 28:8288–8295. https://doi.org/10.1002/adma.201601595
Bin H, Gao L, Zhang Z-G, Yang Y, Zhang Y, Zhang C et al (2016) 11.4% efficiency non-fullerene polymer solar cells with trialkylsilyl substituted 2d-conjugated polymer as donor. Nat Commun 7:13651. https://doi.org/10.1038/ncomms13651
Fei Z, Eisner FD, Jiao X, Azzouzi M, Röhr JA, Han Y et al (2018) An alkylated indacenodithieno[3,2-b]thiophene-based nonfullerene acceptor with high crystallinity exhibiting single junction solar cell efficiencies greater than 13% with low voltage losses. Adv Mater 30:1705209. https://doi.org/10.1002/adma.201705209
Lin Y, He Q, Zhao F, Huo L, Mai J, Lu X et al (2016) A facile planar fused-ring electron acceptor for as-cast polymer solar cells with 8.71% efficiency. J Am Chem Soc 138:2973–2976. https://doi.org/10.1021/jacs.6b00853
Lin Y, Zhao F, Wu Y, Chen K, Xia Y, Li G et al (2016) Mapping polymer donors toward high-efficiency fullerene free organic solar cells. Adv Mater. https://doi.org/10.1002/adma.201604155
Holliday S, Ashraf RS, Wadsworth A, Baran D, Yousaf SA, Nielsen CB et al (2016) High-efficiency and air-stable p3ht-based polymer solar cells with a new non-fullerene acceptor. Nat Commun 7:11585. https://doi.org/10.1038/ncomms11585
Baran D, Kirchartz T, Wheeler S, Dimitrov S, Abdelsamie M, Gorman J et al (2016) Reduced voltage losses yield 10% and >1v fullerene free organic solar cells. Energy Environ Sci 9:3783–3793. https://doi.org/10.1039/c6ee02598f
Liu F, Zhou Z, Zhang C, Vergote T, Fan H, Liu F et al (2016) A thieno[3,4-b]thiophene-based non-fullerene electron acceptor for high-performance bulk-heterojunction organic solar cells. J Am Chem Soc 138:15523–15526. https://doi.org/10.1021/jacs.6b08523
Lin Y, Zhang Z-G, Bai H, Wang J, Yao Y, Li Y et al (2015) High-performance fullerene-free polymer solar cells with 6.31% efficiency. Energy Environ Sci 8:610–616. https://doi.org/10.1039/c4ee03424d
Yao H, Chen Y, Qin Y, Yu R, Cui Y, Yang B et al (2016) Design and synthesis of a low bandgap small molecule acceptor for efficient polymer solar cells. Adv Mater 28:8283–8287. https://doi.org/10.1002/adma.201602642
Li S, Ye L, Zhao W, Zhang S, Mukherjee S, Ade H et al (2016) Energy-level modulation of small-molecule electron acceptors to achieve over 12% efficiency in polymer solar cells. Adv Mater 28:9423–9429. https://doi.org/10.1002/adma.201602776
Yang Y, Zhang ZG, Bin H, Chen S, Gao L, Xue L et al (2016) Side-chain isomerization on n-type organic semiconductor itic acceptor make 11.77% high efficiency polymer solar cells. J Am Chem Soc 138:15011–15018. https://doi.org/10.1021/jacs.6b09110
Li Y, Qian D, Zhong L, Lin J-D, Jiang Z-Q, Zhang Z-G et al (2016) A fused-ring based electron acceptor for efficient non-fullerene polymer solar cells with small homo offset. Nano Energy 27:430–438. https://doi.org/10.1016/j.nanoen.2016.07.019
Li Y, Liu X, Wu F-P, Zhou Y, Jiang Z-Q, Song B et al (2016) Non-fullerene acceptor with low energy loss and high external quantum efficiency: Towards high performance polymer solar cells. J Mater Chem A 4:5890–5897. https://doi.org/10.1039/C6TA00612D
He Q, Shahid M, Jiao X, Gann E, Eisner FD, Wu T et al (2020) Crucial role of fluorine in fully alkylated ladder-type carbazole-based nonfullerene organic solar cells. ACS Appl Mater Interfaces 12:9555–9562. https://doi.org/10.1021/acsami.0c00981
Chen YN, Li M, Wang Y, Wang J, Zhang M, Zhou Y et al (2020) A fully non-fused ring acceptor with planar backbone and near-IR absorption for high performance polymer solar cells. Angew Chem Int Ed 59:22714–22720. https://doi.org/10.1002/anie.202010856
Feng L, Yuan J, Zhang Z, Peng H, Zhang Z-G, Xu S et al (2017) Thieno[3,2-b]pyrrolo-fused pentacyclic benzotriazole-based acceptor for efficient organic photovoltaics. ACS Appl Mater Interfaces 9:31985–31992. https://doi.org/10.1021/acsami.7b10995
Wu J-S, Cheng S-W, Cheng Y-J, Hsu C-S (2015) Donor–acceptor conjugated polymers based on multifused ladder-type arenes for organic solar cells. Chem Soc Rev 44:1113–1154. https://doi.org/10.1039/C4CS00250D
Zhang Z, Li Y, Cai G, Zhang Y, Lu X, Lin Y (2020) Selenium heterocyclic electron acceptor with small urbach energy for as-cast high-performance organic solar cells. J Am Chem Soc 142:18741–18745. https://doi.org/10.1021/jacs.0c08557
Qi F, Jiang K, Lin F, Wu Z, Zhang H, Gao W et al (2021) Over 17% efficiency binary organic solar cells with photoresponses reaching 1000 nm enabled by selenophene-fused nonfullerene acceptors. ACS Energy Lett 6:9–15. https://doi.org/10.1021/acsenergylett.0c02230
Gao W, Fan B, Qi F, Lin F, Sun R, Xia X et al (2021) Asymmetric isomer effects in benzo[c][1,2,5]thiadiazole-fused nonacyclic acceptors: Dielectric constant and molecular crystallinity control for significant photovoltaic performance enhancement. Adv Funct Mater. https://doi.org/10.1002/adfm.202104369
Hong L, Yao H, Wu Z, Cui Y, Zhang T, Xu Y et al (2019) Eco-compatible solvent-processed organic photovoltaic cells with over 16% efficiency. Adv Mater 31:1903441. https://doi.org/10.1002/adma.201903441
Liang J, Pan M, Wang Z, Zhang J, Bai F, Ma R et al (2022) Branched alkoxy side chain enables high-performance non-fullerene acceptors with high open-circuit voltage and highly ordered molecular packing. Chem Mater 34:2059–2068. https://doi.org/10.1021/acs.chemmater.1c03311
Wang L, An Q, Yan L, Bai H-R, Jiang M, Mahmood A et al (2022) Non-fullerene acceptors with hetero-dihalogenated terminals induce significant difference in single crystallography and enable binary organic solar cells with 17.5% efficiency. Energy Environ Sci 15:320–333. https://doi.org/10.1039/d1ee01832a
Zhang X, Li C, Qin L, Chen H, Yu J, Wei Y et al (2021) Side-chain engineering for enhancing the molecular rigidity and photovoltaic performance of noncovalently fused-ring electron acceptors. Angew Chem Int Ed 60:17720–17725. https://doi.org/10.1002/anie.202106753
Liu W, Xu S, Lai H, Liu W, He F, Zhu X (2022) Near-infrared all-fused-ring nonfullerene acceptors achieving an optimal efficiency-cost-stability balancing in organic solar cells. CCS Chem. https://doi.org/10.31635/ccschem.022.202201963
He Q (2020) Synthesis and optoelectronic properties of electron accepting materials in organic photovoltaics. Thesis, Imperial College London. https://doi.org/10.25560/87494
Wang J, Zhan X (2022) From perylene diimide polymers to fused-ring electron acceptors: a 15-year exploration journey of nonfullerene acceptors. Chin J Chem 40:1592–1607. https://doi.org/10.1002/cjoc.202200027
Zhang C, Tan WL, Liu Z, He Q, Li Y, Ma J et al (2022) High-performance unipolar n-type conjugated polymers enabled by highly electron-deficient building blocks containing F and CN groups. Macromolecules. https://doi.org/10.1021/acs.macromol.2c00870
Li Y, Li T, Lin Y (2021) Stability: next focus in organic solar cells based on non-fullerene acceptors. Mater Chem Front 5:2907–2930. https://doi.org/10.1039/D1QM00027F
Speller EM, Clarke AJ, Luke J, Lee HKH, Durrant JR, Li N et al (2019) From fullerene acceptors to non-fullerene acceptors: prospects and challenges in the stability of organic solar cells. J Mater Chem A 7:23361–23377. https://doi.org/10.1039/C9TA05235F
Funding
We would like to thank the Engineering and Physics Science Research Council (EPSRC) (EP/V048686/1 and EP/T028513/1) and the Royal Society and Wolfson Foundation for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, Q., Kafourou, P., Hu, X. et al. Development of non-fullerene electron acceptors for efficient organic photovoltaics. SN Appl. Sci. 4, 247 (2022). https://doi.org/10.1007/s42452-022-05128-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-022-05128-3