
Abstract
In this study, (E)-5-((4,6-dichloro-1,3,5-triazin-2-yl)amino)-4-hydroxy-3-(phenyldiazenyl)naphthalene-2,7-diylbis(hydrogen sulfite), a cyanurated H-acid (CHA) azo dye, was synthesized and characterized using FT-IR spectrophotometer and GC-MS spectroscopy. A density functional theory (DFT) based B3LYP and CAM-B3LYP method with 6–311 + G (d,p) basis set analysis was computed for HOMO-LUMO, natural bonding orbitals (NBO), UV-Vis absorptions and excitation interactions, in order to understand its molecular orbital excitation properties. A low Energy gap (Eg) of 2.947 eV was obtained from the molecular orbital analysis, which showed that HOMO to LUMO transition is highly feasible; hence CHA is adequate for diverse electronic and optic applications. Studies of the first five excitations (S0 → S1/S2/S3/S4/S5) of CHA revealed that S0 → S1 and S0 → S3 are π → π* type local excitations distributed around the –N=N– group; S0 → S2, a Rydberg type local excitation; S0 → S4, a highly localized π → π* excitation; while S0 → S5 is an n → π* charge transfer from a benzene ring to –N=N– group. From NBO analysis, we obtained the various donor–acceptor orbital interactions contributing to the stabilization of the studied compound. Most significantly, some strong hyper-conjugations (n → n*) within fragments, and non-bondingand anti-bonding intermolecular (n → n*/π* and π → n*/π*) interactions were observed to contribute appreciable energies. This study is valuable for understanding the molecular properties of the azo dyes compounds and for synthesizing new ones in the future.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Azo dyes are compounds that are characterized by the functional group (–N=N–) bonding two symmetrical and/or asymmetrical undistinguishable or non-azo alkyl or aryl radicals [1]. These compounds are the further most utilized dyes and nearly 70% of entire dyes used in industry are azo dyes [2,3,4,5]. Benkhaya and coworkers presented an extensive and detailed review of the different classifications of azo dyes, chemical properties of these dyes and finally summarized some recent syntheses of azo dyes along with the mechanism of azo dye/polymer conjugation [6]. Sener et al. [7], reported the synthesis of aminopyrazole-based dye derivative compounds from 2-aminobenzothiazol.Zhao and companions synthesized and characterized a family of bi-heterocyclic dyes by using heterocyclic pyridone and benzothiazole components [8]. Figure 1 presents the structure of a novel cibacron azo dye.

Structure of novel cibacron azo dye
Density Functional Methods (DFT) and Time-dependent density functional theory (TD-DFT) were used to perform theoretical calculations on the experimentally synthesized compounds often done by comparing the experimental spectroscopic data (FT-IR, UV-Vis spectra, X-ray diffraction, NMR, and GC-MS technique) with the theoretically calculated spectroscopic results so as to further validate the chemistry of the synthesized compounds. These studies are valuable for understanding the molecular properties of the azo dyes compounds and for synthesizing new ones in the future [9,10,11,12]. Maliyappa et al., [13] conducted a combined experimental and theoretical studies of 6-Substituted benzothiazole based dispersed azo dyes. The quantum chemical studies (DFT) was utilized to further explain the structural properties of the compounds by using the DFT/B3LYP/6–311 ++ (d, p) level of theory [14,15,16,17]. Osman in 2017 theoretically studied the structure, reactivity, and optical properties of thiazole azo dyes by applying B3LYP, CAM-B3LYP, and wB97XD functionals with 6–311 ++ G** and aug-cc-pvdz basis sets [12, 18]. Shinde and Sekar employed computational DFT to predict the stability of tautomeric forms of the 4-hydroxyl-1-methyl-2(1H)-quinolone azo dyes and found out the electrophilicity index of prepared dyes using TD-DFT/6-31 g(d) theory level [19, 20].
In this research, Density Functional Methods (DFT) was used to perform theoretical calculations on the title compound and was characterized by FT-IR, UV-Vis spectra, and GC-MS technique. The title compound was investigated in comparison with theoretical calculations and experimental data. These studies are valuable for understanding the molecular properties of the azo dyes compounds and for synthesizing new dyes in the future. Also, it will expose the applicability of the synthesized dye in the photo-energy based industries, like in organo-batteries and photovoltaics.
2 Experimental
2.1 Diazotization of aniline
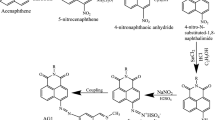
About 10 mL (0.11 mol) of aniline was dissolved in 25 mL (2.5 equivalent) of HCl and rapidly cooled to a temperature of 0–5 °C using an ice bath, after which, 10 mL (1 equivalent) of sodium nitrite (2.5 g, 0.036 mol) was added over a period of 30 min. This process is outlined in Fig. 2.

Diazotization of aniline using HCl and NaNO2 at very low temperature
2.2 Preparation of the coupling components
Cyanuric chloride (1 g, 0.005 mol) was stirred in 25 mL acetone at a temperature below 5 °C for a period of 1 h. A neutral solution of 1-naphthol-8-amino-3,6-disulphonic acid (H-acid); 1.7 g, 0.005 mol) in aqueous solution of Na2CO3 (10% w/v) was added in small amount over a period of 1 h. The pH was kept neutral by simultaneous addition of Na2CO3 solution (1% w/v). The reaction mixture was stirred at 0–5 °C for further 4 h in order to obtain a clear solution; the resulting product was then used directly.
2.3 Dye synthesis
A previously prepared diazonium solution of aniline (0.6 g, 0.005 mol) in 5 mL of water was added drop wise to an ice cooled, well stirred solution of cyanurated H-acid (1.7 g, 0.005 mol) over a period of 10–15 min. The pH was maintained at 7.5–8.5 during the addition by simultaneous addition of 10% w/v Na2CO3. Stirring was then carried out for 4 h, maintaining the temperature at 5 °C. The mixture was then heated to 60 °C, and NaCl (5 g) was added to the mixture and stirred for 1 h. A solid precipitate was formed, which was filtered, and washed with minimum amount of NaCl (5% w/v). The solid was dried at a temperature of 80–90 °C and extracted with dimethylformaldehyde, DMF. The dye was precipitated by diluting the DMF-extract with excess chloroform; it was filtered, washed with chloroform and dried at 60 °C (Fig. 3).

CHA dye synthesis from diazotized anilines and the coupling components
2.4 Infrared spectroscopy of the synthesized dye
The IR absorption spectra of the dyes were acquired using FTIR- 8400S Fourier Transform Infrared Spectrophotometer in order to determine the functional groups present in the newly synthesized reactive dyes.
2.5 GC-MS spectroscopy of the synthesized dye
The GC-MS spectroscopy of the synthesized dye was carried out at National Research Institute for Chemical Technology (NARICT), Zaria, Nigeria using the GCMS-QP2010 PLUS Series in order to elucidate the molecular weights of the representative dyes.
2.6 Computational details
The ground state geometry optimization was conducted by means of B3LYP density functional theory (DFT) method with the 6–311 + G(d,p) basis set [21, 23]. This DFT computational approach has been widely used in the theoretical investigations of organic dyes for dye-sensitized solar cell applications because it is prompt and appropriate in recovery of dynamic electron correlations [22, 23]. Frequency calculations were also done with the same level of theory and none of the frequency calculations generated imaginary frequencies indicating that the optimized geometries are true energy minima. Vertical excitation energy calculations were done for the first 5 singlet states by using the TD-SCF and long-range CAM-B3LYP/6–311 + G(d,p) DFT method in gas phase [21, 24]. Electronic excitation energies were determined using the CAM-B3LYP Coulomb-attenuated functional, which provides by far the best overall performance; no correlation is observed between excitation energy errors and reflecting the good quality, balanced description of all categories of excitation energies [24]. All the computational calculations were accomplished with density functional theory (DFT) using the Gaussian 09 and GaussView 06 suite of programmes [25]. Surface and structural analysis were carried out with the Multiwfn 3.7 (dev) function analyzer [26] which can be freely obtained from Http/sobereva.com/multiwfn. The NBO computation was conducted using the in-built Gaussian 3.1 method available in Gaussian 09 software [25]. The NBO analysis of the second order perturbation energy was conducted by considering all possible and most significant interactions between filled donor and virtual acceptor orbitals through their energy contribution to all the ground states D-\(\pi\)-A systems.
The excitation study was carried out by deducing the hole and electron distribution in space, in order to identify the types of electron excitations for the first five (5) excitations. Some important indices (Sr, D, H, t, hole delocalization index and electron delocalization index) used in this study were calculated at CAM-B3LYP/6–311 + G(d,p).
Sr index defines the overlapping extent of hole and electron,
Centroid is determined to expose the most representative position of hole and electron distribution considering the x,y or z coordinates. For instance, X coordinate of centroid of electron is written as;
where x is X component of position vector r, ρhole and ρele are the densities of hole and electron respectively.
D is defined as the distance between the centroids of hole and electron (Chole and Cele). The length of charge transfer can be determined from the distance between centroid of electron and hole in different coordinates. For instance, Dx is given in Eq. 3., and is also applicable to y and z axis.
D index; the net magnitude of charge transfer (CT) length is given in Eq. 4.
H index is the overall measure of the degree of spatial extension of hole and electron distribution. It is calculated from the root mean square distribution of hole (|σhole |) and electron (|σele |);
t index measures the separation degree of hole and electron in CT direction. It is the arithmetic difference between of H in CT direction from D index. HDI and EDI are functions of hole and electron densities respectively, presented in Eq. 6 and 7.
All equations are adapted from Le Bahers et al. (2011).
3 Results and discussions
3.1 Fourier transform infrared spectroscopy (FTIR) results of the synthesized dyes.
The studied compound was obtained using aniline as diazo component, and cyanurated H-acid coupling component. The peak due to O–H group was at 3426 cm−1, SO3H at 615 cm−1, N=N peak at 1492 cm−1, C=C stretch at 1402 cm−1, C–N at 1184 cm−1, and N–H stretch at 1630 cm−1 as displayed in figure S1, when cyanurated H-acid was used with diazotized aniline. The experimental and theoretical results are summarized in Table 1.
3.2 Assessment of mass spectra
The spectra show the structures of the specimen in suitable solvents, (methanol in this experiment). For the purpose of interpretation, it is important to note that the molecular weight of the combined structures of each of the dyes goes well beyond the calibrated range of the GC-MS machine used; hence, the results are segmented. These different segments have to be combined to have a full structure of the samples. Also, the structures obtained are a representation of the chemical composition of the samples under study, providing the basic points and fragments. The studied compound as shown in Figure S2, with identified mass-to-charge ratio (m/z) are 27, 41, 57,106, and 284 corresponding respectively to C2H3, C3H5, C2H3N, C4H9, C7H9N and C18H36O2. The experimental value was found to be 515: M+, and the theoretical value of 517.13 g/mol with no much deviation of + 2. This increment might be due to isotopy in carbon atom.
3.3 Frontier molecular orbital (FMO) analysis
The HOMO-1, HOMO, LUMO and LUMO + 1 diagrams were plotted to give pictorial explanation of the energy gaps (Eg) and the distributions of the aforementioned orbitals. From Fig. 4, Eg is 2.95 eV, which is relatively low [27,28,29,30,31,32], hence excitation from the HOMO to LUMO is easily feasible; this is a basic property of a reactive molecule. Also, the energy gap between HOMO-1 and LUMO + 1 orbitals was determined for a more explicit information about the studied molecule; we obtained Eg to be 4.94 eV.

HOMO -1, HOMO, LUMO and LUMO + 1 diagrams of cyanurated H-acid with aniline (CHA) using RB3LYP method with 6-31G (d,p) basis set
Also, the locations and distributions of these orbitals (HOMO-1, HOMO, LUMO and LUMO + 1) were determined abreast the pictorial representations in Fig. 4, in order to understand the orbital properties of the valence orbitals [28]. The highest occupied molecular orbital (HOMO) is concentrated around C18, and it is basically from a p-type orbital on C18 (18.69%). The penultimate orbital to HOMO (HOMO-1) is identified to be distributed around C19 (10.99%) and C10 (8.59) atoms, it is also from a p-orbital. The lowest unoccupied molecular orbitals (LUMO) and LUMO + 1 are part of the receiving end on electron transition. The LUMO is contributed basically by a p-orbital around N12 (22.18%) and N13 (22.37%), which are good electron accepting regions of the molecule. It was established that the LUMO + 1 exist on the p-orbitals C21 (16.7%). It is inferable from the low Eg value of the studied compound, that the compound possesses good electronic chemistry.
3.4 Excitation studies
The first five excitations of the studied compound were studied in acetone, ethanol, water, and also in gas phase (no solvent) in order to understand the various interactions between molecular orbitals in its system at excited state. In this excitation study, we employed calculated D, Sr, H, t, HDI and EDI indices in the determination of the properties of different excitations. A combinational consideration of the various indices, coupled with the visualization of plotted isosurfaces for excitations provides a clear view of the various interactions [25,26,27].
The nature of excitation is defined by D values, which is deducible from the distance between the centroids of Choleand Cele. From Table 2, it is evident that the highest D values are recorded for S0 → S5 in all systems (3.92 Å for all solvents and 3.51 Å in no solvent system), which depicts a possible charge transfer (CT) excitation which can be visualized from the hole and electron isosurfaces, where there is high shift between blue (hole) and green (electron) lobes. Relatively average values of D (1.07 Å ≤ D ≤ 1.22 Å) are obtained for S0 → S3; this is also evident in the hole and electron isosurface diagrams, where the distance between the blue and green lobes are a bit obvious. S0 → S1, S0 → S2 and S0 → S4 can be proposed to be local or Rydberg excitations, owing to their relatively low D values (0.32 Å ≤ D ≤ 0.54 Å), (0.68 Å ≤ D ≤ 0.94 Å) and (0.64 Å ≤ D ≤ 0.65 Å) respectively. It is worthy to note the insignificance of the differences in the effect of solvents on the hole and electron separation distance, D values for various excitations in various media is closely dispersed.
The Sr index is a crucial parameter in the assignment of excitation types when studying the hole-electron interaction of a chemical system. The quantitative amount of hole and electron distribution in the available space is defined by Sr values [33,34,35,36]. From Table 2, it is observed that the least Sr values are recorded for S0 → S5, in all media; interpreted as an n → π* type excitation. Also, visualizing the hole and electron isosurface of S0 → S5 in Fig. 5, it is obvious that they are located around the benzene ring attached to the fused –N=N– group of the studied molecule. Relatively, higher values of Sr are recorded for S0 → S1 (0.66 a.u. in all media), S0 → S3 (0.65 a.u. in all media except water with 0.66 a.u.) and S0 → S4 (0.72 a.u. in all media except in no solvent with 0.74 a.u.), hence are regarded as localized π → π* excitations. The Sr isosurfaces of S0 → S1/S2/S3/S4 show that the hole and electron are distributed around the –N=N– group of the molecule.

Visualized hole and electron, hole and electron centroids, and Sr isosurfaces of cyanurated H-acid with aniline (CHA) usingRB3LYP method with 6-31G (d,p) basis set
H index is employed in the assignment of the breath of the approximate distribution of hole and electron of a particular excitation [33, 37]. No significant differences in the H indices were observed for the various excitations under consideration, as evident in Table 2. One can also study the separation of hole and electron for the various excitations by considering the polarity and magnitude of the t indices. Positive t index represents a significant hole and electron separation; hence CT type excitation is predictable for such systems. On the contrary, an extremely low separation between hole and electron counterparts yields a negative t value [35]. Inferring from Table 2, positive values of t indices are recorded for only S0 → S5 excitations in all studied media, showing that S0 → S5 is actually a CT type excitation. On the other hand, S0 → S1/S2/S3/S4 are perceived as local excitations (LE), owing to the negative values of their t indices.
HDI and EDI values directly expose the spatial distributions of the various excited hole and electron during the excitation process. Very high values of EDI and HDI are indicative of highly localized excitations, while delocalization is representative of low HDI and EDI values. We can infer from Table 2, that the hole and electron in S0 → S2 and S0 → S5 excitations are distributed at delocalized orientations, this is traceable to their very high HDI and EDI values in all media. In relation to the first five (5) excitations studied in this work, it is notable that S0 → S4 excitation is a highly localized π → π* type, considering the low HDI (5.25–5.30 in all media) and EDI (5.26–5.56 in all media) distributions. Fairly low values of HDI were obtained for S0 → S1 (6.53–7.01 range in all media) and S0 → S3 (5.51–5.55 in all media) excitations, which represents localized distributions.
Considering the effects of solvent on the first five (5) excitations of the studied compound, we observed that a little or no change occurred in the excitation mode when solvents are substituted for another. Close similarity is confirmed for acetone, ethanol and water as solvents [38]. This can be deduced from the almost unchanging values of D, Sr, H, and t indices for various excitation types with change in solvent. A significant change in excitation behaviour is noted when solvents are introduced to the isolated molecule of the studied compound, especially in the quantitative amount of hole and electron distribution (defined by the Sr index) as highlighted in Table 2.
The excitation behaviour of the studied compound from the hole and electron transition studies of the first five excitations is summarized in Table 3. This summary neglects the effect of solvent (because no significant contribution from the various solvents was recorded), while the distinctive values for the various determination indices are presented in Table 2.
3.5 Electrostatic potentials (ESP) studies
In order to understand the distribution of electronic density around the various regions of the studied compound, a plot of the ESP was carried out using the Gaussview software. In the plotted ESP, red colouration represents regions with high electron densities, blue colour is for regions with very minimal electron density distributions, while greenish-yellowish trend is indicative of a transition from a low electron density region (blue) to a high density region (red). From the obtained plot presented in Fig. 6, it can be visualized that high electron densities are distributed around the highly electronegative terminals at ground state [1].

The Electrostatic Potential (ESP) Diagram of Cyanurated H-acid with Aniline (CHA) using RB3LYP method with 6-31G (d,p) basis set
4 Natural bond orbital (NBO) analysis
NBO analysis has been established as a vital instrument, facilitating the interpretation of electronic structure calculations in a chemically intuitive approach. NBOs majorly consist of natural atomic hybrid orbitals which are orthonormal sets of localized orbitals with maximum occupancy (highest percentage of total electron density), which gives the most precise plausible natural Lewis structure. NBO analysis makes available substantial information regarding the nature of bonding orbitals, their occupancies as well as the type and nature of interaction (inter or intramolecular, hyper-conjugative or charge transfer) existing between virtual and occupied Lewis orbitals. The analysis is conducted by considering all probable interactions between donor (occupied Lewis NBOs) and acceptors (vacant) non-Lewis NBOs, and approximating their energetic significance by second order perturbation theory of Fock matrix [39]. These interactions are referred to as delocalization since they result from a decrease in electron density form the localized NBOs of the idealized Lewis structure and are promoted into the virtual non-Lewis orbitals. This decrease in electron density in the form of delocalization between occupied and vacant orbitals agrees with the interaction energy resulting in the system’s stabilization reported in the form of E(2) [40] estimated as;
where \(\hat{F}\) is the Fock operator, \(\varepsilon _{\sigma }\) and \(\varepsilon _{{\sigma *}}\) are the energies of the NBO and \(\eta _{{\sigma ~}}\) is the orbital occupancy.
The computed most significant second order perturbation energy of interactions between the donor and acceptor NBOs within and between the various ring fragments and substituents atoms of the studied compound along with their occupancy (percentage electron density over bonded atoms) is presented in Table 4. The intramolecular interactions which results from the orbital overlap between bonding (σ/π) and anti-bonding (σ*/π*) orbitals resulting in intramolecular charge transfer and system’s stabilization which are observed as increase in electron density in the C–C, N–N, nC antibonding orbitals are: the strong intramolecular interaction within fragment one between πC1–C6, πC4–C5 and π*C2–C3, π*C1–C6 has a significant stabilization energy of 19.43 and 19.28 kcal/mol respectively. The hyperconjugative interaction of πC4–C5 with adjacent acceptor (π*N12–N13) which has a stabilization energy of 19.30 kcal/mol serves as a link to extend the conjugation from the first ring fragment to the double ring fragment, such extended conjugation further enhances molecular stability and confirms the intense coloration of the molecule. The most important interaction within fragment 2 is the donation from πC21–C23 to n*C17 which has a stabilization energy of 40.88 kcal/mol and in the third fragment, the strongest hyperconjugative interaction in the form of \({\text{n}} \to {\text{n}} *\) and \({\text{n}} \to {\pi }*\) are observed between the heteroatoms (the terminal O27, the bridge N13, N28 and the sulphate O43, O44 etc.) and the antibonding carbon acceptors, these interactions give a strong stabilization energy of 87.43 kcal/mol for \({\text{n}}\left( {{\text{O27}}} \right) \to {\text{n}} * {\text{C15}}\), 67.53 kcal/mol from \({\text{n}}\left( {{\text{N}}28} \right) \to {{\pi }} * \left( {{\text{C}}32 - {\text{N}}33} \right)\) and are responsible for intermolecular charge transfer. Also, the strongest non-bonding interaction from \({\text{nC}}14~ \to {{\pi }} * \left( {{\text{N}}12 - {\text{N}}13} \right)\) and the antibonding to bonding lone pair \({\text{n*C}}15 \to {\text{nC}}16\) gives the highest stabilization as high as 127.60 and 1726.93 kcal/mol respectively; such strong interactions enhance the stability of the system. Since part of the lone pair electrons on the oxygen atoms of the sulphate fragment has been used for bonding (coordination with thering and the Sulphur atom) the lone pairs therefore could not give any strong interaction by donation but acted more as acceptors in the form of \({\text{n}} \to {\sigma } *\) transitions with minimal stabilization energies as reported in Table 4. The NBO analysis of the studied compound gives an intuitive perception about the resonance stabilization present in the dye molecule as revealed by the delocalization of electron density (intramolecular hyperconjugative interactions) from fragment one to the second double ring fragment through the bridge nitrogen atoms and extending the conjugation to the third ring via N24 signifying that these strong interactions confer enormous stabilization to the molecule. The result also shows that the probable transitions are \({\text{n}} \to {{\sigma }} *\), \({\text{n}} \to {\text{n}} *\), \({\text{n}} \to {{\pi }} *\) and \({{\pi }} \to {{\pi }} *\) which are confirmed from the UV-Vis spectroscopic analysis in Fig. 7.

Simulated UV-vis spectra of the studied compound calculated with B3LYP/6-31G(d,p) basis sets
The ultraviolet–visible (UV-vis) spectroscopic analysis of the studied compound computed using the TD-DFT at the B3LYP/631 G (d, p) level of theory in four different solvents for the first five excited state is being exploited in this paper. The TD-DFT is a well-established technique commonly used as a quantum mechanical method in treating the electronic excitation of large systems including organic and inorganic systems [41]; it is highly efficient and convenient due to its low computational cost. This technique is particularly accurate in predicting the excitation energies for singlet excitation mostly when the right functional is included in the exchange–correlation functional, hence, the utilization of the B3LYP hybrid functional. The selected level of theory is able to depict the geometries of the \(S0 \to S1\) ground and singlet excited state. For the optimized S0 geometry of the studied compound to be excited, five lowest singlet excitations were considered and the corresponding vertical excitation energy, oscillator strength and main configuration assignment as well as contribution to the excitation were extracted to Table 5 and the plot is depicted in Fig. 7. The absorption maxima in all the solvents utilized is being compared, the primary absorption maxima for the first excited state (\(S0 \to S1\)) occurs at 431.08, 431.12, 423.58, and 430.62 nm respectively in acetone, ethanol, gas phase and water respectively, because acetone, ethanol, and water are polar solvents, the absorption maxima are almost similar. However, the variation in λmax in gas phase is due to the variation in polarity of the solvent. These excitations (\({\text{S}}0 \to {\text{S}}1\)) result from the HOMO to LUMO transition and have the highest orbital contribution respectively and are assigned to \({{\pi }} \to {{\pi *}}\). The secondary vertical excitation with high oscillator strength and average molecular orbital contribution results from \({\text{H}} - 1 \to {\text{L~}}\) and \({\text{H}} \to {\text{L}} + 1\) in all the solvents and gas phases respectively and corresponds to (\({\text{S}}0 \to {\text{S}}3\)) and (\({\text{S}}0 \to {\text{S}}4\)) transitions, its absorption maxima appears at 330.09 nm, (acetone), 330.06 nm (ethanol), 329.96 nm (gas), 329.76 nm (water), for the (\({\text{S}}0 \to {\text{S}}3\)) excitation; and 291.05 nm (acetone), 291.07 nm (ethanol), 288.40 nm (gas), and 290.89 nm (water) in the case of (\({\text{S}}0 \to {\text{S}}4\)) transition. These transitions are assigned to \({\text{n}} \to {{\pi }}*\) transitions respectively.
S0 → S2 and \({\text{S}}0 \to {\text{S}}5\) transitions are considered as weak absorptions due to their low oscillator strength values and their corresponding spectra are characterized by a weak band located within 390.06 to 390.21 nm in solvents and 391.29 nm gas phase for \({\text{S}}0 \to {\text{S}}2{\text{~}}\) transition and 280.26 to 283.90 nm in both gas and solvents respectively for the \({\text{S}}0 \to {\text{S}}5\) excitation. These weak absorptions are characterized by low molecular orbital contribution from \({\text{H}} - 5 \to {\text{L}}\) in solvents and \({\text{H}} \to {\text{L}}\) in gas phase.
4.1 Density of State
The DOS and PDOS of the studied compound and the associated fragments; benzenediazonium, trichloro-triazine, and amino-hydroxynaphthalene sulfonic acid, are reported in Fig. 8. As demonstrated in Fig. 8a which is the TDOS and PDOS plot of the benzenediazonium fragment, s- and p-orbitals have the broad molecular orbitals (MOs) contribution in the energy range of − 0.55 ~ − 0.35 a.u. with p-orbital having the highest energy contributions. The dash line represents the HOMO energy of the fragment which corresponding to − 0.25454 a.u. having p-orbital as the major MO contribution to the HOMO energy of the fragment. The TDOS and PDOS of the 2,4,6-trichloro-1,3,5-triazine fragment is reported in Fig. 8b. Herein, the MOs contributions arise from p-orbital only and range from − 0.62 a.u. to the HOMO values at − 0.32 a.u. Similarly, the p-orbital MO contribution is maximum at the HOMO energy level. The 4-amino-5-hydroxynaphthalene sulfonic acid fragment TDOS and PDOS plot is shown in Fig. 8c. While p-orbital remains the primary contributing orbital of the MO, the s-orbital has a minimal contribution within energy range of − 0.65 ~ − 0.3 a.u. Surprisingly, none of the contributing orbitals (s- and p-orbitals) has contribution to the HOMO energy level having an energy of − 0.18882 a.u. Figure 8d depicts the TDOS and PDOS of the main studied compound. The p-orbital is observed to have the major contribution to the MOs having a broad energy range from − 0.60 a.u. and diminishes at the HOMO energy point of − 0.223 a.u.

Density of State (DOS) Plots for the s, p and d orbitals for a benzenediazonium fragment b 2,4,6-trichloro-1,3,5-triazine fragment c 4-amino-5-hydroxynaphthalene sulfonic acid fragment and d main studied compound using using RB3LYP method with 6-31G (d,p) basis set
5 Conclusion
(E)-5-((4,6-dichloro-1,3,5-triazin-2-yl)amino)-4-hydroxy-3-(phenyldiazenyl)naphthalene-2,7-diylbis(hydrogen sulfite) was successfully synthesized through a three (3) step (diazotization, coupling component preparation and finally, dye synthesis) process, and characterized using FT-IR and UV-Vis spectrophotometry, and mass spectrometry, from which the structure was deduced. DFT methods were employed in the computational studies of CHA in order to understand its electronic and spectra characteristics. From the FTIR analysis, the major functional groups figure S1 of the supporting information. The mass spectra show a very narrow range between the experimental value of 515 M + and theoretical value of 517 M + respectively. The electrostatic potential, ESP shows a very high electron density around the more electronegative terminals whereas there was a decrease in the electron density between the vacant orbitals. We can conclude from the low Eg obtained from calculations that CHA is very suitable for various optical and cellular applications (e.g. in the photovoltaic industry) like other dyes [38]. CHA also showed good light absorption properties, coupled with interesting stabilizing inter-fragment orbital interactions. From excitation studies, we observed that both local excitations and charge transfers occur about the azo group (–N=N–). From the results, one can see that CHA is a great asset in the organo-photo energy generation industry.
Data availability
Raw data are available, and can be obtained through request from the correspondence.
References
Shankarling GS, Deshmukh PP, Joglekar AR (2017) Process intensification in azo dyes. J Environ Chem Eng 5:3302–3308
Berradi M, Hsissou R, Khudhair M, Assouag M, Cherkaoui O, El Bachiri A, El Harfi A (2019) Textile finishing dyes and their impact on aquatic environs. Heliyon 5:e02711
McLaren K (1983) The colour science of dyes and pigments, Adam Hilger Ltd.
Gürses A, Açıkyıldız M, Günes K, Gürses MS (2016) Classification of dye and pigments. Dyes and Pigments. Springer, pp 31–45
Benkhaya S, El Harfi A (2017) Classifications, properties and applications of textile dyes: a review. Appl J Environ Eng Sci 3:00000–00003
Benkhaya S, M’rabet S, El Harfi A (2020) Classifications, properties, recent synthesis and applications of azo dyes. Heliyon 6(1):e03271
Şener İ, Şener N, Gür M (2018) Synthesis, structural analysis, and absorption properties of disperse benzothiazol-derivative mono-azo dyes. J Mol Struct 1174:12–17
Zhao XL, Qian HF, Huang W (2018) Construction of benzothiazole/pyridone based bi-heterocyclic dyes and their NiII and CuII complexes. Dyes Pigm 149:796–803
Ali A, Rafiq MI, Zhang Z, Cao J, Geng R, Zhou B, Tang W (2020) TD-DFT benchmark for UV-visible spectra of fused-ring electron acceptors using global and range-separated hybrids. Phys Chem Chem Phys 22(15):7864–7874
Fecková M, Kalis IK, Roisnel T, Le Poul P, Pytela O, Klikar M, Le Guen FR, Bureš F, Fakis M, Achelle S (2021) Photophysics of 9, 9-dimethylacridan-substituted phenylstyrylpyrimidines exhibiting long-lived intramolecular charge-transfer fluorescence and aggregation-induced emission characteristics. Chem A Eur J 27(3):1145–1159
Reibnegger G (2018) A DFT study on the one-electron reduction/oxidation of biologically relevant pteridine derivatives. ChemistrySelect 3(39):10925–10931
Omri N, Bu Y (2020) TD-DFT Studies on sp-and sp2-hybridized single vacancy-defected [60] fullerene: electronic excitation and nonlinear optical properties of C59 [9–4] and C59 [8–5] isomers. J Phys Chem A
Maliyappa MR, Keshavayya J, Mahanthappa M, Shivaraj Y, Basavarajappa KV (2020) 6-Substituted benzothiazole based dispersed azo dyes having pyrazole moiety: synthesis, characterization, electrochemical and DFT studies. J Mol Struct 1199:126959
Ahmed S, Kalita DJ (2020) End-capped group manipulation of non-fullerene acceptors for efficient organic photovoltaic solar cells: a DFT study. Phys Chem Chem Phys 22(41):23586–23596
Dutta R, Ahmed S, Kalita DJ (2020) Theoretical design of new triphenylamine based dyes for the fabrication of DSSCs: a DFT/TD-DFT study. Mater Today Commun 22:100731
Ezhov AV, Aleksandrov AE, Zhdanova KA, Zhdanov AP, Klyukin IN, Zhizhin KY, Mironov AF, Tameev AR (2020) Synthesis of Zn (II) porphyrin dyes and revealing an influence of their alkyl substituents on performance of dye-sensitized solar cells. Synth Met 269:116567
El-Mansy MA, Suvitha A, Ibrahim M (2021) Quantum chemical studies on structural, spectroscopic, thermochemistry, photo-physical and bioactivity properties of m-cresol purple dye
Osman OI (2017) DFT study of the structure, reactivity, natural bond orbital and hyperpolarizability of thiazole azo dyes. Int J Mol Sci 18(2):239
Shinde S, Sekar N (2019) Synthesis, spectroscopic characteristics, dyeing performance and TD-DFT study of quinolone based red emitting acid azo dyes. Dyes Pigm 168:12–27
Ramugade SH, Warde US, Sekar N (2019) Azo dyes with ESIPT core for textile applications and DFT study. Dyes Pigm 170:107626
Divya VV, Suresh CH (2020) Density functional theory study on the donating strength of donor systems in dye-sensitized solar cells. New J Chem 44(17):7200–7209
El Mzioui S, Bouzzine SM, Sidir İ, Bouachrine M, Bennani MN, Bourass M, Hamidi M (2019) Theoretical investigation on π-spacer effect of the D–π–A organic dyes for dye-sensitized solar cell applications: a DFT and TD-BH and H study. J Mol Model 25(4):92
Tripathi A, Ganjoo A, Chetti P (2020) Influence of internal acceptor and thiophene based π-spacer in DA-π-A system on photophysical and charge transport properties for efficient DSSCs: a DFT insight. Sol Energy 209:194–205
Yanai T, Tew DP, Handy NC (2004) A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem Phys Lett 393(1–3):51–57
Trucks GW, Frisch MJ, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta Jr. JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowskiand J, Fox DJ (2013) Gaussian09, Revision D. 01, Gaussian, Inc., Wallingford CT
Le Bahers T, Adamo C, Ciofini I (2011) A qualitative index of spatial extent in charge-transfer excitations. J Chem Theory Comput 7(8):2498–2506
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33(5):580–592
Edim MM, Enudi OC, Asuquo BB, Louis H, Bisong EA, Agwupuye JA, Chioma AG, Odey JO, Joseph I, Bassey FI (2021) Aromaticity indices, electronic structural properties, and fuzzy atomic space investigations of naphthalene and its aza-derivatives. Heliyon 7(2):06138
Lai TY, Guo JD, Fettinger JC, Nagase S, Power PP (2019) Facile insertion of ethylene into a group 14 element-carbon bond: effects of the HOMO–LUMO energy gap on reactivity. Chem Commun 55(3):405–407
Shimizu A, Ishizaki Y, Horiuchi S, Hirose T, Matsuda K, Sato H, Yoshida JI (2020) HOMO–LUMO energy-gap tuning of π-conjugated zwitterions composed of electron-donating anion and electron-accepting cation. J Org Chem
Parvathy G, Kaliammal R, Velsankar K, Kumar MK, Sankaranarayanan K, Sudhahar S (2020) Studies on structural, optical, homo-lumo and mechanical properties of piperazinium p-hydroxybenzoate monohydrate single crystal for nonlinear optical applications. Chem Phys Lett 758:137934
Bisong EA, Louis H, Unimuke TO, Odey JO, Ubana EI, Edim MM, Tizhe FT, Agwupuye JA, Utsu PM (2020) Vibrational, electronic, spectroscopic properties, and NBO analysis of p-xylene, 3, 6-difluoro-p-xylene, 3, 6-dichloro-p-xylene and 3, 6-dibromo-pxylene: DFT study. Heliyon 6(12):05783
Liu Z, Lu T, Chen Q (2020) An sp-hybridized all-carboatomic ring, cyclo [18] carbon: electronic structure, electronic spectrum, and optical nonlinearity. Carbon 165:461
Kitagawa Y, Asaoka M, Miyagi K, Matsui T, Nihei M, Oshio H, Okumura M, Nakano M (2015) DFT and TD-DFT studies of electronic structures and one-electron excitation states of a cyanide-bridged molecular square complex. Inorg Chem Front 2(8):771–779
Mallah R, Sreenath MC, Chitrambalam S, Joe IH, Sekar N (2018) Excitation energy transfer processes in BODIPY based donor-acceptor system-Synthesis, photophysics, NLO and DFT study. Opt Mater 84:795–806
Zhang Y, Shen C, Lu X, Mu X, Song P (2020) Effects of defects in g-C3N4 on excited-state charge distribution and transfer: potential for improved photocatalysis. Spectrochim Acta Part A Mol Biomol Spectrosc 227:117687
Liu Z, Hua S, Wu G (2018) Extended first hyperpolarizability of quasi-octupolar molecules by halogenated methylation: whether the iodine atom is the best choice. J Phys Chem C 122(37):21548–21556
Odey JO, Louis H, Agwupuye JA, Moshood YL, Bisong EA, Brown OI (2021) Experimental and theoretical studies of the electrochemical properties of mono azo dyes derived from 2-nitroso-1-naphthol, 1-nitroso-2-naphthol, and CI disperse yellow 56 commercial dye in dye-sensitive solar cell. J Mol Struct 1241:130615
Dunnington BD, Schmidt JR (2012) Generalization of natural bond orbital analysis of periodic systems: applications to solids and surfaces via plane-wave density functional theory. J Chem Theory Comput 8:1902–1911
KariyottuKuniyil MJ, Pandmanaban R (2019) theoretical insights into the structural, photophysical and Nonlinear optical properties of Phenoxazin-3-one Dyes. New J Chem 43:13616
Laurent AD, Adamo C, Jacquemin D (2014) Dye chemistry with time-dependent density functional theory. Phys Chem Chem Phys 16:14334–14356
Acknowledgements
The authors want to acknowledge Louis Hitler, the head of the Computational and Bio-simulation research group for his supervision and visionary role in the activities of this group. Also, to our academic colleagues, for their support and prayers which led to the success of this job.
Author information
Authors and Affiliations
Contributions
HL; conceived and supervised the project and ensured the completion of this manuscript. OCE; carried out the frontier molecular orbital and excitation studies and wrote part of the manuscript. TOU; carried out the DOS and NBO analyses, and wrote some part of the manuscript. JOO; carried out the ESP analysis and reviewed this manuscript after the compilation process. ATI; carried out the synthesis and characterization of CHA. Tabe N. Ntui; helped in the synthesis of CHA and the characterizations as well. IBO; compiled and reviewed this manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Louis, H., Enudi, O.C., Odey, J.O. et al. Synthesis, characterization, DFT, and TD-DFT studies of (E)-5-((4,6-dichloro-1,3,5-triazin-2-yl)amino)-4-hydroxy-3-(phenyldiazenyl)naphthalene-2,7-diylbis(hydrogen sulfite). SN Appl. Sci. 3, 712 (2021). https://doi.org/10.1007/s42452-021-04688-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-021-04688-0